

Abstract
Inhibition of mutant IDH1 is being evaluated clinically as a promising treatment option for various cancers with hotspot mutation at Arg132. Having identified an allosteric, induced pocket of IDH1R132H, we have explored 3-pyrimidin-4-yl-oxazolidin-2-ones as mutant IDH1 inhibitors for in vivo modulation of 2-HG production and potential brain penetration. We report here optimization efforts toward the identification of clinical candidate IDH305 (13), a potent and selective mutant IDH1 inhibitor that has demonstrated brain exposure in rodents. Preclinical characterization of this compound exhibited in vivo correlation of 2-HG reduction and efficacy in a patient-derived IDH1 mutant xenograft tumor model. IDH305 (13) has progressed into human clinical trials for the treatment of cancers with IDH1 mutation.
Keywords: Mutant IDH1, inhibition of 2-HG production, in vivo anticancer activity, brain penetration, clinical candidate
Isocitrate dehydrogenase 1 (IDH1) is a NADP+, metal-dependent oxidoreductase active in cellular metabolism.1−5 Wild-type IDH1 catalyzes the oxidative decarboxylation of isocitrate to give α-ketoglutarate (α-KG), while NADP+ is converted to NADPH. Heterozygous mutations in IDH1 at Arg132 have been tightly linked to various cancers including glioma, glioblastoma, AML, chondrosarcoma, and cholangiocarcinoma.6−9 This gain-of-function mutant reduces α-KG to R-2-hydroxyglutarate (2-HG) resulting in significantly elevated levels of this metabolite.10 Recently, both preclinical11−17 and clinical18,19 inhibition of mutant IDH1 have shown efficacy in various cancers, potentially offering a new treatment option to many patients with IDH1 mutation.
Previously, we have reported hit-to-lead optimization efforts to identify a mutant specific inhibitor of IDH1.20 Compound 1 (IDH889) was shown to bind to an allosteric, induced-fit pocket of IDH1R132H with potent inhibition of 2-HG production as well as selectivity over the wild-type protein. Oral dosing of 1 in a mouse xenograft model demonstrated robust in vivo reduction of 2-HG tumor tissue levels in engineered HCT116 colon carcinoma cells expressing mutant IDH1R132H. While 1 is a potent and selective inhibitor that modulates 2-HG in xenograft models, it showed relatively high in vitro intrinsic clearance (Clint) across different species (rat/mouse/dog/human Clint 588/143/548/205 μL min–1 mg–1), high plasma protein binding (rat/mouse/human plasma protein binding 97/98/98%), and poor solubility (39 μM at pH 6.8), which we anticipated to entail a very high human efficacious dose (>10 g BID). These considerable challenges to clinical development required additional optimization to identify a viable clinical candidate. Efforts focused on maintaining mutant IDH1 potency while optimizing the overall in vitro profile and subsequent translation to in vivo activity. The high unmet medical needs for glioma and glioblastoma patients justified additional selection criteria to identify inhibitors with potentially efficacious brain exposure.
Initial modifications were focused on the amine side chain. It was rationalized that increased polarity may reduce the brain penetration observed with 1,20 whereas reduction of the polarity would increase the lipophilicity, resulting in increased clearance and higher predicted dose for efficacy. Concomitant maintenance of the optimized biaryl system of 1(20) necessitated either retaining two nitrogens in the first aryl ring and none in the second or transposing one nitrogen from the pyrimidine ring to the second ring to give a bipyridyl moiety. Pyrazine 2 was identified as a tolerated replacement for the 2,5-pyrimidine ring, but this did not attenuate the high Clint or improve the solubility (<5 μM at pH 6.8). Alternative six-membered ring systems containing two nitrogens as the internal aryl ring gave significant loss of potency. The crystal structure of 1(20) showed a hydrogen bond between one of the ortho nitrogens of the pyrimidine to Ser278, so one ortho nitrogen was maintained in the first pyridine ring for subsequent exploration of bipyridyl systems. While terminal rings without a meta-substituent lost potency, the various 3,2′-, and 3,3′-bipyridyl systems exhibited increased Clint in rat liver microsomes. However, the 3,4′-bipyridine analogue 3 lost about 10-fold cellular potency compared to 1 but showed significant reduction of Clint in rat and improvement of solubility (430 μM at pH 6.8). Modifications of the meta-substituent on the terminal ring were explored to regain the activity. Larger substituents generally led to improved potency but also increased Clint in rat likely due to the increased lipophilicity. For example, the bulky tert-butyl group of 4 increased both biochemical and cellular potency, however, at the expense of increased rat Clint. The CF3 group provided the best balance of potency and in vitro clearance, as shown with compound 5. Reducing Clint in rat liver microsomes through this stage of the optimization resulted in an ∼8-fold increase of AUC after a single oral dose of 5 in rats at 10 mg/kg when compared to 1 (1.6 vs 0.2 μM·h, respectively). Mutant IDH1 inhibitory potency was further boosted by installation of a methyl at the 2- or 4-position of the internal pyridine ring (6 and 8, respectively), but not at the 5-position (7) without significant impact on rat Clint.
