

Abstract
NOD2 (nucleotide-binding oligomerization domain-containing protein 2) is an internal pattern recognition receptor that recognizes bacterial peptidoglycan and stimulates host immune responses. Dysfunction of NOD2 pathway has been associated with a number of autoinflammatory disorders. To date, direct inhibitors of NOD2 have not been described due to technical challenges of targeting the oligomeric protein complex. Receptor interacting protein kinase 2 (RIPK2) is an intracellular serine/threonine/tyrosine kinase, a key signaling partner, and an obligate kinase for NOD2. As such, RIPK2 represents an attractive target to probe the pathological roles of NOD2 pathway. To search for selective RIPK2 inhibitors, we employed virtual library screening (VLS) and structure based design that eventually led to a potent and selective RIPK2 inhibitor 8 with excellent oral bioavailability, which was used to evaluate the effects of inhibition of RIPK2 in various in vitro assays and ex vivo and in vivo pharmacodynamic models.
Keywords: RIPK2, NOD2, kinase inhibitors, structure-based drug design
Activation of NOD2 in myeloid cells by pathogenic bacteria and genetic variants in NOD21 lead to increased cytokine secretion suggesting that a NOD2 inhibitor would be beneficial for autoinflammatory disorders, such as inflammatory arthritis,2,3 early onset sarcoidosis, Blau Syndrome,4−7 multiple sclerosis,8 and allergic inflammation.9 However, NOD2 has been an elusive target for pharmacological inhibition for many years. Therefore, downstream targets of the pathway can provide an alternative approach for modulating NOD2 pathway activity. Receptor-interacting protein kinase 2 (RIPK2, RICK, or CARDIAK), a key downstream modulator of NOD2 signaling possessing serine/threonine and tyrosine kinase activity, has been recently discovered.10−13 Upon intracellular bacterial exposure, NOD2 binds to RIPK2 and triggers nuclear factor κB (NF-κB) mediated cytokine responses. Thus, RIPK2 represents a critical mediator of NOD2 signaling and becomes a promising target for developing novel chemical inhibitors. A number of agents have been described to inhibit RIPK2, including Gefitinib,13 SB203580,14,15 WEHI-345,16 OD36 and OD38,17 Ponatinib,18 and recently GSK58319 (Figure 1). Among these reported RIPK2 inhibitors, the EGFR tyrosine kinase inhibitor Gefitinib and the p38 inhibitor SB203580 were chemical tools used to demonstrate the function of RIPK2 in NOD pathways but lacked kinase specificity. WEHI-345, OD36, and OD38 were reported to be more selective RIPK2 inhibitors. WEHI-345 was shown to specifically delay NF-κB activation upon NOD stimulation and consequently prevent cytokine production in vitro and in vivo. OD36 demonstrated inhibition of recruitment of inflammatory cells in an in vivo peritonitis model. In a separate study,18 three FDA approved drugs, Ponatinib, Sorafenib, and Regorafenib, also showed promising results in terms of inhibiting RIPK2 and reducing inflammation. In the same study, the crystal structure of RIPK2 complexed with Ponatinib revealed an allosteric site that holds promise for the identification of more selective inhibitors. GSK583 was reported to be a potent, selective, and bioavailable RIPK2 inhibitor. It demonstrated the role of RIPK2 in modulation of intestinal inflammation in both murine and human systems. Collectively, these findings identified RIPK2 as a promising target for the treatment of inflammatory diseases in which NOD2:RIPK2 activity is aberrantly high (i.e., sarcoidosis, Blau syndrome, asthma, multiple sclerosis, and Crohn’s disease when patients do not harbor the loss-of-function mutation). Herein we describe a novel RIPK2 inhibitor, which is potent, selective, and bioavailable. The RIPK2 inhibitor identified here significantly decreased pro-inflammatory cytokine secretion in both cellular and in vivo models. In addition, we describe phosphorylation of ribosomal protein S6 in monocytes as a result of NOD2 activation, an event attenuated upon RIPK2 inhibition. Hence, phosphorylation of S6 could be potentially used as a biomarker for predicting the activity of the RIPK2 inhibitors in patients.
Figure 1.

Selected small molecule RIPK2 inhibitors.
To search for novel selective RIPK2 inhibitors, a virtual library screen (VLS) was performed with ∼11 million commercial and Novartis proprietary compounds using a 2D profile QSAR method developed at Novartis20 and a 3D pharmacophore search. Hits (∼100,000) were then evaluated by flexible docking (Glide, Schrödinger, Inc.) to an in-house obtained RIPK2 protein structure. Approximately 10,000 hits with best docking scores were then clustered and analyzed. A subset from VLS was tested for RIPK2 inhibition in a RIPK2 HTRF ADP transcreener assay.21 Various scaffolds with IC50 ranging from nanomolar to micromolar were identified.
