

Abstract
Selective inhibitors of phosphoinositide 3-kinase delta are of interest for the treatment of inflammatory diseases. Initial optimization of a 3-substituted indazole hit compound targeting the kinase PIM1 focused on improving selectivity over GSK3β through consideration of differences in the ATP binding pockets. Continued kinase cross-screening showed PI3Kδ activity in a series of 4,6-disubstituted indazole compounds, and subsequent structure–activity relationship exploration led to the discovery of an indole-containing lead compound as a potent PI3Kδ inhibitor with selectivity over the other PI3K isoforms.
Keywords: Phosphoinositide-3-kinase delta inhibitor, PI3Kδ inhibitor, kinase cross-screening
Kinases play a pivotal role in nearly every aspect of cellular function and represent an important class of biological targets in the development of novel drugs.1 Kinases modify substrates by chemically adding a phosphate group from adenosine triphosphate (ATP), and this phosphorylation event controls many cellular processes, making them attractive therapeutic targets.2
Identification of starting hits for biological targets can be based on approaches such as high throughput screening (HTS) for diversity or structure-based drug design (SBDD) as a focused design approach,3 but in a target class such as kinases an efficient way of finding hits is from focused or knowledge-based screening. Screening a focused set containing compounds that have previously been shown to have activity against kinases has proven to be a highly successful strategy in discovering hits against new kinases.4 This approach predominantly targets small molecules that bind to the ATP-binding pocket, which is well-conserved across the whole kinome.5 Therefore, a resultant issue in translating hits into candidates and medicines has been selectivity over other undesired kinase targets, thus avoiding unwanted side effects.
Extensive in-house kinase cross-screening is routine within GSK and ensures any unexpected compound activities against off-targets are identified. This extensive cross-profiling has the benefit of providing unexpected opportunities for other targets. This letter discusses how, by considering a hit compound for PIM1 and issues of selectivity over GSK3β, kinase cross-screening and subsequent structure–activity relationship (SAR) exploration led to the discovery of a selective lead series for PI3Kδ.
The PIM (provirus insertion site of Moloney murine leukemia virus) family of serine/threonine protein kinases is represented by PIM1, PIM2, and PIM3, which are all highly conserved in vertebrates.6 PIM proteins have major functions in cell survival, proliferation, and differentiation in response to growth factors and cytokines,7 and PIM1 was the first PIM kinase discovered to have a key role as a proto-oncogene.8 The overexpression of PIM1 has been found in a wide range of human tumors, including B-cell lymphomas and various types of human leukemia as well as solid tumor cancers.9 Therefore, the development of PIM1 inhibitors as new anticancer drugs is of considerable interest.10
Initial screening of a focused kinase compound set against PIM1 identified indazole compound 1 as a moderately potent inhibitor with a pIC50 of 6.3 (Figure 1).11
Figure 1.

Initial indazole hit compound 1.
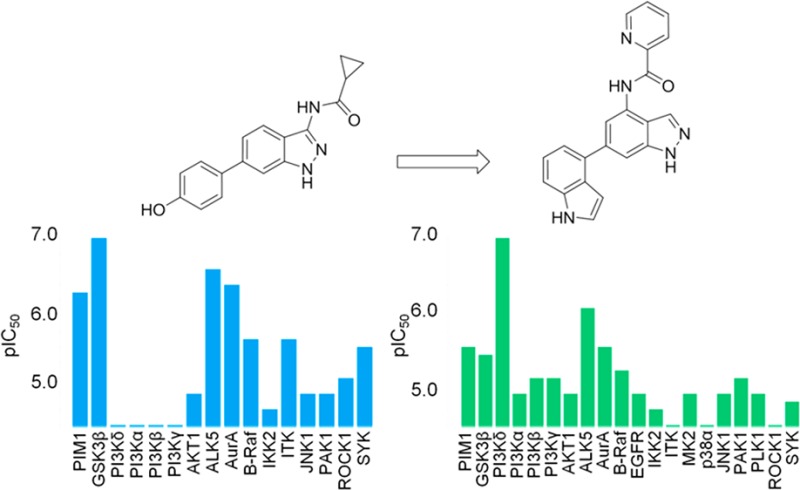
Compound 1 was as an attractive hit with a potential to rapidly expand SAR. However, selectivity data from cross-screening against a panel of in-house kinases revealed a moderate selectivity profile, with inhibition of GSK3β being one of the most significant off-target activities (Figure 2). GSK3β is a multifunctional serine/threonine protein kinase, which regulates a wide range of cellular processes, involving various undesirable signaling pathways.12,13 ALK5 and AurA are serine/threonine kinases that were also inhibited by compound 1, and this would also need to be addressed; however, with X-ray crystallography available in-house for GSK3β, our initial SAR exploration sought to improve selectivity for PIM1 over GSK3β.
