

Abstract

A substantial improvement of potency and selectivity of imidazoline-based inhibitors of hCA VII (a promising target for the treatment of seizures and neuropathic pain) was achieved by simply switching the position of the benzenesulfonamide moiety from N1 (as in the earlier reported series) to C2. Selectivity indices vs the off-target isoforms (hCA I, I, I and IV) greater than 100 were reached, which is exceedingly rare for hCA VII inhibitors. The drastic profile improvement of the new series has been rationalized by an additional hydrogen bonding with the nonconserved Q69 residue in the active site of hCA VII (absent in the other three isoforms studied), which also results in a favorable accommodation of the inhibitor’s lipophilic periphery in the nearby hydrophobic pocket. The robustness of the docking simulations was tested and confirmed by molecular dynamics simulations.
Keywords: Privileged scaffold, 2-imidazolines, N-arylimidazolines, carbonic anhydrase inhibitors, isoform selectivity, zinc binding group, primary sulfonamide, docking simulation, hydrogen bonding, nonconserved residue, molecular dynamics
Human carbonic anhydrases (hCAs, EC 4.2.1.1) regulate the pH inside and outside the living cell by catalyzing reversible hydration of carbon dioxide to produce bicarbonate anion and proton. The fundamental significance of their catalytic function links hCAs to many important physiological processes and therefore allows considering these 15 enzymes (hCA I-XIV, including hCA VA and VB isoforms) as drug targets for a range of diseases such as glaucoma, cancer, epilepsy, obesity, and others.1−5 Development of isoform-selective inhibitors of hCA is desirable from the standpoint of delineating the therapeutic effect caused by inhibition of the isoform in question from that resulting from the inhibition of the “off target” ones.6 Moreover, the ongoing validation of certain hCA isoforms as targets for therapeutic intervention requires reliable, selective tool compounds.7 These can be employed to study, at the cellular or whole-organism levels, the consequences of inhibiting a particular hCA isoform, to gain structural insights into their binding mode via X-ray crystallography and thus serve as well-characterized leads for the development of future therapeutic agents.8
One hCA isoform, whose potential as a drug target is currently being unveiled, is hCA VII. First characterized in 1991,9 it is mostly expressed in various regions of the brain where it is involved in GABAergic neuronal excitation.10 hCA VII has been stipulated as a target for the treatment of epileptic seizures11 and neuropathic pain.12 Additionally, it has been suggested that cysteine-rich regions of the enzyme may be involved in cellular protection against oxidative damage.13 The crystal structure of mutant hCA VII obtained with nonselective inhibitor acetazolamide14 has been successfully employed to understand the binding mode and the origin of isoform selectivity of newly designed inhibitors15 as well as for virtual screening for new compounds in the commercial domain, for which selective hCA VII inhibition was later confirmed experimentally.16
Potent inhibition of hCA VII isoform with high selectivity over other isoforms (particularly, the abundant cytosolic hCA I and II, which have a high catalytic activity and are the usual off-targets in this case17) is extremely rare; hCA VII selectivity indices (SIs) greater than 100 are virtually absent in the current literature. Low-nanomolar18 and even subnanomolar19 levels of hCA VII inhibition are certainly achievable but typically accompanied by hCA I and II inhibition in the single- to double-digit nanomolar range. Another isoform primarily localized in the central nervous system, for which a clean profile of hCA VII inhibitors would be desirable, is the membrane-bound hCA IV.20 The latter has been shown to be involved in supporting the functioning and nutrition of the glial cells.21 Being an approach to the treatment neurologic disorders on its own, hCA IV inhibition should be separated from that of hCA VII for the sake of targeted therapy development.