With the best amine side chain in-hand (8), the C(4) substituent of the oxazolidinone was evaluated to determine whether it could be used as a handle to reduce the clearance further. Truncation of the isopropyl to a methyl (9) and ethyl (10) reduced rat Clint effectively. However, this came with a loss of potency compared to the isopropyl 8. Increasing polarity by the addition of a hydroxy to 10 resulted in reduced cellular potency without any further improvement of metabolic stability as shown with 11. The addition of a fluorine to 9 enhanced potency (12) while maintaining low in vitro clearance. A further boost in potency was observed with 13 (IDH305). Having previously established chirality preference on C(4) of the oxazolidinone and the S-amine,20 we examined a C(1′)-R diastereomer (14) and found that 14 lost not only potency in both biochemical and cellular assays but also rat microsomal stability. Overall, 13 demonstrated an improved balance of mutant IDH1 inhibitory potency, rat in vitro clearance, and solubility (130 μM at pH 6.8) compared to 1. We also observed lower plasma protein binding of compound 13 (rat/mouse/human 83/88/83%) than that of 1, which we attributed to reduction of logD at pH 7.4 (1, 3.4, vs 13, 2.8). While similar optimization with 6 also resulted in reduced clearance, overall cellular potency of the analogues was not retained (data not shown).
Synthesis of 13 is described in Scheme 1. Reduction of methyl ester 15 and cyclization followed by PMB protection gave oxazolidinone 16 in 96% yield over two steps. Removal of tert-butyl and conversion of the resulting hydroxy into a fluorine using perfluoro-1-butanesulfonyl fluoride and NEt3(HF)3 provided 17 efficiently.22 Removal of PMB and subsequent SNAr reaction with 2,4-difluoropyrimidine yielded 18 in 59% overall yield. Amine 19 was protected with Boc and subjected to palladium-catalyzed cross-coupling reaction with (2-(trifluoromethyl)pyridin-4-yl)boronic acid,23 following deprotection of the amine to give 20. Coupling of 18 and 20 in DIPEA and DMSO provided 13 in 28% yield.
Scheme 1. Synthesis of 13 (IDH305).

Compound 13 was evaluated for specificity against mutant IDH1 relative to wild-type (WT) IDH1 in a biochemical assay measuring consumption (mutant) or production (WT) of NADPH.20,21 Compound 13 exhibited greater than 200-fold selectivity for mutant IDH1 isoforms vs WT (IDH1R132H IC50, 0.027 μM; IDH1R132C IC50, 0.028 μM; IDH1WT IC50, 6.14 μM). This potent in vitro biochemical activity correlated with efficient 2-HG reduction in IDH1R132H/+ cells. Furthermore, in the IDH1R132H/+ cell line, which depends on IDH1R132H activity for growth in EGF-depleted conditions,21 2-HG inhibition was correlated with inhibition of EGF-independent proliferation with an IC50 of 0.020 μM (Figure 1). The effects of 13 on proliferation are not due to off-target activity, as 13 had no effect on the EGF-independent growth of a mutant PIK3CA-dependent derivative of MCF10A at doses up to 10 μM (maximum dose tested). Compound 13 was not effective at lowering 2-HG concentration in engineered HCT116 cells with IDH2 mutation of R172K and R140Q (IC50 10 and 3.8 μM, respectively), demonstrating selectivity for mutant IDH1 vs IDH2 (Table 1).
Figure 1.
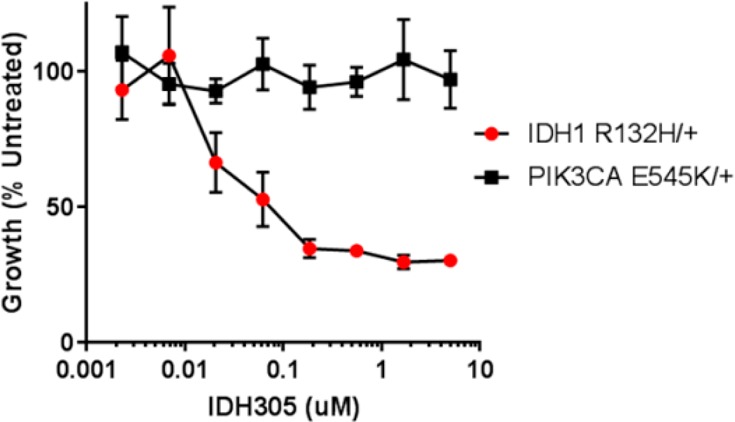
Cell growth inhibition of MCF10A-IDH1R132H/+ by 13 (IDH305) (mean ± SD).