Figure 2.
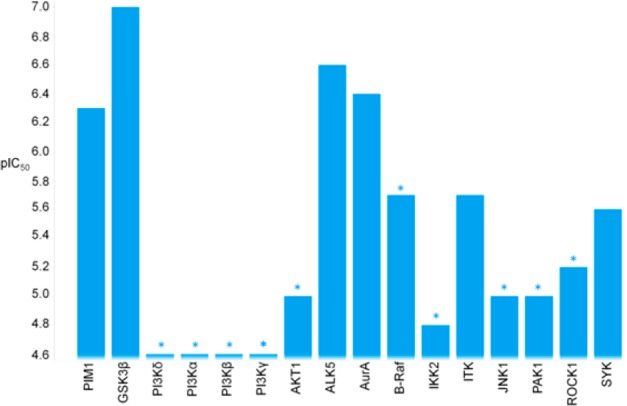
Selectivity profile of compound 1. *Asterisks designate a less than significant value was observed on at least one occasion.
Cocrystallization of compound 1 with PIM1 demonstrated that the proton-donor center N1–H of the indazole forms a hydrogen bond with Glu121 (Figure 3).
Figure 3.
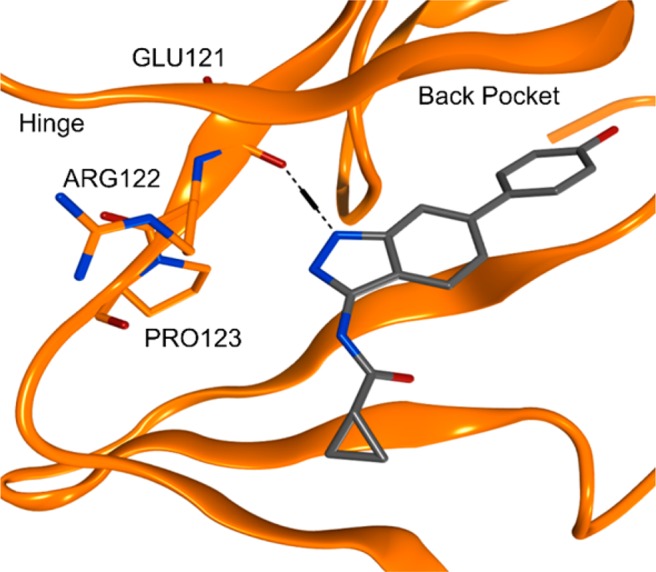
X-ray cocrystal structure of compound 1 in PIM1 with PIM1. Images generated using PyMol.
The greater affinity of compound 1 for GSK3β over PIM1 can be rationalized through consideration of the hinge interactions that are made upon binding to the different kinases. A commonly conserved H-bonding interaction to inhibitors in the hinge region of the ATP site of kinases is that of an NH on the protein backbone interacting with an acceptor motif on the inhibitor. PIM1 is unusual because the presence of a proline residue (Pro123, Figure 3) in the hinge region prevents the formation of this hydrogen bond.
A cocrystal structure of a close analogue of compound 1 with GSK3β confirmed that the two commonly conserved hydrogen bonding interactions are made with Asp133 and Val135 (Figure 4).14 The accessible carbonyl group of Val135 in fact enables a donor–acceptor–donor interaction between the amido-indazole ring and the protein (Figure 4), which is not possible with PIM1.
Figure 4.