Recently, we described a series of imidazoline-based primary sulfonamides 1, which were synthesized via a Pd-catalyzed N-arylation22 followed by sulfonamide deprotection. They displayed preferential inhibition of hCA VII over hCA I and IV (however, with modest selectivity) while also inhibiting hCA II in the same concentration range as they did hCA VII.23 The observed separation of hCA VII potency from its usual off-targets was viewed as a valuable, chemotype-related finding. However, we sought to improve the desired potency of inhibitors 1 and also eliminate or lower the inhibition of hCA II, which has a wide tissue distribution and, preferably, should not be affected in the course of neurologic drug intervention. 2-Imidazolines24 and, more specifically, N-arylimidazolines25 are privileged scaffolds for drug design, and therefore, we were initially not keen to depart from this chemotype and decided to take a more palliative approach and investigate a different presentation of the vicinal aromatic groups on the imidazoline scaffold. Compounds 2, where the pharmacophoric benzenesulfonamide moiety (acting as prosthetic zinc binding group, or ZBG, in hCA inhibitors of this chemical class) is at position 2 of the imidazoline core, would be difficult to access using the Pd-catalyzed (Buchwald–Hartwig) protocol as it only allows introducing electron-deficient (hetero)aromatic moieties.22 To circumvent this obstacle, we recently developed an alternative imidazoline N-arylation approach using the Chan–Evans–Lam procedure26 and expected to efficiently employ it to append various aromatic groups to protected benzenesulfonamide imidazoline 3 on reaction with boronic acids 4 (Figure 1).
Figure 1.
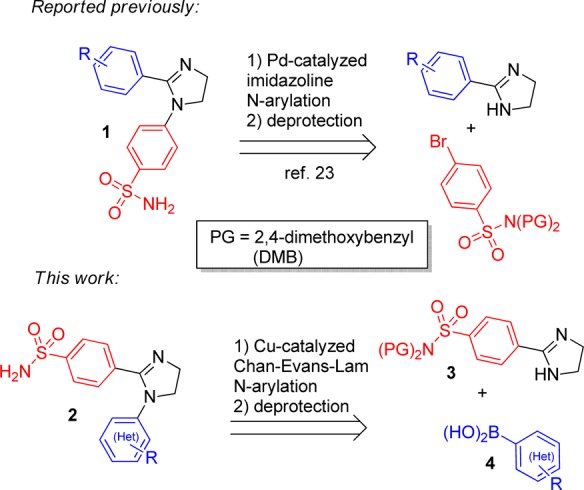
Approaches to imidazoline-based sulfonamides 1 and 2.
The starting imidazoline 3 was synthesized from nitrile 5 using the CS2-catalyzed reaction with ethylenediamine. The Chan–Evans–Lam arylation with boronic acids 4 in DMSO using the open-flask reaction format26 furnished DMB-protected compounds 6a–i. The latter were transformed into target primary sulfonamides 2a–i on brief treatment with TFA in ice-cold dichloromethane (Scheme 1).
Scheme 1. Synthesis of Compounds 2a–i.

Reagents and conditions: (a) (DMB)2NH, TEA, CH2Cl2, r.t., overnight (93%); (b) ethylenediamine, cat. CS2, 120 °C, 2 h (80%); (c) (Het)ArB(OH)2, K2CO3, Cu(OAc)2·H2O, DMSO, open flask, r.t., overnight (33–43%); (d) TFA, CH2Cl2, 0 °C, 30 min (52–68%).
Having synthesized compounds 2a–i, we proceeded to interrogate hCA VII and other isoforms with this set of tools. Biochemical testing of compounds 2a–i against the same panel of hCA as was described for compounds 1 (hCA I, II, IV, and VII)23 using hCA pan-inhbitor acetazolamide (AAZ) as a comparator revealed a promising (and desired in the context of present study) SAR trend (Table 1).
Table 1. Inhibitory Profile of Compounds 2a–i against hCA I, II, IV, and VIIa.
| Ki (nM) |
|||||
|---|---|---|---|---|---|
| compd | (Het)Ar | hCA I | hCA II | hCA IV | hCA VII |
| 2a | Ph | 466.1 | 376.5 | 3912.1 | 6.8 |
| 2b | 4-ClC6H4 | 453.0 | 182.1 | 608.2 | 0.96 |
| 2c | 4-FC6H4 | 428.7 | 742.2 | 1036.5 | 9.4 |
| 2d | 4-MeOC6H4 | 317.9 | 53.3 | 3643.4 | 0.97 |
| 2e | 2-MeC6H4 | 70.7 | 61.7 | 961.2 | 0.93 |
| 2f | 2-EtOC6H4 | 216.3 | 866.5 | 2299.7 | 0.84 |
| 2g | 3,4-(MeO)2C6H4 | 847.6 | 723.6 | 8302.6 | 0.87 |
| 2h | 3-pyridyl | 316.0 | 848.3 | 1826.6 | 0.84 |
| 2i | 5-pyrimidinyl | 274.5 | 703.2 | 2296.0 | 0.83 |
| 1a | 4-FC6H4 | 2343.0 | 76.1 | 9726.0 | 303.5 |
| 1b | 4-ClC6H4 | 960.5 | 46.7 | 8142.0 | 269.0 |
| AAZ | 250.0 | 12.1 | 74.0 | 6.0 | |
Mean Ki values from three different stopped-flow assays (errors were in the range of ±5–10% of the reported values). Inhibitory profile of compounds 1a–b23 is provided for comparison.