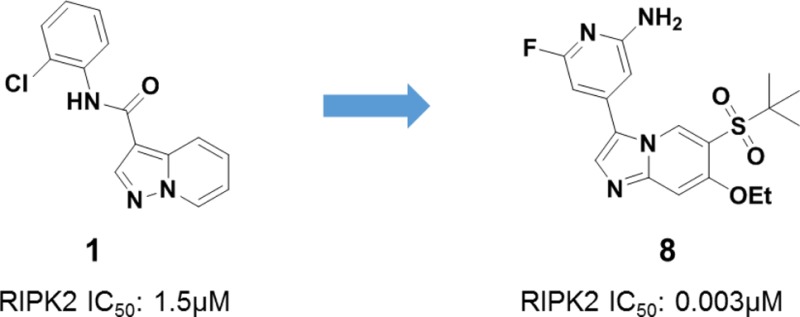
After further evaluation of the hits, including ligand efficiency (LE), lipophilic efficiency (LiPE), kinase selectivity, and binding mode, compound 1 was considered for further characterization (Figure 2A). Although it was a compound with micromolar activity (IC50 = 1.5 μM), it exhibited attractive ligand efficiency (LE = 0.32) and lipophilic efficiency (LiPE = 3.5). In addition, the cocrystal structure revealed compound 1 bound to the ATP binding site with DFG motif adopting a DFG-in conformation (Figure 2B), highlighting compound 1 as a type I inhibitor with clearly defined interactions with the protein. As shown in Figure 2B, there is a single hinge interaction between N1 of compound 1 and the amide from Met98 in the hinge region of RIPK2. The gatekeeper Thr95-OH acts as a H-bond donor to form a H-bond with the backbone carbonyl of Glu96 and a H-bond acceptor to form a H-bond with the amide NH of compound 1. The phenyl ring is twisted out of plane by ∼50° and fits into the back pocket with the chlorine atom occupying a small hydrophobic pocket. Importantly, it was noted that the region around the 5- and 6-positions of the molecule was open and near the unique Ser25 of RIPK2. We speculated that chemical modification around this region could potentially impact potency and kinase selectivity.
Figure 2.
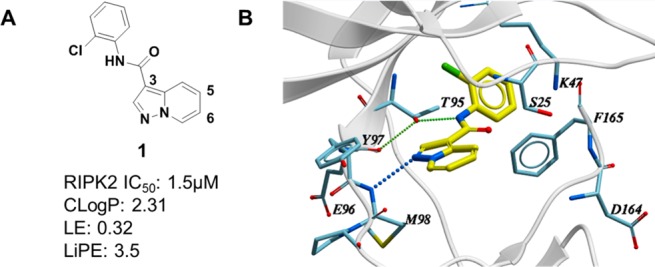
(A) VLS confirmed hit 1. (B) Cocrystal structure of compound 1 in RIPK2 kinase domain (2.9 Å resolution). Hydrogen bonds are indicated by dashed lines.
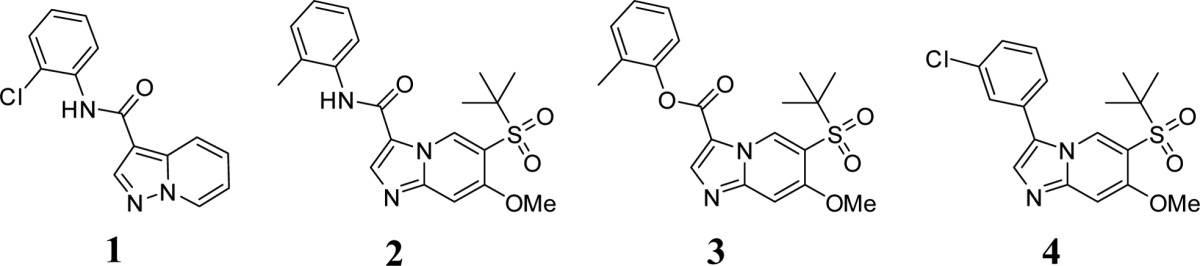
Early structure–activity relationship (SAR) led to the discovery of compound 2. By adding a t-butyl sulfone group at the 5-position and a methoxy group at the 6-position, the potency of 2 was dramatically improved. Meanwhile, the metabolic stability (liver microsomal extraction ratio) was also improved by changing the core from pyrazolopyridine to imidazopyridine. Evaluation of 2 in a small in-house enzymatic kinase profiling panel revealed activity on many other kinases, especially the Src family of kinases, such as Lck and Lyn (Table 1).
Table 1. Early SAR Evolution.
| compd | RIPK2 ADP transcreener IC50 (μM)* | Lyn/Lck IC50 (μM)* | liver microsomal ER h/m |
|---|---|---|---|
| 1 | 1.5 | NA/9.2 | 0.92/0.99 |
| 2 | 0.024 | 0.05/0.13 | <0.21/0.61 |
| 3 | 0.038 | 0.15/0.51 | 0.44/0.86 |
| 4 | 0.09 | 9.7/NA | 0.80/0.88 |
Data are the mean of three measurements, SEM < ±30%.
Further analysis of early SAR identified compound 3, an ester analogue of 2. Although compound 3 can not act as H-bond donor to form H-bond interaction with gate keeper Thr95, its potency was still comparable to compound 2. This implied that, whereas this H-bond interaction may not determine compound 3 activity on RIPK2, it did contribute to kinase target specificity. Indeed, compound 3 activity on Lck and Lyn was significantly reduced (∼3-fold). Compound 4, with a simple C–C bond replacing the amide bond, was thus designed and synthesized. As expected, compound 4 demonstrated potent inhibitory activity on RIPK2, while its activities over Src family kinases were significantly reduced (Table 1). A docking study of compound 4 in RIPK2 ATP binding site (Supporting Information, Figure SI-4) indicated that the chlorobenzene moiety occupied the back pocket region differently when compared to the hit compound 1, which might contribute to the improved kinase selectivity. A systematic SAR study of the imidazolepyridine core was carried out to optimize potency (Supporting Information for synthetic protocols), as well as ADME-PK properties for in vivo investigations.
