

Abstract

(+)-Negamycin, isolated from Streptomyces purpeofuscus, shows antimicrobial activity against Gram-negative bacteria and readthrough activity against nonsense mutations. Previously, we reported that two natural negamycin analogues, 5-deoxy-3-epi-negamycin and its leucine adduct, have more potent readthrough activity in eukaryocytes (COS-7 cells) than negamycin but possess no antimicrobial activity and no in vitro readthrough activity in prokaryotic systems. In the present study, on leucyl-3-epi-deoxynegamycin, a structure–activity relationship study was performed to develop more potent readthrough agents. In a cell-based readthrough assay, the derivative 13b with an o-bromobenzyl ester functions as a prodrug and exhibits a higher readthrough activity against TGA-type PTC than the aminoglycoside G418. This ester (13b) shows an in vivo readthrough activity with low toxicity, suggesting that it has the potential for treatment of hereditary diseases caused by nonsense mutations.
Keywords: Duchenne muscular dystrophy, Leucyl-3-epi-deoxynegamycin, (+)-Negamycin, Readthrough
In recent years, more than 1,800 hereditary diseases have been reported,1 and it is known that 5–20% of these are attributable to nonsense mutations.2 Because these nonsense mutations provide congenital premature termination codons (PTCs) in mRNA, normal translation of the corresponding functional proteins is disrupted. Specifically, either protein translation is terminated at the PTC site with the production of a nonfunctional truncated protein or the mRNA with a PTC is easily recognized and degraded by the inherent nonsense-mediated mRNA decay (NMD) system. Consequently, the corresponding protein is largely not produced following the emergence of PTC in mRNA, and this is a basis of hereditary diseases. As a fundamental treatment to regenerate the intact protein from the mRNA that has lost its normal translational ability, gene transfer using virus vectors and stem cell transplantation have been studied extensively.3,4 However, there is currently no accepted clinical treatment for nonsense-associated diseases, although an antisense-mediated exon skipping drug, eteplirsen (AVI-4658), received accelerated approval from the US Food and Drug Administration in 2016 after controversy over its efficacy for the treatment of Duchenne muscular dystrophy (DMD).5 Recently, another strategy, application of “readthrough drugs”, has been attracting attention as a new therapeutic method with which to treat nonsense-associated diseases.6 It has been reported that readthrough compounds can skip PTCs and lead to the production of functional full-length proteins. Most studies concerning readthrough drugs have been directed against two serious hereditary diseases, DMD and cystic fibrosis (CF). DMD is a serious progressive X-linked recessive genetic disorder occurring in 1 of ∼3,500 newborn boys.7 Approximately 20% of DMD cases are caused by a nonsense mutation of the dystrophin gene.8 Dystrophin protein plays an essential role in maintenance of the structure of muscle cells, and its deficiency causes dysfunction and necrosis of these cells.9
Approximately 10% of CF cases are caused by a nonsense mutation of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Hypoactivity of CFTR causes intestinal malabsorption, obstruction, and respiratory tract infection as a result of chloride ion defective secretion in digestive and respiratory organs.10 Therefore, promotion of readthrough drugs by chemotherapy is a promising challenge.
To date, many small molecules that exhibit readthrough-promoting activity have been reported and include various aminoglycoside antibiotics, such as gentamicin,11 G418,12 arbekacin13 and their analogues.14 Barton-Davis et al. reported that, after administration of gentamicin, the dystrophin expression increases in the mdx mouse, an animal model of DMD,11 and Sabbavarapu et al. reported that gentamicin-derived novel aminoglycoside derivatives enhance suppression of diseases that cause nonsense mutations.14 However, it is generally known that there are serious side effects such as nephrotoxicity15 and ototoxicity16 from long-term treatment with aminoglycosides. Recently, a nonaminoglycoside 1,2,4-oxadiazole derivative Ataluren,17 which was found by chemical library screening, has been conditionally approved from the European Medicines Agency (EMA) for DMD.18
(+)-Negamycin (1, (2-(2-(3R,5R)-3,6-diamino-5-hydroxyhexanoyl)-1-methylhydrazinyl)acetic acid) is a dipeptide-like antibiotic that was isolated in 1970 from Streptomyces purpeofuscus and that exhibits efficacious antimicrobial activity against Gram-negative microorganisms.19 In 2003, Arakawa et al. reported that (+)-negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice with toxicity lower than that of gentamicin.20 Therefore, 1 was regarded as a new readthrough drug candidate for treatment of DMD and other diseases caused by nonsense mutations.