Table 1. Biochemical, Cellular Potency, and Clint in Rat Liver Microsomes.

| compd | biochemical IDH1R132Hhomodimer IC50 (μM) | cellular HCT116-IDH1R132H/+ IC50 (μM) | rat liver microsomal Clint (μL min–1 mg–1) |
|---|---|---|---|
| 1 | 0.020 | 0.014 | 588 |
| 2 | 0.062 | 0.047 | 641 |
| 3 | 0.339 | 0.145 | 68 |
| 4 | 0.004 | <0.002 | 235 |
| 5 | 0.077 | 0.041 | 101 |
| 6 | 0.015 | 0.013 | 141 |
| 7 | 0.084 | 0.067 | 128 |
| 8 | 0.013 | 0.014 | 125 |
| 9 | 0.116 | 0.258 | 36 |
| 10 | 0.035 | 0.042 | 58 |
| 11 | 0.033 | 0.102 | 84 |
| 12 | 0.028 | 0.069 | 27 |
| 13 | 0.018 | 0.024 | 45 |
| 14 | 0.073 | 0.316 | 157 |
An X-ray structure of 13 bound to IDH1R132H homodimer revealed similar interactions as observed with 1 in the allosteric binding pocket and subsequent stabilization of the catalytically inactive conformation (Figure 2).20,21 Consistent with 1, the aminopyrimidine moiety forms a pair of hydrogen bonds with the backbone atoms of Ile128, while the carbonyl of the oxazolidinone forms a hydrogen bond with the backbone amide of Leu120, and the alpha-methyl fits into the methyl nook. Compound 13 demonstrated a hydrophobic collapse conformation in which the fluoroethyl is in van der Waals contact with the pyridine of the amine side chain. The bipyridine moiety adopts a similar trajectory as the phenyl-pyrimidine of 1, extending out toward the dimer interface. The regulatory segment (α10) is folded next to 13, with the side chain of Ser278 positioned in the vicinity of the ortho nitrogen of the pyridine ring, potentially forming a hydrogen bond.
Figure 2.
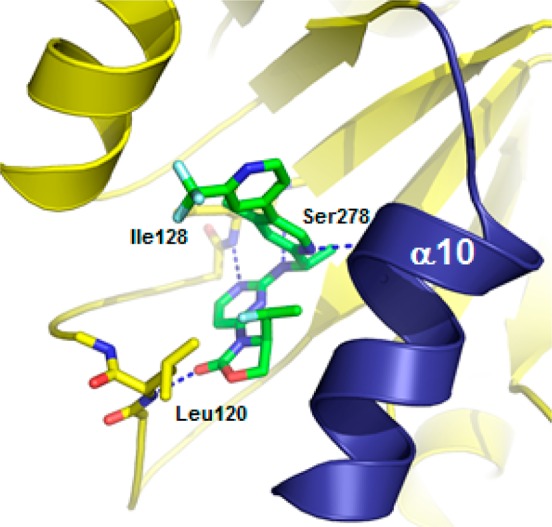
X-ray cocrystal structure of 13 (IDH305) in IDH1R132H homodimer (PDB 6B0Z).
Pharmacokinetic profiling of 13 across multiple species was performed, including assessment of brain penetrant exposure (Table 2). Moderate clearance both in vitro and in vivo was observed in all species examined. In response to a single 10 mg/kg oral dose in rats, plasma exposure of 13 (AUC 6.2 μM·h) was improved ∼30-fold compared to 1. The brain to plasma (total/unbound) ratio of 13 in mouse was 0.29/0.17 at 1 h post-oral dosing at 30 mg/kg. Following intravenous administration of 13 at 5 mg/kg in rats, the brain to plasma exposure ratio was 0.61, suggesting 13 is brain penetrant (AUCbrain 3.4 μM·h vs AUCplasma 5.5 μM·h). High passive permeability of compound 13 in both an artificial membrane (PAMPA calculated %FA: 99.3%) and a cellular system (Caco-2 Papp(B-A)/(A-B): 23/19) seemed sufficient to limit the impact of the p-glycoprotein driven efflux mechanism (MDR1-MDCK efflux ratio 4.9) and demonstrate rodent brain exposure.