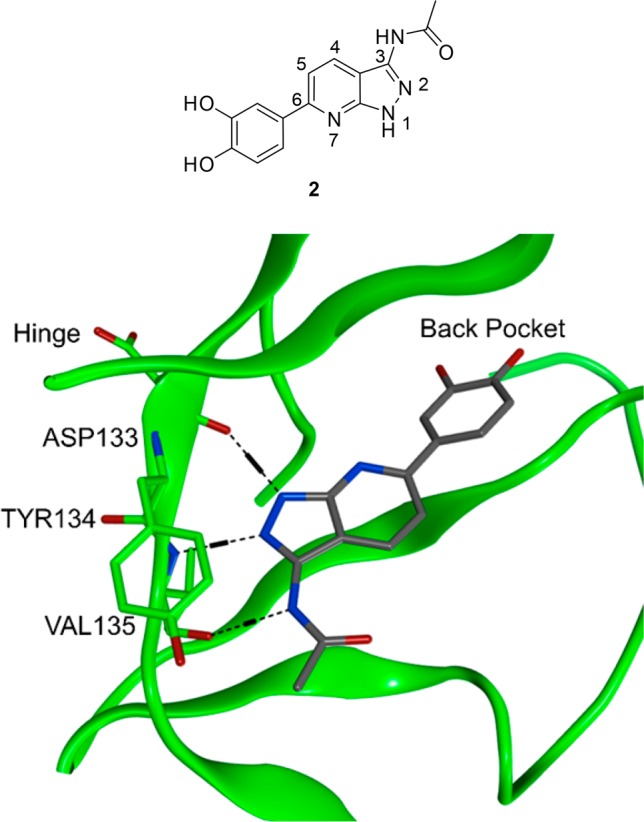
X-ray cocrystal structure of compound 2 with GSK3β. Images generated using PyMol.
We looked to alter the PIM1/GSK3β selectivity profile in favor of PIM1 by disrupting the additional H-bonding interaction between Val135 and the amido-NH observed in GSK3β (Figure 4) by moving the amido-substituent to the indazole 4-position.
To access the desired 4,6-disubstituted indazoles, a routine synthesis was employed that began with conversion of the commercially available 4-nitro-6-bromoindazole (3) to the corresponding THP-protected indazole intermediate 4 (general synthesis shown in Scheme 1). Installation of the 6-aryl groups under standard palladium-catalyzed cross-coupling conditions with the required boronic acid or boronate ester yielded compounds of structure 5 (Scheme 1). The THP-protected phenolic boronic acids were employed when introducing phenol-substituted aromatic groups (see Supporting Information). Reduction of the nitro group afforded aromatic amine intermediates of structure 6, which allowed the introduction of different amide substituents by coupling with the corresponding acid chlorides. Subsequent THP-deprotection gave the target compounds 8a, 8b, 8d, 8e, and 8f (see Table 1 for the range of substituents). Compound 8c was synthesized using an analogous route with SEM-protection of the indazole (see Supporting Information).
Scheme 1. Synthetic Route for Compounds 8a, 8b, 8d, 8e, and 8f.

Reagents and conditions: (a) 3,4-dihydropyran, TFA; (b) boronic acid/ester, Pd(dppf)Cl2, aq. NaHCO3, IPA, 150 °C, microwave; (c) H2, Pd/C, EtOAc; (d) acid chloride, DIPEA, DCM or acid, HATU, DIPEA, DMF; (e) 4 M HCl in dioxane, MeOH. See Table 1 for range of substituents.
Table 1. SAR of 4,6-Disubstituted Indazoles.
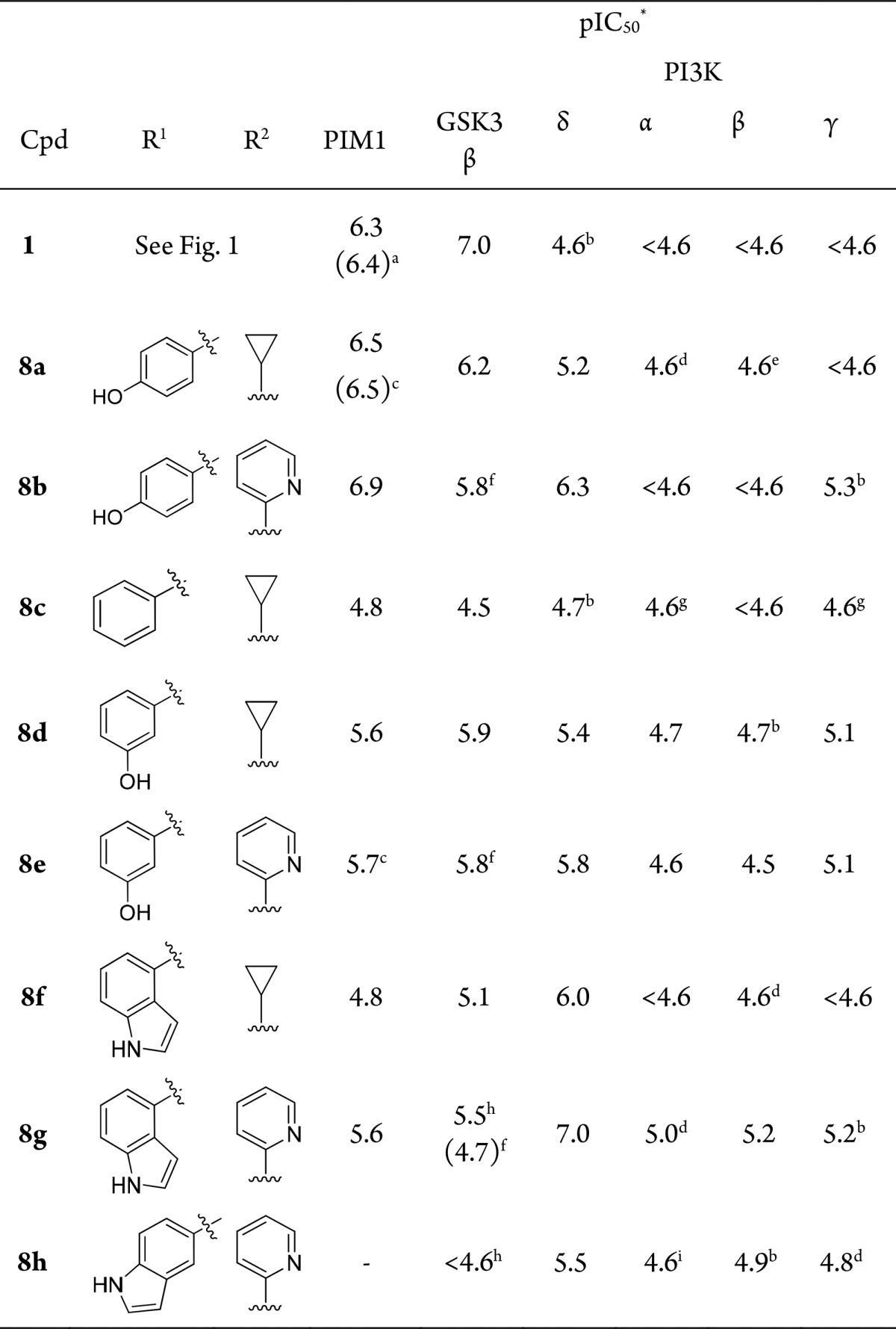
Data is mean of at least three separate test occasions unless stated.
Data from in-house PIM1 assay, n = 1.
Tested <4.6 on one occasion.
Data from in-house PIM1 assay.
Tested <4.6 on two occasions.
Tested <4.6 on three occasions.
GSK3β data was generated at Reaction Biology Corporation.23
Tested <4.6 on four occasions.
n = 1.
n = 2.