To date, using 1 as a lead compound in a structure–activity relationship (SAR) study, we have obtained several compounds with more potent readthrough activity than 1.21,22 Particularly, as shown in Figure 1, we found that native 5-deoxy-3-epi-negamycin analogues23 such as 3-epi-deoxynegamycin (2) and leucyl-3-epi-deoxynegamycin (3) show potent readthrough activity with no antimicrobial activity,21 suggesting that these analogues have no effect on the prokaryotic ribosomal system. We demonstrated that these analogues fail to show any readthrough activity in a prokaryotic cell-free translation system and were therefore thought to be more suitable leads for the development of readthrough drugs with a lower risk of emergence of antibiotic-resistant bacterial strains. Moreover, from a SAR study focused on the length of main chain in 2, the derivative 4 (TCP-112) with one less carbon atom showed a higher readthrough activity than 1 or 2.22 Further optimization of 4 demonstrated that the o-bromo- and m-chlorobenzyl ester derivatives (5 and 6, respectively) as prodrugs exhibit more potent readthrough activity than 4. This is probably due to the enhanced hydrophobic properties leading to an improved cell penetration ability and increased intracellular production of parent drug 4.
Figure 1.

Structures of readthrough compounds.
As leucyl-3-epi-deoxynegamycin (3) has an additional leucine residue connected to the 3-amino group of 2 through an amide bond, the amino function that had been thought to be important for the readthrough activity is masked. However, because 2 and 3 exhibit similar readthrough activities, the role of this residue in the readthrough activity was of interest. Accordingly, a SAR study based on a substitution of the Leu residue at the 3-position was performed. Ester derivatives of 3 as prodrugs were also investigated in an effort to develop more potent readthrough compounds. A prodrug (13b) containing an o-bromobenzyl moiety was found to exhibit a higher readthrough activity than the aminoglycoside G418 in a cell-based readthrough assay. The in vivo readthrough activity of 13b, using transgenic readthrough evaluation and assessment by dual reporter (READ) mice,24 and its in vivo acute toxicity were also evaluated.
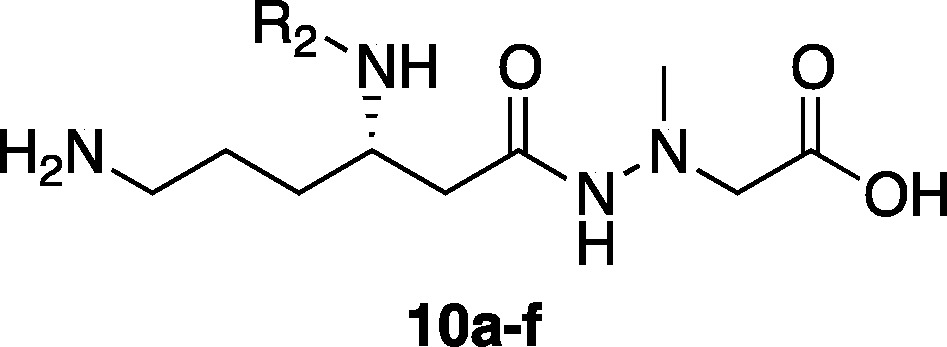
A series of derivatives (10a–f) with different amino acid residues at the 3-position of 3 were synthesized using the procedure previously reported for 3.21 Briefly, as illustrated in Scheme 1, the intermediate hydrazide (8) was prepared in 14% overall yield from commercially available Boc-Orn(Cbz)-OH (7) in 6 steps. The resulting hydrazide (8) was treated with 4 M HCl/dioxane to deprotect the N-Boc group at the 3-position followed by coupling with several Boc-protected amino acids (for 9a, Boc-d-Leu-OH; 9c, Boc-Phe-OH; 9d, Boc-Tyr-OH; 9e, Boc-Val-OH; 9f, Boc-Gly-OH) and 9b ((S)-2-hydroxy-4-pentanoic acid) by a 1-ethyl-3-(3-(dimethyl amino)propyl)carbodiimide/1-hydroxy-benzotriazole (EDC/HOBt) method25 to give the protected derivatives (9a–f) in 50–95% yields. The Cbz and benzyl protective groups of 9a–f were removed by hydrogenation in the presence of 10% Pd/C under H2 at 1 atm. Such treatment of 9b gave a crude product that was purified by RP-HPLC to afford 10b in 65% yield. In other derivatives (9a, 9c–f), the crude products obtained were further treated with 4 M HCl/dioxane to remove the N-Boc group, and this was followed by purification by RP-HPLC to afford the desired derivatives (10a, 10c–f) in 30–65% yields.