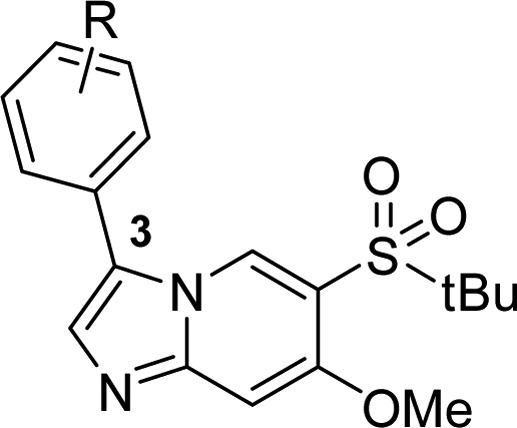
As listed in Table 2, a series of aromatic derivatives at the 3-position with different substitution patterns and electronic properties were prepared to probe the back pocket. It was noted that ortho substitutions consistently reduced activity (5a, 5c, and 5f), presumably, the larger torsional angle resulting from the ortho substitutions as well as its clash with the gatekeeper side chain kept the benzene ring from fitting optimally into the back pocket. Electron donating groups seemed more favorable than the electron withdrawing groups (5d vs 4, 5g and 5k). Both meta- and para-substituted analogues in general showed more favorable activities, and the two phenol analogues (5i and 5j) demonstrated the best RIPK2 activities. Building upon these important findings, a series of disubstituted analogues were generated to see if there is a synergistic effect (5l to 5p). We were pleased to see that the disubstituted analogues were much more potent than their monosubstituted counterparts.
Table 2. SAR Exploration at the 3-Position*.
| compd | R | RIPK2 ADP transcreener IC50 (μM)** | liver microsomal ER m/h |
|---|---|---|---|
| 5a | 2-Cl | >10 | NA |
| 4 | 3-Cl | 0.09 | NA |
| 5b | 4-Cl | 0.52 | NA |
| 5c | 2-MeO | 0.46 | NA |
| 5d | 3-MeO | 0.04 | NA |
| 5e | 4-MeO | 0.13 | NA |
| 5f | 2-CONH2 | >10 | NA |
| 5g | 3-CONH2 | 0.43 | NA |
| 5h | 4-CONH2 | 0.16 | NA |
| 5i | 3-OH | 0.035 | 0.84/0.34 |
| 5j | 4-OH | 0.016 | 0.74/0.4 |
| 5k | 3-CN | 0.18 | NA |
| 5l | 3-Cl-4-CONH2 | 0.07 | NA |
| 5m | 3-MeO-4-OH | 0.011 | 0.82/0.48 |
| 5n | 3-Cl-4-OH | 0.005 | 0.83/0.56 |
| 5o | 3-Cl-5-MeO | 0.045 | NA |
| 5p | 3-Cl-5-OH | 0.009 | 0.90/0.67 |
Refer to Supporting Information for synthetic protocols.
Data are the mean of three measurements, SEM < ±30%.
Although compound 5n and 5p showed single digit nanomolar enzymatic activity, these phenols demonstrated poor metabolic stability (Table 2) and low oral exposure (5n, F% = 2; 5p, F% = 0) in mouse pharmacokinetics (PK) studies, presumably due to their susceptibility to oxidation and phase II metabolism.22 A bioisostere approach was then executed to search for replacement of the phenols in these potent analogues (Table 3, Supporting Information for synthetic protocols). It was found that the 6-indazole (6a) and the 5-benzimidazole (6c) analogues were much less potent compared to their phenol counterparts (5i and 5j); 5-indazole (6b) was equally potent to its phenol analogue 5j. However, the reduced cellular activity (MDP-stimulated human peripheral blood mononuclear cell (hPBMC) IL8 secretion assay) and poor metabolic stability (liver microsomal ER in mouse, rat and human = 0.95, 0.83, and 0.5, respectively) indicate that 6b is not an ideal phenol replacement either. We were encouraged to see that both the 3-N-acyl-phenyl (6d) and the 2-aminopyridin-4-yl (6e) derivatives exhibited good biochemical and cellular potency as well as metabolic stability. In addition, consistent to synergistic effects observed from 5l to 5p, introducing meta-substitution in the same aromatic ring (6g and 6h) further boosted the potency by 5–10-fold. Compound 6h, with the best metabolic stability (liver microsomal ER in mouse, rat, and human = 0.63/0.49/0.51), was then chosen for PK study in mice. The compound demonstrated good bioavailability (81%), but relatively high clearance (52 mL/min/kg).
Table 3. Bioisosteres for Phenol Replacement*.