Table 2. In Vitro and in Vivo PK Parameters of 13 (IDH305).
| PK parameter | mouse | rat | dog | human |
|---|---|---|---|---|
| in vitro liver microsomal Clint (μL min–1 mg–1) | 61 | 45 | 28 | 56 |
| in vitro predicted liver microsomal Cl (mL min–1 kg–1) | 65 | 33 | 18 | 14 |
| in vivo plasma clearance (mL min–1 kg–1) | 29 | 34 | 24 | n.d.a |
| plasma protein binding (%) | 88 | 83 | 89 | 83 |
| brain homogenate binding (%) | 93 | 95 | n.d. | n.d. |
| brain to plasma ratio (total/unbound) | 0.29/0.17 | 0.61/0.18 | n.d. | n.d. |
n.d., not determined
Compound 13 was profiled in the HCT116-IDH1R132H/+ mechanistic xenograft model to evaluate whether robust in vivo inhibition of 2-HG production could be observed. Following a single oral dose of 200 mg/kg, AUC48h and Cmax of 13 were 149 μM·h and 16.2 μM, respectively. Mouse plasma protein binding of 88.4% was used to approximate unbound plasma AUC48h of 17.3 μM·h and Cmax of 1.88 μM, which suggested unbound plasma concentrations above the HCT116-IDH1R132H/+ cellular IC50 for greater than 12 h. This resulted in reduction of tumor 2-HG concentration (Figure 3A) with maximal 2-HG inhibition of 87.2 ± 1.5% (mean ± SEM) relative to the vehicle treated tumors 8 h post-treatment (Table SI-1). Tumor 2-HG levels recovered above baseline by 48 h post-treatment. The relationship between unbound plasma exposure of 13 and percent inhibition of 2-HG suggested an estimated unbound EC50 value of 0.02–0.03 μM, similar to the in vitro IC50 of 0.024 μM (Figure 3B). This was consistent with our previous observation that inhibition of 2-HG production was correlated with unbound plasma drug concentration. Additional dose escalation studies demonstrated dose-dependent exposure and 2-HG inhibition (Figure 3C) and further supported our PK/PD model. Pharmacokinetic parameter (AUC, Cmax) values were not calculated for this study due to insufficient data points. Notably, 300 mg/kg dosed orally (po) achieved greater than 95% 2-HG production inhibition 12 h post-treatment (Table SI-2). These data suggested a BID dosing regimen would achieve maximal and sustained 2-HG inhibition.
Figure 3.
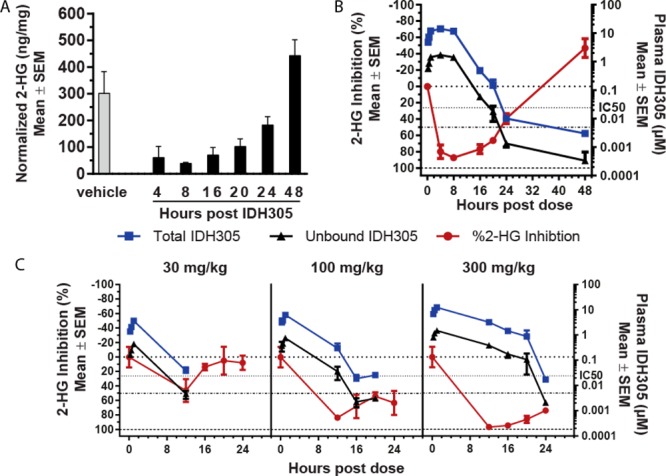
2-HG reduction in 13 (IDH305) treated HCT116-IDH1R132H/+ xenograft tumor tissue. (a) Tumor 2-HG levels following a 200 mg/kg dose. (b) Percent 2-HG inhibition in tumors relative to vehicle treatment in correlation with total and calculated unbound 13 plasma concentration following a 200 mg/kg dose. (c) Percent 2-HG inhibition in tumors in correlation with total and calculated unbound 13 plasma concentration following a 30, 100, or 300 mg/kg dose.