Scheme 1. Synthesis of Derivatives 10a–f.

Reagents and conditions: (a) 4 M HCl/dioxane, rt, 1 h; (b) Boc-protected amino acid (for 9a, 9c–f) or (S)-2-hydroxy pentanoic acid (for 9b), EDC·HCl, HOBt·H2O, Et3N, DMF, rt, overnight, 50–95% (2 steps); (c) H2, Pd/C, MeOH, rt, 3 h, quant, then RP-HPLC, 65% (for 10b); (d) 4 M HCl/dioxane, rt, 1 h, then RP-HPLC, 30–47% (for 10a, c–f). R2 substituents of derivatives 10a–f are shown in Table 1.
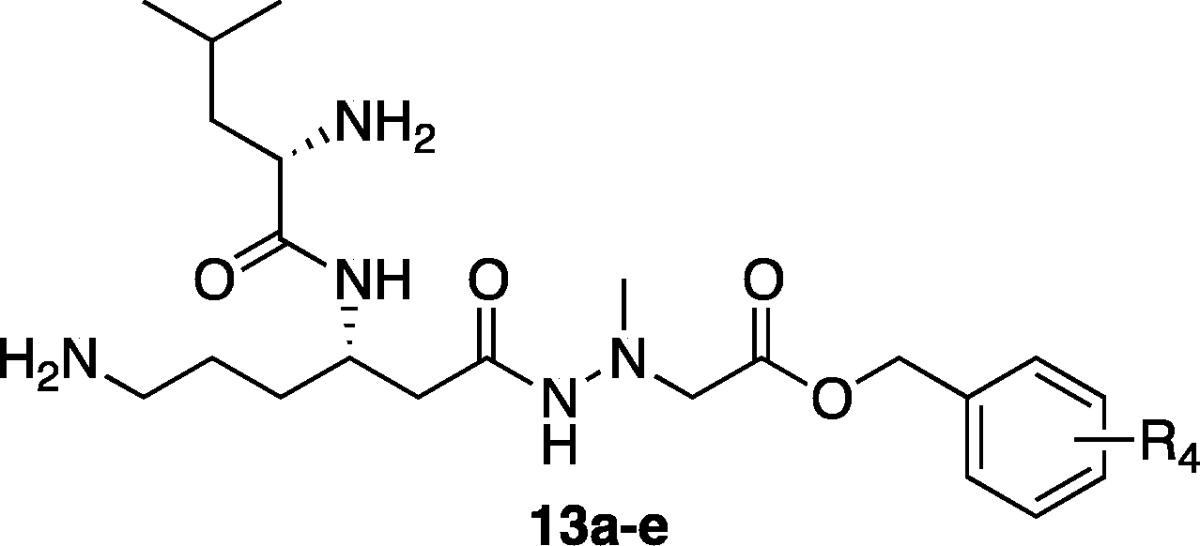
The esters derivatives 13a–e were synthesized from intermediate 11(21) in four steps as illustrated in Scheme 2. After the Cbz and benzyl protecting groups were removed by hydrogenation, the resultant 6-amino group was protected by treatment with Boc2O in the presence of Et3N in DMF. Subsequently, without further purification, the crude product was directly condensed with benzyl alcohol or substituted benzyl alcohol using N,N′-dicyclohexylcarbodiimide (DCC) in the presence of a catalytic amount of 4-dimethylaminopyridine (DMAP) to give corresponding ester (12a–e) in 43–82% yields after three steps. Finally, deprotection of two N-Boc groups was performed with 4 M HCl/dioxane, and subsequent purification with RP-HPLC afforded the desired derivatives (13a–e) in 23–55% yields.
Scheme 2. Synthesis of Ester Derivatives 13a–e.

Reagents and conditions: (a) H2, Pd/C, MeOH, rt, 35–60 min; (b) Boc2O, Et3N, DMF, rt, 1–2.5 h; (c) benzyl alcohol or substituted benzyl alcohol, DCC, DMAP, DMF, rt, 3.5 h–overnight, 43–82% (3 steps); (d) 4 M HCl/dioxane, rt, 1 h, then RP-HPLC purification, 23–55%. R4 substituents of derivatives 13a–e are shown in Table 2.