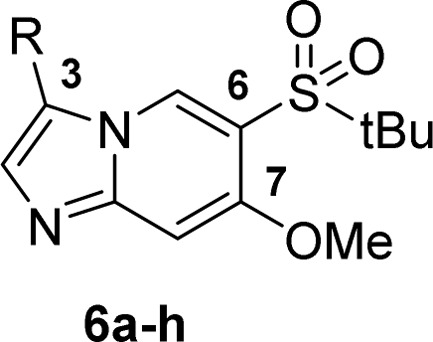
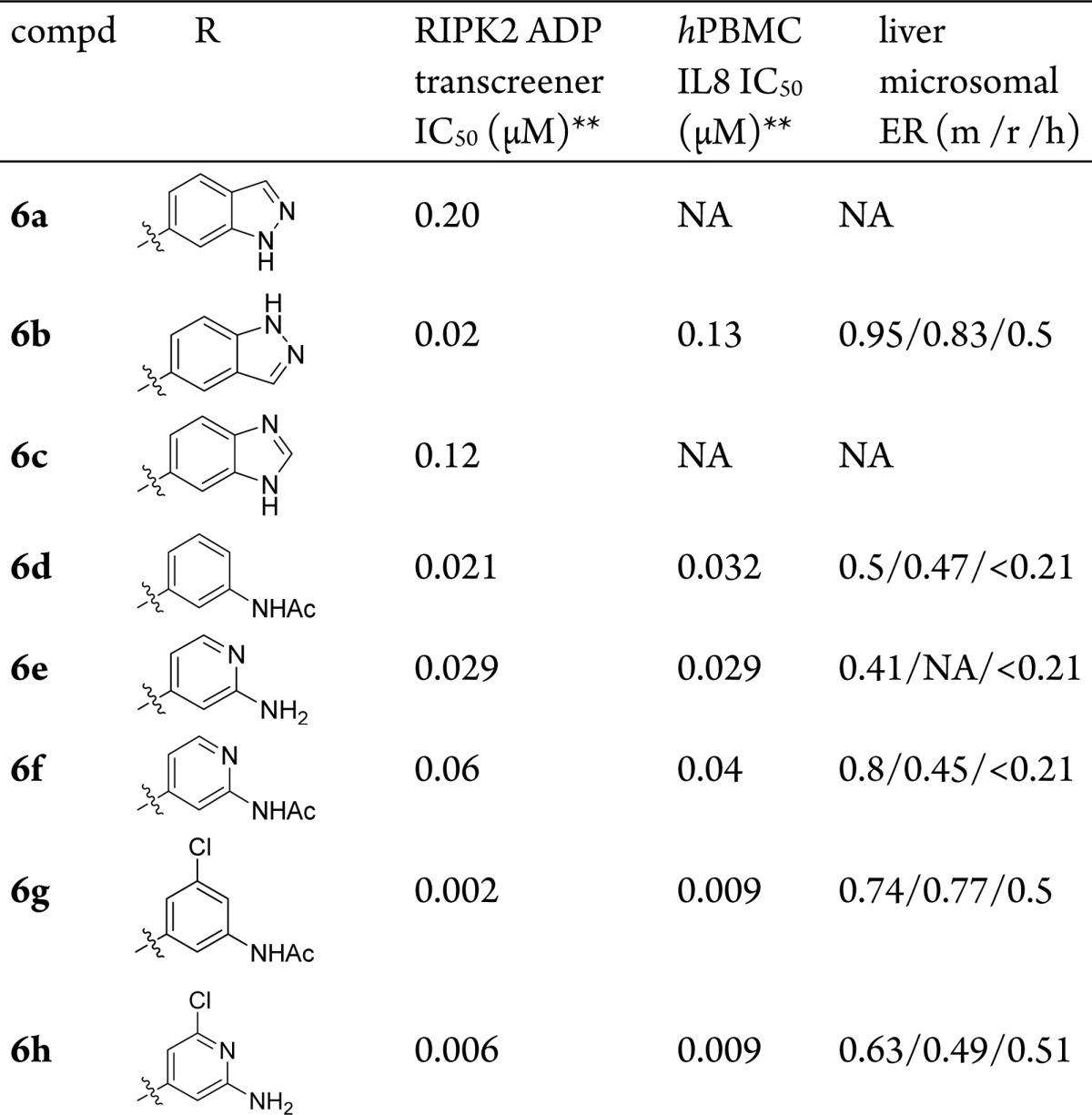
Refer to Supporting Information for synthetic protocols.
Data are the mean of three measurements, SEM < ±30%.
To further optimize the metabolic stability and pharmacokinetic profile, a metabolite identification study was conducted on compound 6h in rodent and human liver microsomes. The study revealed that O-demethylation at position 7, as well as oxidation of the aromatic core and at the t-butyl group were the primary metabolic pathways. Optimization was then carried out accordingly.
As exemplified in Table 4, to overcome the O-demethylation metabolic pathway, the methoxy group was replaced with various alkoxy groups (Supporting Information for synthetic protocols). All analogues illustrated in Table 4 demonstrated comparable RIPK2 enzymatic activity except the cyclopropyl analogue 7b, probably due to the unique π character of cyclopropane C–C bond as compared to normal alkanes. Different metabolic stabilities were observed, with compounds 7d and 7f improving the most and 7a maintaining a similar profile to 6h. Both 7c and 7e showed reduced metabolic stability across species. Murine PK studies of these compounds revealed that, despite the good in vitro metabolic stability (liver microsomal ER m/h = 0.49/0.55), 7f showed extremely high clearance in vivo (109 mL/min/kg). Moderate improvement of clearance was observed for compounds 7a and 7d.
Table 4. Optimization for metabolic stability and clearance*.