A shortened synthesis from commercially available 4-amino-6-bromoindazole (9), where amide formation was carried out as the first step before introduction of the required aryl groups via palladium-catalyzed cross-coupling, was followed for the synthesis of compounds 8g and 8h (Scheme 2, see Table 1 for range of substituents).
Scheme 2. Synthesis of Compounds 8g and 8h.

Reagents and conditions: (a) 2-pyridinecarbonyl chloride hydrochloride, DIPEA, DCM; (b) boronic acid, Pd(dppf)Cl2, Na2CO3, 1,4-dioxane, H2O. See Table 1 for the range of substituents.
Consideration of the residue differences in the hinge region of PIM1 and GSK3β quickly led to the design of compound 8a. The initial data for compound 8a was encouraging, with GSK3β potency reduced but PIM1 activity maintained (Table 1). The introduction of a picolinamide substituent at the indazole 4-position in compound 8b showed a slight increase in PIM1 activity, while a corresponding increase in potency for GSK3β was not observed (Table 1).
Continued kinase profiling also highlighted some interesting SARs against the PI3Ks. The phosphoinositide 3-kinases (PI3Ks) are a family of enzymes that are central to signaling involving intracellular lipids.15 The Class 1 PI3K family includes phosphoinositide 3-kinase delta (PI3Kδ) and the closely related isoforms α, β, and γ, which convert phosphatidylinositol-4,5-bisphosphate to phosphatidylinositol-3,4,5-trisphosphate in vivo.16 This leads to downstream signaling cascades that control cell growth, cell cycle entry, and cell survival. PI3Kδ inhibition is of interest for oncology indications, with the approval of GS-1101 (idelalisib) recently reported for the treatment of patients with relapsed chronic lymphoid leukemia and non-Hodgkin lymphoma.17 Since PI3Kδ is primarily expressed in leukocytes and plays a key role in immune signaling and inflammatory processes, it is also an attractive target for respiratory diseases such as asthma18 and chronic obstructive pulmonary disease (COPD).19 PI3Kγ is also enriched in leukocytes and has been targeted for the treatment of inflammatory disease,20 whereas the α and β isoforms are ubiquitously expressed and the α and β murine knockout phenotypes are embryonically lethal.21 This demonstrates the potential requirement for isoform selectivity when trying to treat chronic disease.