The readthrough activities of the synthesized derivatives 10a–f were evaluated using a previously described procedure21 in a cell-based reporter assay using COS-7 cells. The reporter consisted of a dual-reporter plasmid encoding β-galactosidase and luciferase genes that were connected with a TGA-containing nucleotide sequence used as a PTC.21 All derivatives were evaluated at a concentration of 200 μM, and the results are shown in Table 1. As previously reported,21 leucyl-3-epi-deoxynegamycin (3) showed a stronger readthrough activity (2.72) than that of (+)-negamycin (1) (1.54). The activity of derivative 10a with d-Leu and of 10b with 2-hydroxyisovaleic acid were approximately half that of 3. This result suggests that the appropriate configuration at the α-position and the existence of α-amino group in the Leu side chain are important for potent activity. The conversion of Leu to aromatic amino acids l-phenylalanine (10c) or l-tyrosine (10d) to understand the effect of hydrophobic interaction with an aromatic group failed to increase the readthrough activity.
Table 1. Cell-Based Readthrough Activity of Derivatives 10a–f.
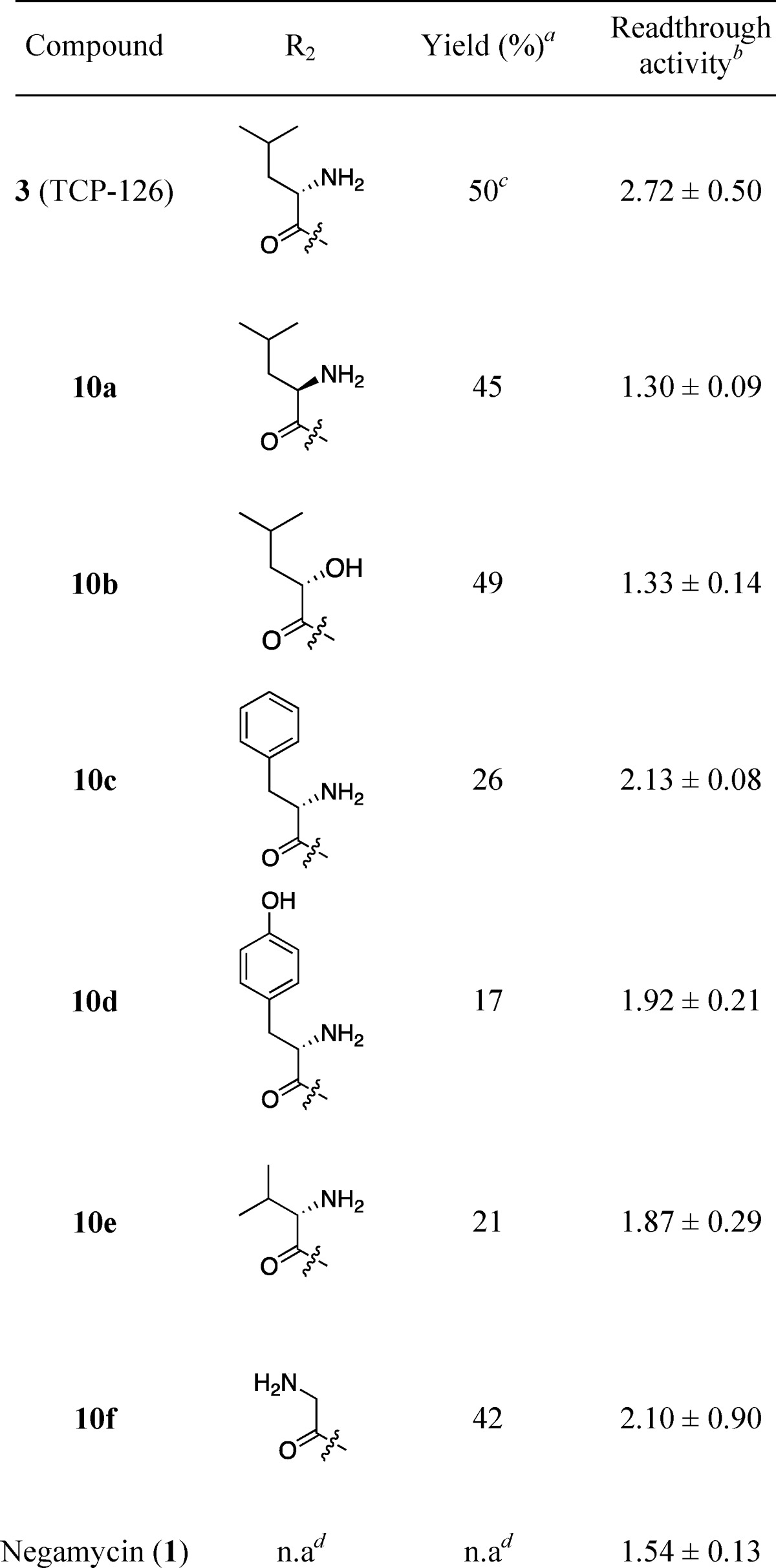
Yields were calculated from intermediate 8 for analogues 3 and 10a–f, respectively.