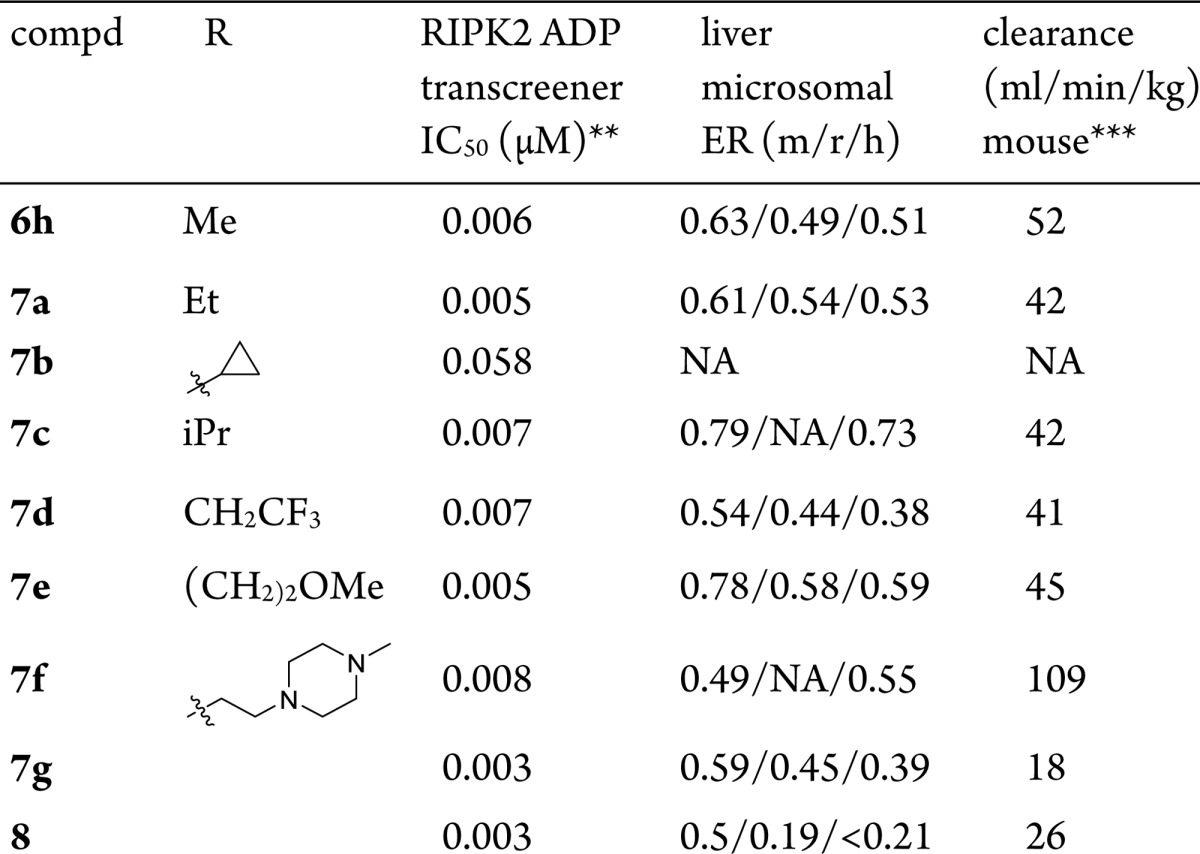
Refer to Supporting Information for synthetic protocols.
Data are the mean of three measurements, SEM < ±30%.
Data obtained from mouse in vivo PK study.
X-ray cocrystal structures of 7f complexed to the kinase domain of RIPK2 was obtained (Figure 3), albeit with relatively low resolution of 2.9 Å. Similar to compound 1, compound 7f binds to ATP binding site of the kinase and forms single hinge interaction with Met98. There is a hydrogen bond interaction between the sulfone group and the hydroxyl side chain of the RIPK2’s unique Ser25 residue; another visible interaction is the hydrogen bond between the pyridyl amino group and the side chain of catalytic residue Asp16,4 which is involved in the hydrogen bond networks with Lys47 and Gly66. We speculated that the hydrogen bond with the pyridyl amino group contributes to the binding activity, while the orientation of the aromatic ring in the back pocket impacts the kinase selectivity. The piperazine side chain extends toward the edge of the ATP binding pocket and is exposed to solvent, which explains that modification around this region exhibits no impact on binding affinity (7a to 7f).
Figure 3.
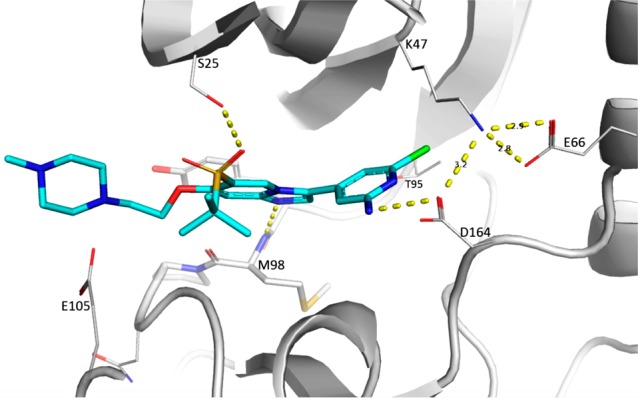
Cocrystal structure of 7f with RIPK2 kinase domain (2.9 Å resolution). Hydrogen bonds are indicated by dashed lines.
Combining the cocrystal structure information with the results from the metabolic identification study, we proposed further strategies to minimize core oxidation. One strategy was to reduce electron density of the pyridine ring while intensifying the H-bond interaction with Asp164. This strategy was exemplified by compound 7g. By replacing Cl with F in the 3-pyridinyl ring of 7a, 7g improved the potency, the metabolic stability (liver microsomal ER m/r/h = 0.59/0.45/0.39), and in vivo clearance (18 mL/min/kg).
A second strategy for further optimization was to replace the metabolically labile t-butyl sulfone group with an isopropyl sulfone group. Compound 8 was obtained from this effort, which not only retained the activity of 7g but also demonstrated a better metabolic stability profile.
A PK study revealed that compound 8 exhibited low clearance and good oral exposure in mice. Moreover, it displayed low clearance and high oral bioavailability in rats and dogs as well (Table 5). The compound also demonstrated dose proportionality from 10 to 300 mg/kg in rat PK studies (data not shown). Kinase selectivity of 8 was confirmed by a broad kinase panel profiling (Nanosyn 250 kinases kinome profile, Supporting Information). At 5 μM, 90% percent of kinases showed <50% inhibition, while at 0.5 μM, 98% of kinases showed <50% inhibition. All of these findings highlighted compound 8 as a promising tool compound for mechanistically investigating the role of RIPK2 inhibition in vivo.