Compound 8a was the first in the series to show significant PI3Kδ activity (pIC50 = 5.2) compared with the original hit compound 1, which was inactive. The increased potency for PI3Kδ over the other class 1 PI3K isoforms was a key observation, as this was the first compound to show this selectivity profile from many compound series screened in-house against the PI3Ks. These initial results from kinase cross-screening and recognition of the therapeutic potential of selective PI3Kδ inhibitors led to a shift in target emphasis from PIM1 to PI3Kδ for this series of compounds.
A significant increase in PI3Kδ potency was observed for the picolinamide compound 8b, and subsequent SAR exploration has shown that the internal hydrogen bond was key to maintaining the planarity of the amide leading to stabilization of the preferred binding conformation to PI3Kδ, which was confirmed through crystallography as described by Down et al.22
Removal of the phenolic hydroxyl group in compound 8c caused a reduction in potency against PIM1, GSK3β, and PI3Kδ, but moving the phenol from the para- to the meta-position in compounds 8d and 8e was more detrimental to PIM1 activity than to that of GSK3β and PI3Kδ.
Replacement of the phenol with a bioisoteric indole moeity in compound 8f resulted in reduced activities for PIM1 and GSK3β but increased activity for PI3Kδ. Combining the previously recognized 4-position picolinamide with 6-position indole moieties led to compounds 8g and 8h, where the 4-substituted indole displayed much more potent PI3Kδ inhibition than the 5-substituted analogue. Significantly, there was a continued lack of activity of the 4,6-disubstituted indazole compounds against the other PI3K isoforms.
Due to the lack of a suitable crystal structure of PI3Kδ at this time, a homology model was built to aid the rationalization of the SAR.24 The model suggested that the indazole nitrogen engaged in the hinge-binding interaction but that the core was shifted significantly when compared with the indazole binding in GSK3β and PIM1 (overlay of PI3Kδ homology model with GSK3β cocrystal structure shown in Figure 5). This movement is hypothesized to be due to changes in the gatekeeper residue and nearby back pocket residues between the three kinases. The gatekeeper residue is a single residue in the ATP pocket that has been shown to control sensitivity to small molecule kinase inhibitors since it influences the size and shape of the back cavity that is not occupied by ATP.5 The PI3Kδ Ile825 gatekeeper residue (PIM1 = Leu120, GSK3β = Leu132) and Tyr813 residue (PIM1 = Ile104, GSK3β = Val110) extend further into the back pocket than the corresponding residues of PIM1 and GSK3β. The movement of the indazole core, to avoid clashing with the aryl substituent of Tyr813, orients the back pocket substituent such that a key H-bond interaction with Asp787 can be made, and this is achieved when the indole substituent is substituted at the 4-position (Figure 5). This differs from the para-phenol substituents that are preferred for PIM1 and GSK3β, which give the optimal orientation for an H-bond interaction with a glutamic acid residue in both kinases (Glu97 of GSK3β shown in Figure 5).
Figure 5.
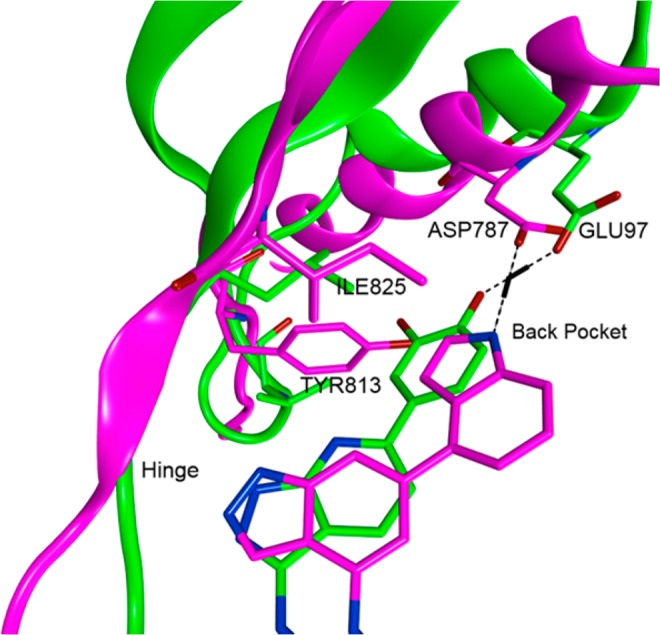
Close-up of the hinge and back pocket region of compound 8f docked in PI3Kδ (magenta) and overlay with compound 2 cocrystallized in GSK3β (green).