Cell-based readthrough activity (ratio) relative to control (D-MEM) in COS-7 cells; compounds are evaluated at a concentration of 200 μM. Values are expressed as the mean ± SD (n = 3).
See ref (21).
N.a: not applicable.
Next, for understanding the effect on the activity of the side chain aliphatic group, the readthrough activity of derivative with one less carbon atom 10e (l-valine) and derivative lacking a carbon atom (glycine, 10f) was evaluated. However, these derivatives showed limited activity compared to 3, and these results suggest that the isobutyl group existing in 3 provides a more favorable side chain structure for the potent readthrough activity.
Because in the previous study the benzyl esters of derivative 4 as a prodrug showed increased activity in cell-based assays,22 we synthesized five different benzyl esters (13a–e) derived from the most potent compound (3) obtained in the present SAR study. As shown in Table 2, the readthrough activity of 13a–e is higher than that of the original analogue 3 (2.58). In particular, the readthrough activity of unsubstituted (13a), o-bromo (13b), and o-nitrobenzyl esters (13d) (8.92, 8.84, and 9.15, respectively) were noteworthy. On the other hand, no significant cytotoxicity of these derivatives was observed in COS-7 cells and human dermal fibroblasts (Figure S1). Furthermore, the calculated log P (ClogP) value, which is an indication of hydrophobicity, highlighted that o-bromobenzyl ester 13b (ClogP; 0.85) showed the best value among them. Considering these results comprehensively, we selected 13b (TCP-199) as a representative prodrug and performed the detailed biological activity of 13b. In a dose-dependent assay (Figure 2), prodrug 13b shows an improved potent readthrough activity (8.89 ± 0.10, 3.95-fold increase) compared to that of parent drug 3 (2.25 ± 0.23) at 200 μM. In particular, this value is higher than that of G418 (7.20 ± 0.48), an aminoglycoside possessing the most potent readthrough activity but also strong toxicity.26 Prodrug 13b exhibits dose-dependent readthrough activity at concentrations ranging from 10 to 200 μM. Moreover, the in vitro readthrough activity of 13b (200 μM) against other PTCs (TAG and TAA) was also evaluated using the previously reported plasmids,21 resulting in good activity with values of 2.55 ± 0.05 and 1.22 ± 0.05, respectively. These activities were more potent than those observed in 3 (1.30 ± 0.06 and 1.11 ± 0.05, respectively). In the TGA sequence, 13b was shown to have a 2.69% readthrough frequency in comparison to the in-frame control containing TGG (tryptophan coding) sequence.
Table 2. . Cell-Based Readthrough Activity of Esters 13a–e.
| compound | R4 | yield (%)a | readthrough activityb | ClogPc |
|---|---|---|---|---|
| 3 (TCP-126) | n.ad | n.ad | 2.58 ± 0.07 | –1.97 |
| 13a | H | 16 | 8.92 ± 0.19 | 0.02 |
| 13b (TCP-199) | o-Br | 28 | 8.84 ± 0.07 | 0.85 |
| 13c | m-Cl | 13 | 5.37 ± 0.22 | 0.58 |
| 13d | o-NO2 | 45 | 9.15 ± 0.21 | –0.23 |
| 13e | m-OMe | 13 | 5.15 ± 0.13 | –0.10 |
Yields were calculated from intermediate 11 for ester derivatives 13a–e.
Cell-based readthrough activity (ratio) relative to control (D-MEM) in COS-7 cells; compounds are evaluated at a concentration of 200 μM. Values are expressed as the mean ± SD (n = 3).
Value of the calculated log P (ClogP) determined by ChemDraw professional 16.0.
N.a: not applicable.
Figure 2.

Cell-based readthrough activity (TGA) of 13b (TCP-199) in the COS-7 cells. Error bars indicate SD (n = 3). NM: (+)-negamycin (1).