Table 5. PK Parameters of 8 in Mouse, Rat, and Dog.
| parameters | mouse | rat | dog |
|---|---|---|---|
| dosage (iv/po, mg/kg) | 2.0/10.0 | 5.0/20.0 | 0.5/2.0 |
| CL (mL/min/kg) | 26.1 | 12.6 | 7.8 |
| Vss(L/kg) | 1.1 | 1.6 | 2.0 |
| T1/2 (h) | 1.0 | 3.2 | 5.0 |
| AUC (h·nM) iv | 3377 | 17539 | 2844 |
| AUC (h·nM) po | 7951 | 91278 | 15391 |
| Cmax (nM) po | 3886 | 14218 | 2343 |
| oral BA (%F) | 47.1 | 130.1 | 135.3 |
Accordingly, we next evaluated the role of RIPK2 in IL-6 secretion by using mouse bone marrow derived macrophages (mBMDM). mBMDM were pretreated with the corresponding RIPK2 inhibitors for 30 min, then stimulated with 5 μg/mL of muramyl dipeptide (MDP, a known activator of the NOD2/RIPK2 pathway) and a very low concentration of lipopolysaccharides (LPS, 1 ng/mL. Under this concentration, LPS alone did not stimulate IL6 secretion; however, it was necessary to stimulate a significant IL6 signal when combined with MDP). Compound 7e and 8 effectively suppressed the secretion of IL6 in a dose-dependent manner with IC50 of 6 and 12 nM, respectively (Figure 4A). In addition, compound 8 demonstrated inhibition of TNFα secretion in both rat and human whole blood stimulated by MDP (Figure 4B,C).
Figure 4.

RIPK2 inhibitor suppresses cytokines secretion. (A) Compounds 7e and 8 suppress MDP stimulated IL6 secretion in mouse BMDM. (B) Compounds 7e and 8 suppress MDP-stimulated TNFα secretion in rat whole blood. (C) Compound 8 suppresses MDP-stimulated IL8 secretion in human whole blood.
In an attempt to identify downstream targets for RIPK2, we employed a mass cytometry method known as CyTOF (Figure 5, see Supporting Information for method details). First, we treated human peripheral blood mononuclear cell (hPBMC) with MDP or LPS and identified a number of proteins that were phosphorylated, including ERK1/2, p38, and S6. We observed increased phosphorylation of the above proteins specifically in monocytes and dendritic cells. S6 was the protein that showed the most profound phosphorylation upon MDP and LPS treatment. Pretreatment of hPMBC with the RIPK2 inhibitor 7e for 15 min blocked S6 phosphorylation only when the cells were stimulated with MDP but not LPS, suggesting a selective effect of the RIPK2 inhibitor on pS6 modulation when the NOD2 pathway is activated.
Figure 5.
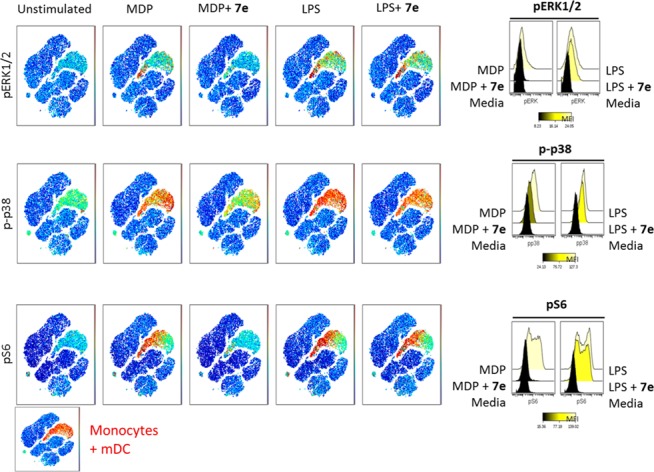
Human PBMC CyTOF experiments. Human PBMC were treated with MDP and LPS, respectively; increased phosphorylation for ERK1/2, p38, and S6 was observed in monocytes and dendritic cells. RIPK2 inhibitor 7e blocks phosphorylation of ERK1/2, p38, and S6 induced by MDP and not by LPS.
We also confirmed the inhibition of phosphorylation of S6 in hPBMC by FACS (Supporting Information, Figure SI-1), further supporting specificity for the NOD2:RIPK2 pathway. We observed that both 7e and 8 significantly reduced phosphorylation of S6 only upon MDP treatment.
Assessment of the effect of 8 in a rat ex vivo PK/PD model of cytokine secretion is shown in Figure 6A. Female Wistar rats were dosed with 8 at 30 mg/kg. Blood was collected at different time points (0, 0.5, 3, 5, and 24 h) and challenged ex vivo with MDP. It was observed that blood samples treated with 8 significantly attenuated MDP-induced cytokine secretion at all time points.
Figure 6.

RIPK2 inhibitor 8 suppresses cytokine release in rat ex vivo and in vivo models. (A) Rats (n = 6/group) were dosed with 8 at 30 mg/kg, blood was collected at different time points (0, 0.5, 3, 5, and 24 h), plasma concentration of 8 at different time points is shown in the left panel; MDP was added in plasma samples of each time point and incubated for 23 h to show that 8 inhibits MDP-stimulated cytokine (IL6 and TNFα) release in all time points. (B,C) Rats (n = 6/group) were dosed with either vehicle or 8 at 30 mg/kg followed by intraperitoneal injection of MDP. Colon (B) was collected 4 h post-MDP stimulation for measurement of cytokines (IL6, IL8, and TNFα) secretion. Blood (C) was collected 4 h post-MDP stimulation for measurement of cytokines (IL6, IL8, and TNFα) secretion. Error bars represent the standard deviation of six animals.