Since the compounds were not expected to extend into areas of the protein where there are significant residue differences between the PI3K isoforms, the PI3K selectivity profile was unexpected. Subsequent SAR investigation showed that the PI3K activities were greatly affected by the back pocket substituent.21 Since the residues are conserved in this region, it suggested a more complex mechanism of achieving selectivity than could be explained by modeling. Potentially, a subtle change in the extended H-bonding network could be responsible for the isoform selectivity observed.25
In addition to the highly encouraging selective PI3Kδ inhibitory characteristics of compound 8g, in-house kinase cross-screening indicated a good overall kinase selectivity profile compared with the initial 4-substituted indazole compounds 8a and 8b (Figure 6). This identified the indole-substituted indazole as a potentially novel PI3Kδ selective template. ALK5 remained the only kinase where moderate selectivity (<10-fold) was observed and would need to be addressed by further optimization.22
Figure 6.
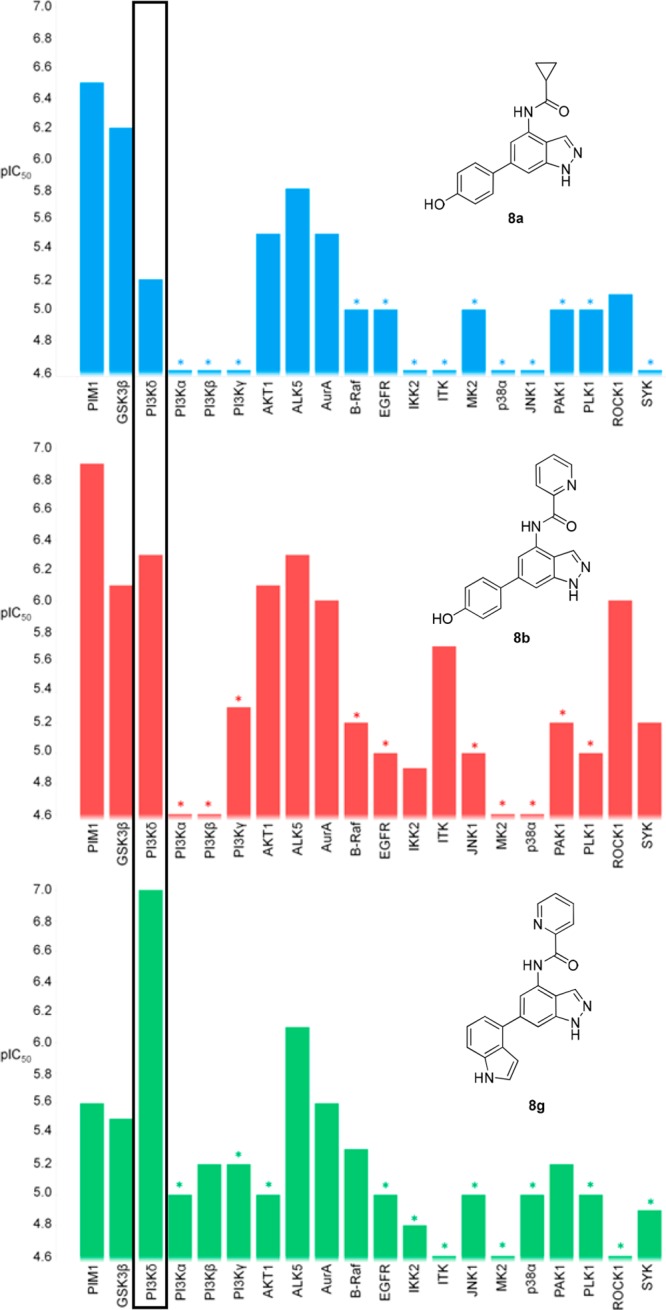
Kinase selectivity profiles of compounds 8a, 8b, and 8g. *Asterisks designate a less than significant value was observed on at least one occasion.