For confirming that 13b behaves as a prodrug, it was treated with porcine liver esterase (110 units of enzyme/mL) in a 100 mM phosphate buffer (pH 7.4) at 37 °C, a process based on a previously reported procedure.22 After incubation, the reaction mixture was analyzed by RP-HPLC, and the conversion of 13b to the parent compound (3) by esterase was evaluated. In addition, the peak corresponding to a new metabolite was collected and identified by high-resolution mass spectrometry. As a result, time-dependent production of parent 3 (TCP-126) along with the decrease of 13b (Figure S2) was observed.
Incubated with human plasma, 13b was almost completely hydrolyzed within 30 min. Under similar reaction conditions, 13b was found to be stable in the absence of esterase (99% remaining after a 6 h incubation at 37 °C). This result suggests that ester 13b functions as a prodrug that produces the active parent drug (3) (TCP-126) probably intracellularly.
For the in vivo biological effects of prodrug 13b to be evaluated, its readthrough activity was evaluated using READ mice,24 which express a dual-reporter gene segmentalized with a PTC. Compound 13b was injected subcutaneously in the abdominal region of READ mice at a dose of 1 mg in saline (0.2 mL) day–1 20 g–1 body weight for 7 days (Figure 3A). As a positive control, arbekacin, a clinically used minimally toxic aminoglycoside that is known to have a relatively strong readthrough activity,13 was used. In comparison to the negative control (saline), 13b-treated mice show a significant increase in readthrough activity (1.59 ± 0.11), although this is slightly inferior, but statistically insignificantly so, to that of arbekacin (1.94 ± 0.32). This result indicated that 13b effectively functions as a readthrough agent in vivo.
Figure 3.

(A) In vivo readthrough activity in READ mice. Compound 13b (TCP-199) and ABK (arbekacin) were subcutaneously injected into the abdominal region of READ mice at a dosage of 1 mg day–1 20 g–1 body weight for 7 days. Data are mean ± SD * p < 0.01. (B) Acute toxicity test of 13b. The body weight of 13b-treated B10 mice (n = 4) during 2 weeks was measured in comparison to saline-treated B10 mice (n = 3) as a control. The analogue was subcutaneously injected only once (10 mg day–1 20 g–1 body weight) into the abdominal region of B10 mice.
In an evaluation of in vivo acute toxicity for 2 weeks based on the body-weight change of B10 mice, shown in Figure 3B, after single subcutaneous injection of 13b (10 mg day–1 20 g–1 body-weight) into the abdominal region of mice, the body-weight of 13b-treated mice showed a gradual increased similar to that observed in control mice, although a slight weight decrease was observed just after administration (day 1). The result of this preliminary experiment suggested that the acute toxicity of 13b is low, and consequently, 13b has the potential for long-term treatment of DMD.
In conclusion, we synthesized a series of derivatives focused on negamycin analogue (3) and evaluated their readthrough activities with a cell-based reporter assay. On the basis of the modification of analogue 3, we synthesized o-bromobenzyl ester 13b (TCP-199) as a prodrug. This compound exhibits a higher readthrough activity than those of both the parent (3) and aminoglycoside G418. By treatment with esterase, 13b was converted to the parent (3) in vitro. In in vivo studies using READ mice, 13b exhibited readthrough activity with a low acute toxicity. It was concluded that 13b (TCP-199) has potential for the treatment of nonsense-associated hereditary diseases by readthrough chemotherapy. Further SAR studies of 3 and evaluation of the biological effects of 13b in different nonsense-associated diseases are in progress in an effort to develop a clinical candidate.
Acknowledgments
The authors thank Dr. Cédric Rentier, Tokyo University of Pharmacy and Life Sciences, for proofreading the manuscript and improvement suggestions, as well as Mr. Masaya Kotake and Ms. Noriko Omura, Tokyo University of Pharmacy and Life Sciences for the technical assistance and Dr. Yamato Kikkawa, Tokyo University of Pharmacy and Life Sciences, for kindly providing us with the human dermal fibroblast cell line. This work was supported by the Japan Society for the Promotion of Science (JSPS), KAKENHI including a Grant-in-Aid for JSPS Research Fellow 16J08454, a Grant-in-Aid for Scientific Research (B) 23390029, a Grant-in-Aid for Young Scientists (B) 25860092, a Grant-in-Aid for Research Activity Start-up 25893258, and a Grant-in-Aid for Scientific Research on Innovative Areas “Chemical Biology of Natural Products” from the Ministry of Education, Culture, Sports, Science and Technology, and Platform for Drug Discovery, Informatics and Structural Life Science, and Intramural Research Grant (29-4) for Neurological and Psychiatric Disorders of NCNP. MEXT-supported Program for the Strategic Research Foundation at Private Universities.