In order to demonstrate pharmacodynamic activity in target tissues to treat inflammatory bowel diseases (IBD) such as blood and gut, rat in vivo challenge studies were carried out with pretreatment of 8 at 30 mg/kg followed by intraperitoneal injection of MDP. Blood and colon were collected 4 h post-MDP treatment for measurement of cytokine secretion. As illustrated in Figure 6, compound 8 treatment significantly reversed MDP-induced proinflammatory cytokines in rat colon (Figure 6B). In blood (Figure 6C), we observed moderate but significant reduction in IL6 and TNFα.
In summary, we have identified structurally distinct, potent, and selective RIPK2 inhibitors through extensive structure based design and SAR exploration. Compound 8 was tested in multiple in vitro assays and in vivo models to demonstrate its activity in suppressing cytokine secretion upon activation of the NOD2:RIPK2 pathway. In addition, we described activation of the NOD2 pathway led to a profound increase in phosphorylation of S6 in monocytes, and this event was reversed by RIPK2 inhibitor 8. These findings will definitely encourage additional investigations for validation and could potentially provide a convenient biomarker for RIPK2 activity.
Acknowledgments
The authors would like to thank Paul Calvin and his team (compound management), David Jones (NMR), John Isbell and Todd Groessl (DMPK), as well as Jennifer Shaffer for assistance with crystallization experiments, Ananda Herath for flow chemistry consultation, and staff at Advanced Light Source, BL503 (Berkeley, CA), for beamline assistance. We thank Bryan Laffitte, Jon Loren, and Ignacio Sancho-Martinez for critical reading of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00258.
Experiment details are provided for the preparation of 4, 5a–p, 6a–6h, 7a–g, and 8, analytical data, kinome profile data for compound 8, X-ray crystallography experiments of compound 1 and 7f, docking study of 4, and experimental details for in vitro assay and in vivo animal models (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fritz J. H.; Ferrero R. L.; Philpott D. J.; Girardin S. E. NOD-like Proteins in Immunity, Inflammation and Disease. Nat. Immunol. 2006, 7, 1250–1257. 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- Rosenzweig H. L.; Jann M. J.; Vance E. E.; Planck S. R.; Rosenbaum J. T.; Davey M. P. Nucleotide-binding Oligomeri-zation Domain 2 and Toll-like Receptor 2 Function Independently in a Murine Model of Arthritis Triggered by Intraarticular Peptidoglycan. Arthritis Rheum. 2010, 62, 1051–1059. 10.1002/art.27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig H. L.; Clowers J. S.; Nunez G.; Rosenbaum J. T.; Davey M. P. Dectin-1 and NOD2 Mediate Cathepsin Activation in Zymosan-Induced Arthritis in Mice. Inflammation Res. 2011, 60, 705–714. 10.1007/s00011-011-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henckaerts L.; Vermeire S. NOD2/CARD15 Disease Associations Other Than Crohn’s Disease. Inflamm. Bowel Dis. 2007, 13, 235–241. 10.1002/ibd.20066. [DOI] [PubMed] [Google Scholar]
- Kanazawa N.; Okafuji I.; Kambe N.; Nishikomori R.; Nakata-Hizume M.; Nagai S.; Fuji A.; Yuasa T.; Manki A.; Sakurai Y.; Nakajima M.; Kobayashi H.; Fujiwara I.; Tsutsumi H.; Utani A.; Nishigori C.; Heike T.; Nakahata T.; Miyachi Y. Early-onset Sarcoidosis and CARD15 Mutations with Constitutive Nuclear Factor-KappaB Activation: Common Genetic Etiology with Blau Syndrome. Blood 2005, 105, 1195–1197. 10.1182/blood-2004-07-2972. [DOI] [PubMed] [Google Scholar]
- Okafuji I.; Nishikomori R.; Kanazawa N.; Kambe N.; Fujisawa A.; Yamazaki S.; Saito M.; Yoshioka T.; Kawai T.; Sakai H.; Tanizaki H.; Heike T.; Miyachi Y.; Nakahata T. Role of the NOD2 Genotype in the Clinical Phenotype of Blau Syndrome and Early-onset Sarcoidosis. Arthritis Rheum. 2009, 60, 242–250. 10.1002/art.24134. [DOI] [PubMed] [Google Scholar]
- Schurmann M.; Valentonyte R.; Hampe J.; Muller-Quernheim J.; Schwinger E.; Schreiber S. CARD15 Gene Mutations in Sarcoidosis. Eur. Respir. J. 2003, 22, 748–754. 10.1183/09031936.03.00040602. [DOI] [PubMed] [Google Scholar]