In summary, continued kinase cross-screening of indazole compounds focused on PIM1 antagonism identified the 4,6-disubstituted indazole series to be a novel, potent, and selective class of PI3Kδ inhibitors. The promising selectivity profile of compound 8g for PI3Kδ, specifically against the other PI3K isoforms, led to its further development and enabled the discovery of potent PI3Kδ inhibitors with therapeutic potential.22
Acknowledgments
The authors would like to thank Luke Carter, Maire Convery, and Lisa Shewchuk-Chapman for protein production and crystallography assistance; Geoffrey Cutler, Yvonne Joseph, and Sarah Smith for assay support; and Andrew Perrett for synthetic contributions.
Glossary
ABBREVIATIONS
- AKT1
v-Akt murine thymoma viral oncogene homologue 1
- ALK5
activin receptor-like kinase 5
- AurA
Aurora kinase A
- B-Raf
v-Raf murine sarcoma viral oncogene homologue B
- COPD
chronic obstructive pulmonary disease
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- EGFR
epidermal growth factor receptor
- GSK3β
glycogen synthase kinase 3 beta
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- IKK2
inhibitor of nuclear factor kappa-B kinase subunit beta
- ITK
interleukin-2-inducible T-cell kinase
- IPA
isopropyl alcohol
- JNK1
c-Jun N-terminal kinase 1
- MK2
mitogen-activated protein kinase-activated protein kinase-2
- p38α
p38-mitogen-activated protein kinase alpha
- PAK1
p21-activated kinase 1
- PI3K
phosphoinositide 3-kinase
- PIM
provirus insertion site of Moloney murine leukemia virus
- PLK1
polo-like kinase 1
- ROCK1
Rho-associated protein kinase 1
- SEM
2-(trimethylsilyl)ethoxymethyl
- SYK
spleen tyrosine kinase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00296.
Biological assays and experimental procedures (PDF)
Accession Codes
PDB codes to be despoited on acceptance.
Author Present Address
Δ B.D. B. York Structural Biology Laboratory, University of York, Heslington, York, YO10 5DD.
Author Present Address
○ S.G. Charles River, 8–9 Spire Green Centre, Flex Meadow, Harlow, Essex, CM19 5TR.
Author Present Address
◊ P.F. Elixir Software Ltd., The BioHub, Alderley Park, Alderley Edge, SK10 4TG.
Author Contributions
I.B. oversaw the medicinal chemistry. S.G. and Y.W. designed compounds, and L.I., A.C., and S.G. performed chemical synthesis. B.B. carried out protein crystallography. P.F. and J.L. carried out computational modeling and molecular design. D.T. oversaw and analyzed in vitro assay data. Z.H. and I.B. wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): All authors were employees of GlaxoSmithKline at the time the work was carried out.
Supplementary Material
References
- Cohen P.; Alessi D. R. Kinase Drug Discovery – What’s Next in the Field. ACS Chem. Biol. 2013, 8, 96–104. 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discovery Today 2016, 21, 5–10. 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Jhoti H.; Rees S.; Solari R. High-throughput screening and structure-based approaches to hit discovery: is there a clear winner?. Expert Opin. Drug Discovery 2013, 8, 1449–1453. 10.1517/17460441.2013.857654. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Christopher J. A.; Cutler G. J.; Dickson M. C.; Mellor G. W.; Morey J. V.; Patel C. B.; Shewchuk L. M. 5-(1H-Benzimidazol-1-yl)-3-alkoxy-2-thiophenecarbonitriles as potent, selective, inhibitors of IKK-ε kinase. Bioorg. Med. Chem. Lett. 2006, 16, 6236–6240. 10.1016/j.bmcl.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Zuccotto F.; Ardini E.; Casale E.; Angiolini M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Confomration. J. Med. Chem. 2010, 53, 2681–2694. 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- Mikkers H.; Nawijn M.; Allen J.; Brouwers C.; Verhoeven E.; Jonkers J.; Berns A. Mice deficient for all PIM kinases display reduced body size and impaired responses to haematopoietic growth factors. Mol. Cell. Biol. 2004, 24, 6104–6115. 10.1128/MCB.24.13.6104-6115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault L.; Gasser C.; Bracher F.; Huber K.; Knapp S.; Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 2010, 95, 1004–1015. 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuypers H. T.; Selten G.; Quint W.; Zijlstra M.; Mandaag E. R.; Boelens W. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell 1984, 37, 141–150. 10.1016/0092-8674(84)90309-X. [DOI] [PubMed] [Google Scholar]
- Nawijn M. C.; Alendar A.; Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer 2011, 11, 23–34. 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- Anizon F.; Shtil A. A.; Danilenko V. N.; Moreau P. Fighting tumor cell survival: advances in the design and evaluation of pim inhibitors. Curr. Med. Chem. 2010, 17, 4114–4133. 10.2174/092986710793348554. [DOI] [PubMed] [Google Scholar]
- pIC50 = −log10IC50; where the IC50 is the concentration of compound required to inhibit the kinase activity by 50%.