Glossary
Abbreviations
- PTCs
premature termination codons
- NMD
nonsense-mediated mRNA decay
- DMD
Duchenne muscular dystrophy
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- EMA
European Medicines Agency
- SAR
structure–activity relationship
- READ
readthrough evaluation and assessment by dual reporter
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOBt
1-hydroxy-benzotriazole
- Et3N
triethylamine
- DMF
N,N-dimethylformamide
- Cbz
benzyloxycarbonyl
- RP-HPLC
reversed-phase high-performance liquid chromatography
- Boc
tert-butoxycarbonyl
- DCC
N,N′-dicyclohexylcarbodiimide
- DMAP
4-dimethylaminopyridine
- D-MEM
Dulbecco’s modified Eagle medium
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00269.
Synthesis procedures, characterization of derivatives, biological assay protocols, and NMR data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Keeling K. M.; Bedwell D. M. Pharmacological suppression of premature stop mutations that cause genetic diseases. Curr. Pharmacogenomics 2005, 3, 259–269. 10.2174/157016005774913149. [DOI] [Google Scholar]
- Bidou L.; Hatin I.; Perez N.; Allamand V.; Panthier J. J.; Rousset J. P. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004, 11, 619–627. 10.1038/sj.gt.3302211. [DOI] [PubMed] [Google Scholar]
- Okada T.; Takeda S. Current challenges and future directions in recombinant AAV-mediated gene therapy of Duchenne muscular dystrophy. Pharmaceuticals 2013, 6, 813–836. 10.3390/ph6070813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. H.; Fujimoto N.; Sasakawa N.; Shirai S.; Ohkame T.; Sakuma T.; Tanaka M.; Amano N.; Watanabe A.; Sakurai H.; Yamamoto T.; Yamanaka S.; Hotta A. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A.; Krieg A. M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R.; Mogg A. E.; Heywood L. A.; Nitschke L.; Burke J. F. Aminoglycoside suppression at UAG, UAA and UGA codons in Escherichia coli and human tissue culture cells. Mol. Gen. Genet. 1989, 217, 411–418. 10.1007/BF02464911. [DOI] [PubMed] [Google Scholar]
- Bakker E.; Veenema H.; Den Dunnen J. T.; van Broeckhoven C.; Grootscholten P. M.; Bonten E. J.; van Ommen G. J.; Pearson P. L. Germinal mosaicism increases the recurrence risk for new duchenne muscular dystrophy mutations. J. Med. Genet. 1989, 26, 553–559. 10.1136/jmg.26.9.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima Y.; Yagi M.; Okizuka Y.; Awano H.; Zhang Z.; Yamauchi Y.; Nishio H.; Matsuo M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. 10.1038/jhg.2010.49. [DOI] [PubMed] [Google Scholar]
- Ozawa E.; Yoshida M.; Suzuki A.; Mizuno Y.; Hagiwara Y.; Noguchi S. Dystrophin-associated proteins in muscular dystrophy. Hum. Mol. Genet. 1995, 4, 1711–1716. 10.1093/hmg/4.suppl_1.1711. [DOI] [PubMed] [Google Scholar]
- Du M.; Jones J. R.; Lanier J.; Keeling K. M.; Lindsey J. R.; Tousson A.; Bebok Z.; Whitsett J. A.; Dey C. R.; Colledge W. H.; Evans M. J.; Sorscher E. J.; Bedwell D. M. Aminoglycoside suppression of a premature stop mutateon in a Cftr–/– mouse carrying a human CFTR-G542X transgene. J. Mol. Med. 2002, 80, 595–604. 10.1007/s00109-002-0363-1. [DOI] [PubMed] [Google Scholar]
- Barton-Davis E. R.; Cordier L.; Shoturma D. I.; Leland S. E.; Sweeney H. L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Invest. 1999, 104, 375–381. 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke J. F.; Mogg A. E. Suppression of a nonsense mutation in mammalian calls in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985, 13, 6265–6272. 10.1093/nar/13.17.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda R.; Shiozuka M.; Wagatsuma A.; Takahashi Y.; Ikeda D.; Nonomura Y.; Matsuo M.; Nishida A.. Read through inducer, and therapeutic agent for nonsense-mutation-type genetic diseases containing arbekacin. WO2011096484, 2011.