- Shaw P. J.; Barr M. J.; Lukens J. R.; McGargill M. A.; Chi H.; Mak T. W.; Kanneganti T. D. Signaling via the RIP2 Adaptor Protein in Central Nervous System-Infiltrating Dendritic Cells Promotes Inflammation and Autoimmunity. Immunity 2011, 34, 75–84. 10.1016/j.immuni.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W.; Mehta A. K.; Magalhaes J. G.; Ziegler S. F.; Dong C.; Philpott D. J.; Croft M. Innate Signals from NOD2 Block Respiratory Tolerance and Program Th2-Driven Allergic Inflammation. J. Allergy Clin. Immunol. 2010, 126, 1284–1293. 10.1016/j.jaci.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N.; del Peso L.; Koseki T.; Chen S.; Nunez G. RICK, a Novel Protein Kinase Containing a Caspase Recruitment Domain, Interacts with CLARP and Regulates CD95-Mediated Apoptosis. J. Biol. Chem. 1998, 273, 12296–12300. 10.1074/jbc.273.20.12296. [DOI] [PubMed] [Google Scholar]
- Thome M.; Hofmann K.; Burns K.; Martinon F.; Bodmer J. L.; Mattmann C.; Tschopp J. Identification of CARDIAK, a RIP-like Kinase That Associates with Caspase-1. Curr. Biol. 1998, 8, 885–888. 10.1016/S0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- McCarthy J. V.; Ni J.; Dixit V. M. RIP2 Is a Novel NF-κB-activating and Cell Death-inducing Kinase. J. Biol. Chem. 1998, 273, 16968–16975. 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- Tigno-Aranjuez J. T.; Asara J. M.; Abbott D. W. Inhibition of RIP2′s Tyrosine Kinase Activity Limits NOD2-driven Cytokine Responses. Genes Dev. 2010, 24, 2666–2677. 10.1101/gad.1964410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argast G. M.; Fausto N.; Campbell J. S. Inhibition of RIP2/RICK/CARDIAK Activity by Pyridinyl Imidazole Inhibitors of p38 MAPK. Mol. Cell. Biochem. 2005, 268, 129–140. 10.1007/s11010-005-3701-0. [DOI] [PubMed] [Google Scholar]
- Hollenbach E.; Neumann M.; Vieth M.; Roessner A.; Malfertheiner P.; Naumann M. Inhibition of p38 MAP Kinase- and RICK/NF-κB-signaling Suppresses Inflammatory Bowel Disease. FASEB J. 2004, 18, 1550–1552. 10.1096/fj.04-1642fje. [DOI] [PubMed] [Google Scholar]
- Nachbur U.; Stafford C. A.; Bankovacki A.; Zhan Y.; Lindqvist L. M.; Fiil B. K.; Khakham Y.; Ko H. J.; Sandow J. J.; Falk H.; Holien J. K.; Chau D.; Hildebrand J.; Vince J. E.; Sharp P. P.; Webb A. I.; Jackman K. A.; Muhlen S.; Kennedy C. L.; Lowes K. N.; Murphy J. M.; Gyrd-Hansen M.; Parker M. W.; Hartland E. L.; Lew A. M.; Huang D. C.; Lessene G.; Silke J. A RIPK2 Inhibitor Delays NOD Signalling Events yet Prevents Inflammatory Cytokine Production. Nat. Commun. 2015, 6, 6442. 10.1038/ncomms7442. [DOI] [PubMed] [Google Scholar]
- Tigno-Aranjuez J. T.; Benderitter P.; Rombouts F.; Deroose F.; Bai X.; Mattioli B.; Cominelli F.; Pizarro T. T.; Hoflack J.; Abbott D. W. In Vivo Inhibition of RIPK2 Kinase Alleviates Inflammatory Disease. J. Biol. Chem. 2014, 289, 29651–29664. 10.1074/jbc.M114.591388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P.; Ruan Q.; Schwerd T.; Hrdinka M.; Maki J. L.; Saleh D.; Suebsuwong C.; Ray S.; Brennan P. E.; Cuny G. D.; Uhlig H. H.; Gyrd-Hansen M.; Degterev A.; Bullock A. N. Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem. Biol. 2015, 22, 1174–1184. 10.1016/j.chembiol.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile P. A.; Votta B. J.; Marquis R. W.; Bury M. J.; Mehlmann J. F.; Singhaus R. Jr.; Charnley A. K.; Lakdawala A. S.; Convery M. A.; Lipshutz D. B.; Desai B. M.; Swift B.; Capriotti C. A.; Berger S. B.; Mahajan M. K.; Reilly M. A.; Rivera E. J.; Sun H. H.; Nagilla R.; Beal A. M.; Finger J. N.; Cook M. N.; King B. W.; Ouellette M. T.; Totoritis R. D.; Pierdomenico M.; Negroni A.; Stronati L.; Cucchiara S.; Ziolkowski B.; Vossenkamper A.; MacDonald T. T.; Gough P. J.; Bertin J.; Casillas L. N. The Identification and Pharmacological Characterization of 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine (GSK583), a Highly Potent and Selective Inhibitor of RIP2 Kinase. J. Med. Chem. 2016, 59 (10), 4867–4880. 10.1021/acs.jmedchem.6b00211. [DOI] [PubMed] [Google Scholar]
- Martin E.; Mukherjee P.; Sullivan D.; Jansen J. Profile-QSAR: A Novel meta-QSAR Method that Combines Activities across the Kinase Family to Accurately Predict Affinity, Selectivity, and Cellular Activity. J. Chem. Inf. Model. 2011, 51, 1942–1956. 10.1021/ci1005004. [DOI] [PubMed] [Google Scholar]
- Hong L.; Quinn C. M.; Jia Y. Evaluating the Utility of the HTRF Transcreener ADP Assay Technology: A Comparison with the Standard HTRF Assay Technology. Anal. Biochem. 2009, 391, 31–38. 10.1016/j.ab.2009.04.033. [DOI] [PubMed] [Google Scholar]
- Gao S.; Hu M. Bioavailability Challenges Associated with Development of Anti-Cancer Phenolics. Mini-Rev. Med. Chem. 2010, 10, 550–567. 10.2174/138955710791384081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.