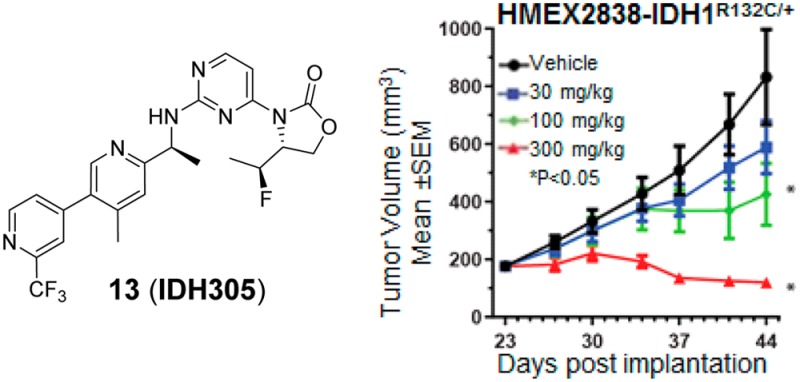
To determine if the exposure-dependent 2-HG reduction observed in the mechanistic HCT116-IDH1R132H/+ model would be similar in an endogenous IDH1R132C or IDH1R132G mutant tumor model, we evaluated 13 induced 2-HG suppression in two patient derived xenograft (PDX) melanoma tumor models propagated in nude mice.24 In the IDH1R132C mutant HMEX2838 PDX model, 13 led to dose-dependent 2-HG inhibition, with greater than 98% maximal inhibition for 16–20 h following a single 300 mg/kg dose (Figure 4A). Plasma exposures were comparable to those observed in the HCT116-IDH1R132H/+ model (Table SI-3), with AUC8h of 10.1 μM·h, AUC8h of 71.9 μM·h, and AUC24h of 212 μM·h following doses of 30, 100, and 300 mg/kg, respectively. Similarly, in the IDH1R132G mutant HMEX2700 melanoma PDX model, 13 at 100 and 300 mg/kg achieved maximal 2-HG inhibition 8 h post-treatment of 92% and 97% relative to vehicle treatment, respectively (Figure SI-1 and Table SI-4).
Figure 4.
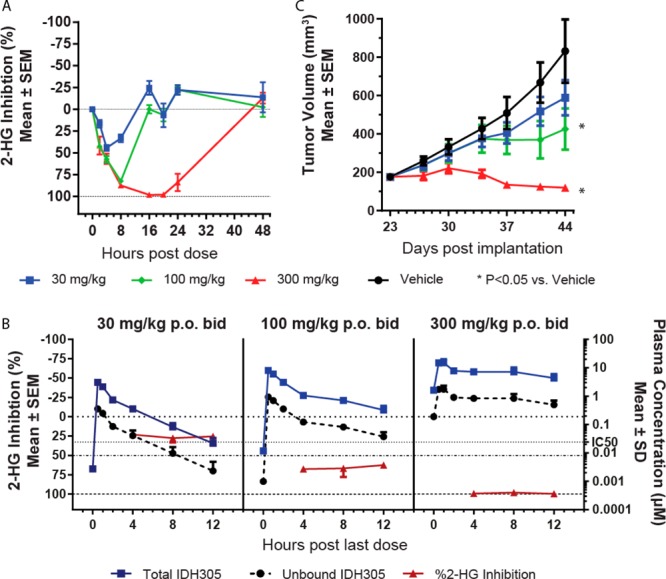
In vivo 2-HG reduction and efficacy of 13 (IDH305) in HMEX2838-IDH1R132C/+. (a) Percent 2-HG inhibition in tumors relative to vehicle treatment by single dose of 13 at indicated doses. (b) Percent 2-HG inhibition in tumors in correlation with total and unbound 13 plasma concentration following 30, 100, or 300 mg/kg BID for 21 days. (c) Tumor volume of HMEX2838 xenograft tumors treated as in (b).
We next evaluated 2-HG reduction in the IDH1R132C HMEX2838 PDX model in a multidose PK/PD/efficacy study. Compound 13 was well tolerated for 21 days of treatment, with no body weight loss at 30, 100, or 300 mg/kg po BID (Figure SI-2). Exposures at the steady state were reduced relative to single dose treatment (Table SI-5), with AUC12h of 5.4 19.2, and 88.9 μM·h at doses of 30, 100, and 300 mg/kg, respectively. Despite reduced exposure, dose-dependent inhibition of 2-HG levels in tumor tissue was achieved (Figure 4B) and correlated with antitumor activity (Figure 4C). A dose of 30 mg/kg po BID resulted in 22–25% 2-HG reduction and no significant tumor growth inhibition (%T/C = 63%). In contrast, 100 mg/kg po BID resulted in 62–67% 2-HG reduction and significant antitumor activity (%T/C = 38%, P < 0.05), consistent with the robust PD observed after a single dose (Figure 3C). Furthermore, 300 mg/kg of 13 dosed po BID resulted in 97–99% 2-HG reduction and partial tumor regression of 32% (P < 0.05). As the HMEX2838 model was propagated by in vivo tumor passage, an in vitro IC50 for 2-HG inhibition for this model was not determined. It is notable that in both the engineered HCT116-IDH1R132H/+ and the endogenously mutant PDX xenograft models, maintenance of unbound plasma concentration of 13 above the in vitro IC50 for the engineered model (0.024 μM) was required to achieve greater than 50% 2-HG inhibition in vivo. This observation suggested that the engineered HCT116-IDH1R132H/+ was useful for predicting efficacious unbound plasma concentrations required to inhibit 2-HG in solid tumor models.