Even a brief comparison of chemotypes 1 and 2 in terms of their inhibitory profiles against the panel of hCAs shown in Table 1 and reported earlier23 allows one to note a remarkable, up to two orders of magnitude on average, improvement of hCA VII potency, with some of the compounds reaching into the subnanomolar range. At the same time, the inhibition profile of 2 against hCA I and hCA IV remains relatively low, similarly to 1. However, we were particularly pleased to find that the potency against hCA II (the principal “off-target” of 1 as hCA VII inhibitors) was significantly lowered, thus even more improving the selectivity of 2 for hCA VII. This is clearly illustrated by comparing the pairs of direct analogues in series 1 (1a–b) and 2 (2b–c) where swapping the positions of sulfonamide- and halogen-substituted aryl groups (1a → 2c and 1b → 2b) leads to dramatic improvement of hCA VII selectivity, particularly for within the latter pair (Figure 2).
Figure 2.
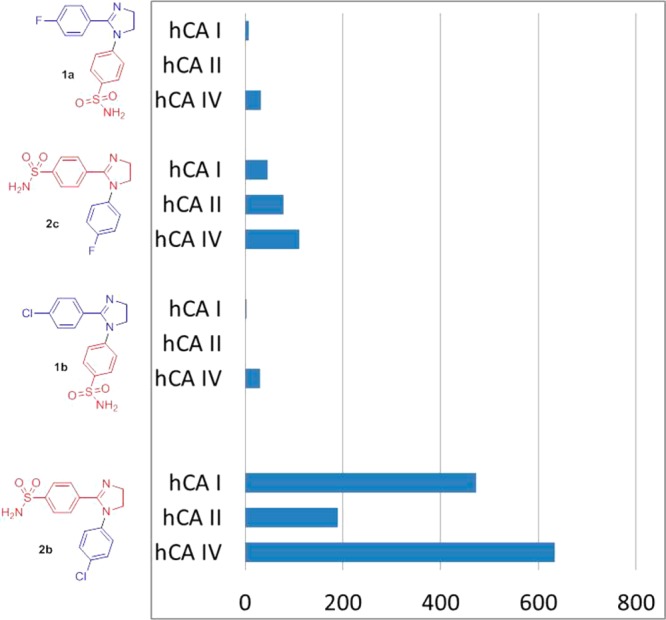
Selectivity indices (Ki(hCA N)/Ki(hCA VII) of hCA I, II, and IV vs hCA VII inhibition by compounds 1a–b and 2b–c.
In order to rationalize the hCA VII selectivity improvement trend observed on going from series 1 to series 2, we performed a back-to-back docking simulation of the binding of compounds 1b and 2b within the active site of hCA VII and compared it to the docking poses of the same compounds in the active site of hCA I, II, and IV. Figure 3 shows the binding site interactions of compound 1b into the four hCA isoforms. In all the binding sites the compound shows almost the same binding disposition: the sulfonamide group acts as a ZBG and forms hydrogen bonds with the protein backbone and the hydroxy group of the threonine residue; the phenyl ring attached to the ZBG does not show important interactions. The 4-chlorophenyl substituent is partially exposed to the solvent, and it is in contact with the external region of the binding site cavity. With regard to the imidazoline ring, in the hCAI, IV, and VII binding sites, it shows the same disposition with the presence of lipophilic interactions with L142 (isoleucine for hCAIV) and L199 (hCAI sequence number). In the hCAII binding site, due to the presence of I91 and F130, the imidazole ring of compound 1b is shifted by about 3 Å and shows lipophilic interactions with these two residues.
Figure 3.

Docking of compound 1b into hCAI (A), hCAII (B), hCAIV (C), and hCAVII (D).