- Leloir L. F.; Olavaria S. H.; Goldenberg S. H.; Carminatti H. Biosynthesis of Glycogen from Uridine Diphosphate Glucose. Arch. Biochem. Biophys. 1959, 81, 508. 10.1016/0003-9861(59)90232-2. [DOI] [PubMed] [Google Scholar]
- Mandarino L.; Wright K.; Verity L.; Nichols J.; Bell J.; Kolterman O.; Beck-Nielsen H. Effects of insulin infusion on human skeletal muscle pyruvate dehydrogenase, phosphofructokinase, and glycogen synthase. Evidence for their role in oxidative and nonoxidative glucose metabolism. J. Clin. Invest. 1987, 80, 655. 10.1172/JCI113118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherington J.; Bordas V.; Gaiba A.; Garton N. S.; Naylor A.; Rawlings A. D.; Slingsby B. P.; Smith D. G.; Takle A. K.; Ward R. W. Bioorg. Med. Chem. Lett. 2003, 13, 3055–3057. 10.1016/S0960-894X(03)00645-0. [DOI] [PubMed] [Google Scholar]
- Cantley L. C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B.; Guillermet-Guibert J.; Graupera M.; Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- Dienstmann R.; Rodon J.; Serra V.; Tabernero J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- Rowan W. C.; Smith J. L.; Affleck K.; Amour A. Targeting phosphoinositide 3-kinase δ for allergic asthma. Biochem. Soc. Trans. 2012, 40, 240–245. 10.1042/BST20110665. [DOI] [PubMed] [Google Scholar]
- Sriskantharajah S.; Hamblin N.; Worsley S.; Calver A. R.; Hessel E. M.; Amour A. Targeting phosphoinositide 3-kinase δ for the treatment of respiratory diseases. Ann. N. Y. Acad. Sci. 2013, 1280, 35–39. 10.1111/nyas.12039. [DOI] [PubMed] [Google Scholar]
- Rückle T.; Schwarz M. K.; Rommel C. PI3Kγ inhibition: towards an ‘aspirin of the 21st century’?. Nat. Rev. Drug Discovery 2006, 5, 903–918. 10.1038/nrd2145. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B.; Ali K.; Bilancio A.; Geering B.; Foukas L. C. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem. Sci. 2005, 30, 194–204. 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Down K. D.; Amour A.; Baldwin I. R.; Cooper A. W. J.; Deakin A. M.; Felton L. M.; Guntrip S. B.; Hardy C.; Harrison Z. A.; Jones K. A.; Jones P.; Keeling S. E.; Le J.; Livia S.; Lucas F.; Luniss C. J.; Parr N. J.; Robinson E.; Rowland P.; Smith S.; Thomas D. A.; Vitulli G.; Washio Y.; Hamblin J. N. Optimization of Novel Indazoles as Highly Potent and Selective Inhibitors of Phosphoinositide 3-Kinase δ for the Treatment of Respiratory Disease. J. Med. Chem. 2015, 58, 7381–7399. 10.1021/acs.jmedchem.5b00767. [DOI] [PubMed] [Google Scholar]
- http://www.reactionbiology.com.
- A homology model of PI3Kδ was developed using an X-ray structure of PI3Kγ (1vgwt) as a template and using the Biopolymer Composer module in Sybyl v7.2.
- Sutherlin D. P.; Baker S.; Bisconte A.; Blaney P. M.; Brown A.; Chan B. K.; Chantry D.; Castanedo G.; DePledge P.; Goldsmith P.; Goldstein D. M.; Hancox T.; Kaur J.; Knowles D.; Kondru R.; Lesnick J.; Lucas M. C.; Lewis C.; Murray J.; Nadin A. J.; Nonomiya J.; Pang J.; Pegg N.; Price S.; Reif K.; Safina B. S.; Salphati L.; Staben S.; Seward E. M.; Shuttleworth S.; Sohal S.; Sweeney Z. K.; Ultsch M.; Waszkowycz B.; Wei B. Potent and selective inhibitors of PI3Kδ: Obtaining isoform selectivity from the affinity pocket and tryptophan shelf. Bioorg. Med. Chem. Lett. 2012, 22, 4296–4302. 10.1016/j.bmcl.2012.05.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.