Based on PK/PD studies of 13 in the HCT116-IDH1R132H/+ mechanistic xenograft mouse model above, maintaining unbound concentrations of 13 above the cell IC50 range (0.024 μM) was anticipated to be sufficient for inhibition of 2-HG. Human pharmacokinetics was predicted from in vitro and preclinical in vivo data based on allometric scaling approaches.25 With predicted clearance of 7.9 mL min–1 kg–1, Vdss of 3.8 L kg–1, and an oral bioavailability value of 70% (average of mouse, rat, dog, and monkey values), the resulting efficacious dose of 13 for a 70 kg human was predicted to be 500 mg QD or 150 mg BID to maintain unbound concentration above the in vitro IC50. Using a 150 mg BID dose, the anticipated Cmax after a single dose was 1.0 μM and the AUC was 6.6 μM·h. For a 500 mg QD dose, Cmax was predicted to be 3.2 μM and AUC was 22 μM. The pharmacokinetics of 13 has been investigated in patients with gliomas, AML, chondrosarcomas, and intrahepatic cholangiocarcinomas, who have documented IDH1R132 mutations. After a single oral dose of 13 at 150 mg in human, the geometric mean Cmax and AUC were 1.2 μM and 6.6 μM·h with a moderate inter-subject variability (CV% of geometric mean: 49% and 57%, respectively), which was within 1.5-fold from the predicted values.
In conclusion, optimization of 1 focusing on potency and PK properties led to the discovery of 13, an oral inhibitor of mutant IDH1 that efficiently inhibited 2-HG production in various xenograft models and inhibited 2-HG-dependent tumor growth of an IDH1 mutant PDX melanoma model. Robust dose-dependent 2-HG inhibition in preclinical models by 13 enabled development of an exposure–response relationship. Based on these data, 13 was selected for evaluation in clinical trials in humans to treat IDH1 mutant tumors, such as AML, chondrosarcoma, and cholangiocarcinoma. Compound 13 also demonstrated improved brain penetration relative to 1, suggesting potential utility in treating patients with IDH1 mutant brain cancers.
Acknowledgments
The authors would like to thank Bill Sellers, Karin Briner, Tim Ramsey, Juliet Williams, and Travis Stams for their support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00342.
Synthetic procedures, analytical data, assay protocols, X-ray data of 13 (IDH305) bound to mutant IDH1, and PK, PK/PD, and efficacy study protocols and data (PDF)
Author Present Address
† Constellation Pharmaceuticals, 215 First Street, Suite 200, Cambridge, Massachusetts 02142, United States.