The analysis of compound 2b suggests that for the hCAI, II, and IV isoforms, it shows the same binding disposition of compound 1b, with the 4-chlorophenyl substituent exposed to the solvent without important interactions with the protein and the interaction of the imidazoline ring with L142 (isoleucine for hCAIV) and L199 (hCAI sequence number) for hCAI and IV and with I91 and F130 for hCAII (Figure 4). Interestingly, in the hCAVII binding site, due to the presence of the nonconserved Q69, the compound shows a completely different binding orientation (Figure 4D). As for the other ligand–enzyme complexes, the sulfonamide group acts as a ZBG and forms hydrogen bonds with the protein backbone and the hydroxy group of the T201, the nitrogen of the imidazole ring forms an H-bond with the nonconserved Q69, and this interaction allows the disposition of the 4-chlorophenyl group in the lipophilic region of the binding site mainly delimited by L143, L200, and F133, thus suggesting this as a likely reason for the high hCAVII inhibition activity observed for this compound.
Figure 4.
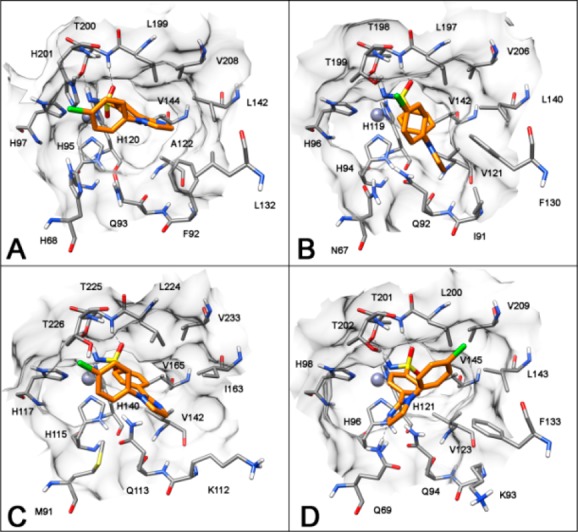
Docking of compound 2b into hCAI (A), hCAII (B), hCAIV (C), and hCAVII (D).
In order to evaluate the reliability of the docking results27 and to carry out an analysis of the ligand–protein interaction, the hCAVII–2b complex was used as a starting structure for 11 ns of molecular dynamics (MD) simulation. As shown in Figure 5, the complex is stable during the simulations, and the analysis of the root-mean-square deviation (rmsd) of all the heavy atoms from the X-ray structures highlights a stabilization of the rmsd value around 1.0 Å. Regarding the geometry of the compound, analyzing the rmsd of the position of the ligand during the simulations with respect to the starting structure, it maintains its starting disposition with an rmsd value between 0.2 and 0.5 Å. Finally, the H-bond between Q69 and the imidazoline nitrogen appears to be very stable as this interaction is maintained for about 90% of the MD simulation.
Figure 5.

Analysis of the MD simulation of 2b complexed with hCAVII. (A) Minimized average structure of the complex. (B) Rmsd analysis of the heavy atoms of the receptor and the ligand from the starting model structure during the simulation.
In summary, we reported a substantial improvement of potency and selectivity of imidazoline-based inhibitors of hCA VII by switching the position of the benzenesulfonamide moiety from the N1 (as in the earlier reported series) to the C2 position of the imidazoline ring. This minor alteration in the chemotype resulted in subnanomolar inhibition of hCA VII, a promising target for the treatment of seizures and neuropathic pain. This eloquently illustrates the privileged character of imidazolines for drug design and a possibility to fine-tune affinity profiles against a panel of closely related targets by altering the positions of the periphery appendage groups around this polar, nonflat scaffold.
Acknowledgments
We are grateful to the Research Centre for Magnetic Resonance and the Centre for Chemical Analysis and Materials Research of Saint Petersburg State University Research Park for the analytical data.
Glossary
ABBREVIATIONS
- hCA
human carbonic anhydrase
- ZBG
zinc-binding group
- DMSO
dimethyl sulfoxide
- DMB
2,4-dimethoxybenzyl
- TEA
triethylamine
- TFA
trifluoroacetic acid
- AAZ
acetazolamide
- SAR
structure–activity relationship
- MD
molecular dynamics
- RMSD
root-mean-square deviation
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00300.
Experimental procedures for the synthesis of 5, 3, 6a–i, 2a–i; docking and molecular dynamics simulations protocol for compounds 1b and 2b (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by the Russian Science Foundation (project grant 14-50-00069).
The authors declare no competing financial interest.
Supplementary Material
References
- Supuran C. T. Diuretics: from classical carbonic anhydrase inhibitors to novel applications of the sulfonamides. Curr. Pharm. Des. 2008, 14, 641–648. 10.2174/138161208783877947. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Scozzafava A.; Mincione F. The development of topically acting carbonic anhydrase inhibitors as antiglaucoma agents. Curr. Pharm. Des. 2008, 14, 649–654. 10.2174/138161208783877866. [DOI] [PubMed] [Google Scholar]
- Di Fiore A.; Supuran C. T.; De Simone G. Are carbonic anhydrase inhibitors suitable for obtaining antiobesity drugs?. Curr. Pharm. Des. 2008, 14, 655–660. 10.2174/138161208783877820. [DOI] [PubMed] [Google Scholar]
- Thiry A.; Dogne J. M.; Supuran C. T.; Masereel B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr. Top. Med. Chem. 2007, 7, 855–864. 10.2174/156802607780636726. [DOI] [PubMed] [Google Scholar]
- Bao B.; Groves K.; Zhang J.; Handy E.; Kennedy P.; Cuneo G.; Supuran C. T.; Yared W.; Rajopadhye M.; Peterson J. D. In vivo imaging and quantification of carbonic anhydrase IX expression as an endogenous biomarker of tumor hypoxia. PLoS One 2012, 7, e50860. 10.1371/journal.pone.0050860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone D. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms?. Chem. Rev. 2012, 112, 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Moeker J.; Mahon B. P.; Bornaghi L. F.; Vullo D.; Supuran C. T.; McKenna R.; Poulsen S.-A. Structural Insights into Carbonic Anhydrase IX Isoform Specificity of Carbohydrate-Based Sulfamates. J. Med. Chem. 2014, 57, 8635–8645. 10.1021/jm5012935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraroni M.; Lucarini L.; Masini E.; Korsakov M.; Scozzafava A.; Supuran C. T.; Krasavin M. 1,3-Oxazole-based selective picomolar inhibitors of cytosolic human carbonic anhydrase II alleviate ocular hypertension in rabbits: potency is supported by X-ray crystallography of two leads. Bioorg. Med. Chem. 2017, 25, 4560. 10.1016/j.bmc.2017.06.054. [DOI] [PubMed] [Google Scholar]
- Montgomery J. C.; Venta P. J.; Eddy R. L.; Fukushima Y. S.; Shows T. B.; Tashian R. E. Characterization of the human gene for a newly discovered carbonic anhydrase, CA VII, and its localization to chromosome 16. Genomics 1991, 11, 835–848. 10.1016/0888-7543(91)90006-Z. [DOI] [PubMed] [Google Scholar]
- Thiry A.; Masereel B.; Dogne J. M.; Supuran C. T.; Wouters J.; Michaux C. Exploration of the binding mode of indanesulfonamides as selective inhibitors of human carbonic anhydrase type VII by targeting Lys 91. ChemMedChem 2007, 2, 1273–1280. 10.1002/cmdc.200700057. [DOI] [PubMed] [Google Scholar]
- Hen N.; Bialer M.; Yagen B.; Maresca A.; Aggarwal M.; Robbins A. H.; McKenna R.; Scozzafava A.; Supuran C. T. Anticonvulsant 4-Aminobenzenesulfonamide Derivatives with Branched-Alkylamide Moieties: X-ray Crystallography and Inhibition Studies of Human Carbonic Anhydrase Isoforms I, II, VII, and XIV. J. Med. Chem. 2011, 54, 3977–3981. 10.1021/jm200209n. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev. Neurother. 2016, 16, 961–968. 10.1080/14737175.2016.1193009. [DOI] [PubMed] [Google Scholar]
- Monti D. M.; De Simone G.; Langella E.; Supuran C. T.; Di Fiore A.; Monti S. M. Insights into the role of reactive sulfhydryl groups of Carbonic Anhydrase III and VII during oxidative damage. J. Enzyme Inhib. Med. Chem. 2017, 32, 5–12. 10.1080/14756366.2016.1225046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore A.; Truppo E.; Supuran C. T.; Alterio V.; Dathan N.; Bootorabi F.; Parkkila S.; Monti S. M.; De Simone G. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg. Med. Chem. Lett. 2010, 20, 5023–5026. 10.1016/j.bmcl.2010.07.051. [DOI] [PubMed] [Google Scholar]