Author Present Address
‡ Ideaya Biosciences, 280 Utah Avenue, Suite 250, South San Francisco, California 94080, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Yen K. E.; Bittinger M. A.; Su S. M.; Fantin V. R. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene 2010, 29, 6409–6417. 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]
- Reitman Z. J.; Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo C. M.; Gameiro P. A.; Bell E. L.; Mattaini K. R.; Yang J.; Hiller K.; Jewell C. M.; Johnson Z. R.; Irvine D. J.; Guarente L.; Kelleher J. K.; Vander Heiden M. G.; Iliopoulos O.; Stephanopoulos G. Reductive glutamine metabolisim by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen A. R.; Wheaton W. W.; Jin E. S.; Chen P. H.; Sullivan L. B.; Cheng T.; Yang Y.; Linehan W. M.; Chandel N. S.; DeBerardinis R. J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise D. R.; Ward P. S.; Shay J. E.; Cross J. R.; Gruber J. J.; Sachdeva U. M.; Platt J. M.; DeMatteo R. G.; Simon M. C.; Thompson C. B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 19611–19616. 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns R. A.; Mak T. W. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discovery 2013, 3, 730–741. 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- Parsons D. W.; Jones S.; Zhang X.; Lin J. C.; Leary R. J.; Angenendt P.; Mankoo P.; Carter H.; Siu I. M.; Gallia G. L.; Olivi A.; McLendon R.; Rasheed B. A.; Keir S.; Nikolskaya T.; Nikolsky Y.; Busam D. A.; Tekleab H.; Diaz L. A. Jr.; Hartigan J.; Smith D. R.; Strausberg R. L.; Marie S. K.; Shinjo S. M.; Yan H.; Riggins G. J.; Bigner D. D.; Karchin R.; Papadopoulos N.; Parmigiani G.; Vogelstein B.; Velculescu V. E.; Kinzler K. W. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H.; Parsons D. W.; Jin G.; McLendon R.; Rasheed B. A.; Yuan W.; Kos I.; Batinic-Haberle I.; Jones S.; Riggins G. J.; Friedman H.; Friedman A.; Reardon D.; Herndon J.; Kinzler K. W.; Velculescu V. E.; Vogelstein B.; Bigner D. D. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschka P.; Schlenk R. F.; Gaidzik V. I.; Habdank M.; Krönke J.; Bullinger L.; Späth D.; Kayser S.; Zucknick M.; Götze K.; Horst H. A.; Germing U.; Döhner H.; Döhner K. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; Vander Heiden M. G.; Su S. M. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Travins J. M.; Yan S.; Zhao F.; Gross S.; Dang L.; Yen K. E.; Yang H.; Straley K. S.; Jin S.; Kunii K.; Fantin V. R.; Zhang S.; Pan O.; Shi D.; Biller S. A.; Su S. M. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012, 3, 850–855. 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D.; Popovici-Muller J.; Palaskas N.; Turcan S.; Grommes C.; Campos C.; Tsoi J.; Clark O.; Oldrini B.; Komisopoulou E.; Kunii K.; Pedraza A.; Schalm S.; Silverman L.; Miller A.; Wang F.; Yang H.; Chen Y.; Kernytsky A.; Rosenblum M. K.; Liu W.; Biller S. A.; Su S. M.; Brennan C. W.; Chan T. A.; Graeber T. G.; Yen K. E.; Mellinghoff I. K. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B.; Yao Y.; Liu Z.; Deng L.; Anglin J. L.; Jiang H.; Prasad B. V. V.; Song Y. Crystallographic investigation and selective inhibition of mutant isocitrate dehydrogenase. ACS Med. Chem. Lett. 2013, 4, 542–546. 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Yao Y.; Kogiso M.; Zheng B.; Deng L.; Qiu J. J.; Dong S.; Lv H.; Gallo J. M.; Li X. N.; Song Y. Inhibition of cancer-associated mutant isocitrate dehydrogenases: synthesis, structure–activity relationship, and selective antitumor activity. J. Med. Chem. 2014, 57, 8307–8318. 10.1021/jm500660f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks E.; Wu X.; Hanel A.; Nguyen S.; Wang J.; Zhang J. H.; Harrison A.; Zhang W. Identification and characterization of small-molecule inhibitors of the R132H/R132H mutant isocitrate dehydrogenase 1 homodimer and R132H/wild-type heterodimer. J. Biomol. Screening 2014, 19, 1193–1200. 10.1177/1087057114541148. [DOI] [PubMed] [Google Scholar]