- Carta F.; Mannelli L. D. C.; Pinard M.; Ghelardini C.; Scozzafava A.; McKenna R.; Supuran C. T. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg. Med. Chem. 2015, 23, 1828–1840. 10.1016/j.bmc.2015.02.027. [DOI] [PubMed] [Google Scholar]
- De Luca L.; Ferro S.; Damiano F. M.; Supuran C. T.; Vullo D.; Chimiri A.; Gitto R. Structure-based screening for the discovery of new carbonic anhydrase VII inhibitors. Eur. J. Med. Chem. 2014, 71, 105–111. 10.1016/j.ejmech.2013.10.071. [DOI] [PubMed] [Google Scholar]
- Güzel Ö.; Innocenti A.; Scozzafava A.; Salman A.; Supuran C. T. Carbonic anhydrase inhibitors. Phenacetyl-, pyridylacetyl- and thienylacetylsubstituted aromatic sulfonamides act as potent and selective isoform VII inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3170–3173. 10.1016/j.bmcl.2009.04.123. [DOI] [PubMed] [Google Scholar]
- Altug C.; Güneş H.; Nocentini A.; Monti S. M.; Buonanno M.; Supuran C. T. Synthesis of isoxazole-containing sulfonamides with potent carbonic anhydrase II and VII inhibitory properties. Bioorg. Med. Chem. 2017, 25, 1456–1464. 10.1016/j.bmc.2017.01.008. [DOI] [PubMed] [Google Scholar]
- Bruno E.; Buemi M. R.; Di Fiore A.; De Luca L.; Ferro S.; Angeli A.; Cirilli R.; Sadutto D.; Alterio V.; Monti S. M.; Supuran C. T.; De Simone G.; Gitto R. Probing Molecular Interactions between Human Carbonic Anhydrases (hCAs) and a Novel Class of Benzenesulfonamides. J. Med. Chem. 2017, 60, 4316–4326. 10.1021/acs.jmedchem.7b00264. [DOI] [PubMed] [Google Scholar]
- Scheibe R. J.; Gros G.; Parkkila S.; Waheed A.; Grubb J. H.; Shah G. N.; Sly W. S.; Wetzel P. Expression of membrane-bound carbonic anhydrases IV, IX, and XIV in the mouse heart. J. Histochem. Cytochem. 2006, 54, 1379–1391. 10.1369/jhc.6A7003.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svichar N.; Esquenazi S.; Waheed A.; Sly W. S.; Chesler W. Functional demonstration of surface carbonic anhydrase IV activity on rat astrocytes. Glia 2006, 53, 241–247. 10.1002/glia.20277. [DOI] [PubMed] [Google Scholar]
- Krasavin M. Novel diversely substituted 1-heteroaryl-2-imidazolines for fragment-based drug discovery. Tetrahedron Lett. 2012, 53, 2876–2880. 10.1016/j.tetlet.2012.03.122. [DOI] [Google Scholar]
- Supuran C. T.; Kalinin S.; Tanç M.; Sarnpitak P.; Mujumdar P.; Poulsen S.-A.; Krasavin M. Isoform-selective inhibitory profile of 2-imidazoline-substituted benzenesulfonamides against a panel of human carbonic anhydrases. J. Enzyme Inhib. Med. Chem. 2016, 31 (Suppl. 1), 197–202. 10.1080/14756366.2016.1178248. [DOI] [PubMed] [Google Scholar]
- Krasavin M. Biologically active compounds based on the privileged 2-imidazoline scaffold: the world beyond adrenergic/imidazoline receptor modulators. Eur. J. Med. Chem. 2015, 97, 525–537. 10.1016/j.ejmech.2014.11.028. [DOI] [PubMed] [Google Scholar]
- Krasavin M. N-(Hetero)aryl 2-imidazolines: an emerging privileged motif for contemporary drug design. Chem. Heterocycl. Compd. 2017, 53, 240–255. 10.1007/s10593-017-2047-3. [DOI] [Google Scholar]
- Dar’in D.; Krasavin M. The Chan-Evans-Lam N-Arylation of 2-Imidazolines. J. Org. Chem. 2016, 81, 12514–12519. 10.1021/acs.joc.6b02404. [DOI] [PubMed] [Google Scholar]
- Sakano T.; Mahamood M. I.; Yamashita T.; Fujitani H. Molecular dynamics analysis to evaluate docking pose prediction. Biophys. Physicobiol. 2016, 13, 181–194. 10.2142/biophysico.13.0_181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.