- Deng G.; Shen J.; Yin M.; McManus J.; Mathieu M.; Gee P.; He T.; Shi C.; Bedel O.; McLean L. R.; Le-Strat F.; Zhang Y.; Marquette J. P.; Gao Q.; Zhang B.; Rak A.; Hoffmann D.; Rooney E.; Vassort A.; Englaro W.; Li Y.; Patel V.; Adrian F.; Gross S.; Wiederschain D.; Cheng H.; Licht S. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 2015, 290, 762–774. 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye-Okafor U. C.; Bartholdy B.; Cartier J.; Gao E. N.; Pietrak B.; Rendina A. R.; Rominger C.; Quinn C.; Smallwood A.; Wiggall K. J.; Reif A. J.; Schmidt S. J.; Qi H.; Zhao H.; Joberty G.; Faelth-Savitski M.; Bantscheff M.; Drewes G.; Duraiswami C.; Brady P.; Groy A.; Narayanagari S. R.; Antony-Debre I.; Mitchell K.; Wang H. R.; Kao Y. R.; Christopeit M.; Carvajal L.; Barreyro L.; Paietta E.; Makishima H.; Will B.; Concha N.; Adams N. D.; Schwartz B.; McCabe M. T.; Maciejewski J.; Verma A.; Steidl U. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 2015, 11, 878–886. 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan B.; Le K.; Manyak E.; Liu H.; Prahl M.; Bowden C. J.; Biller S.; Agresta S.; Yang H. Longitudinal pharmacokinetic/pharmacodynamic profile of AG-120, a potent inhibitor of the IDH1 mutant protein, in a phase 1 study of IDH1-mutant advanced hematologic malignancies. Blood 2015, 126, 1310. [Google Scholar]
- Burris H.; Mellinghoff I.; Maher E.; Wen P.; Beeram M.; Touat M.; Faris J.; Azad N.; Cloughesy T.; Gore L.; Trent J.; Von Hoff D.; Goldwasser M.; Liu H.; Fan B.; Wu B.; Agresta S. The first reported results of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a phase 1 study of patients with advanced IDH1-mutant solid tumors, including gliomas. Mol. Cancer Ther. 2015, 14, PL04-05. 10.1158/1535-7163.TARG-15-PL04-05. [DOI] [Google Scholar]
- Levell J. R.; Caferro T.; Chenail G.; Dix I.; Dooley J.; Firestone B.; Fortin P. D.; Giraldes J.; Gould T.; Growney J. D.; Jones M. D.; Kulathila R.; Lin F.; Liu G.; Mueller A.; van der Plas S.; Slocum K.; Smith T.; Terranova R.; Toure B. B.; Tyagi V.; Wagner T.; Xie X.; Xu M.; Yang F. S.; Zhou L. X.; Pagliarini R.; Cho Y. S. Optimization of 3-pyrimidin-4-yl-oxazolidin-2-ones as allosteric and mutant specific inhibitors of IDH1. ACS Med. Chem. Lett. 2017, 8, 151–156. 10.1021/acsmedchemlett.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Capka V.; Chen J.; Chenail G.; Cho Y. S.; Dooley J.; Farsidjani A.; Fortin P. D.; Kohl D.; Kulathila R.; Lin F.; McKay D.; Sage D.; van der Plas S.; Wright K.; Xu M.; Yin H.; Levell J.; Pagliarini R. A. Allosteric mutant IDH1 inhibitors reveal mechanisms for IDH1 mutant and isoform selectivity. Structure 2017, 25, 1–8. 10.1016/j.str.2016.12.017. [DOI] [PubMed] [Google Scholar]
- Yin J.; Zarkowsky D. S.; Thomas D. W.; Zhao M. M.; Huffman M. A. Direct and convenient conversion of alcohols to fluorides. Org. Lett. 2004, 6, 1465–1468. 10.1021/ol049672a. [DOI] [PubMed] [Google Scholar]
- de Koning P. D.; McAndrew D.; Moore R.; Moses I. B.; Boyles D. C.; Kissick K.; Stanchina C. L.; Cuthbertson T.; Kamatani A.; Rahman L.; Rodriguez R.; Urbina A.; Sandoval A.; Rose P. R. Fit-for-purpose development of the enabling route to crizotinib (PF-02341066). Org. Process Res. Dev. 2011, 15, 1018–1026. 10.1021/op200131n. [DOI] [Google Scholar]
- Gao H.; Korn J. M.; Ferretti S.; Monahan J. E.; Wang Y.; Singh M.; Zhang C.; Schnell C.; Yang G.; Zhang Y.; Balbin O. A.; Barbe S.; Cai H.; Casey F.; Chatterjee S.; Chiang D. Y.; Chuai S.; Cogan S. M.; Collins S. D.; Dammassa E.; Ebel N.; Embry M.; Green J.; Kauffmann A.; Kowal C.; Leary R. J.; Lehar J.; Liang Y.; Loo A.; Lorenzana E.; McDonald R.; McLaughlin M. E.; Merkin J.; Meyer R.; Naylor T. L.; Patawaran M.; Reddy A.; Röelli C.; Ruddy D. A.; Salangsang F.; Santacroce F.; Singh A. P.; Tang Y.; Tinetto W.; Tobler S.; Velazquez R.; Venkatesan K.; Von Arx F.; Wang H. Q.; Wang Z.; Wiesmann M.; Wyss D.; Xu F.; Bitter H.; Atadja P.; Lees E.; Hofmann F.; Li E.; Keen N.; Cozens R.; Jensen M. R.; Pryer N. K.; Williams J. A.; Sellers W. R. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Berellini G.; LaBonte L. R.; Liang G.; Kim S. Systematic evaluation of Wajima superposition (steady-state concentration to mean residence time) in the estimation of human intravenous pharacokinetic profile. J. Pharm. Sci. 2016, 105, 1277–1287. 10.1016/S0022-3549(15)00174-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.