- Sabbavarapu N. M.; Shavit M.; Degani M.; Smolkin B.; Belakhov V.; Baasov T. Design of novel aminoglycoside derivatives with enhanced suppression of diseases-causing nonsense mutations. ACS Med. Chem. Lett. 2016, 7, 418–423. 10.1021/acsmedchemlett.6b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P.; Tulkens P. M. Aminoglycosides: Nephrotoxicity. Antimicrob. Agents Chemother. 1999, 43, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchin T.; Cortopassi G. Proposed Molecular and Cellular Mechanism for Aminoglycoside Ototoxicity. Antimicrob. Agents Chemother. 1994, 38, 2517–2520. 10.1128/AAC.38.11.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch E. M.; Barton E. R.; Zhuo J.; Tomizawa Y.; Friesen W. J.; Trifillis P.; Paushkin S.; Patel M.; Trotta C. R.; Hwang S.; Wilde R. G.; Karp G.; Takasugi J.; Chen G.; Jones S.; Ren H.; Moon Y. C.; Corson D.; Turpoff A. A.; Campbell J. A.; Conn M. M.; Khan A.; Almstead N. G.; Hedrick J.; Mollin A.; Risher N.; Weetall M.; Yeh S.; Branstrom A. A.; Colacino J. M.; Babiak J.; Ju W. D.; Hirawat S.; Northcutt V. J.; Miller L. L.; Spatrick P.; He F.; Kawana M.; Feng H.; Jacobson A.; Peltz S. W.; Sweeney H. L. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- Haas M.; Vlcek V.; Balabanov P.; Salmonson T.; Bakchine S.; Markey G.; Weise M.; Schlosser-Weber G.; Brohmann H.; Yerro C. P.; Mendizabal M. R.; Stoyanova-Beninska V.; Hillege H. L. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul. Disord. 2015, 25, 5–13. 10.1016/j.nmd.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Hamada H.; Takeuchi T.; Kondo S.; Ikeda Y.; Naganawa H.; Maeda K.; Okami Y.; Umezawa H. A new antibiotic, negamycin. J. Antibiot. 1970, 23, 170–171. 10.7164/antibiotics.23.170. [DOI] [PubMed] [Google Scholar]
- Arakawa M.; Shiozuka M.; Nakayama Y.; Hara T.; Hamada M.; Kondo S.; Ikeda D.; Takahashi Y.; Nonomura Y.; Sheykholeslami K.; Kondo K.; Kaga K.; Kitamura T.; Suzuki-Miyagoe Y.; Takeda S.; Matsuda R. Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J. Biochem. 2003, 134, 751–758. 10.1093/jb/mvg203. [DOI] [PubMed] [Google Scholar]
- Taguchi A.; Hamada K.; Kotake M.; Shiozuka M.; Nakaminami H.; Pillaiyar T.; Takayama K.; Yakushiji F.; Noguchi N.; Usui T.; Matsuda R.; Hayashi Y. Discovery of natural products possessing selective eukaryotic readthrough activity: 3-epi-deoxynegamycin and its leucine adduct. ChemMedChem 2014, 9, 2233–2237. 10.1002/cmdc.201402208. [DOI] [PubMed] [Google Scholar]
- Hamada K.; Taguchi A.; Kotake M.; Aita S.; Murakami S.; Takayama K.; Yakushiji F.; Hayashi Y. Structure-activity relationship studies of 3-epi-deoxynegamycin derivatives as potent readthrough drug candidates. ACS Med. Chem. Lett. 2015, 6, 689–694. 10.1021/acsmedchemlett.5b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S.; Yoshida K.; Ikeda T.; Iinuma K.; Honma Y.; Hamada M.; Umezawa H. 3-epi-Deoxynegamycin and leucyl-3-epi-deoxynegamycin produced by Streptomyces. J. Antibiot. 1977, 30, 1137–1139. 10.7164/antibiotics.30.1137. [DOI] [PubMed] [Google Scholar]
- Shiozuka M.; Wagatsuma A.; Kawamoto T.; Sasaki H.; Shimada K.; Takahashi Y.; Nonomura Y.; Matsuda R. Transdermal delivery of a readthrough-inducing drug: a new approach of gentamicin administration for the treatment of nonsense mutation-mediated disorders. J. Biochem. 2010, 147, 463–470. 10.1093/jb/mvp185. [DOI] [PubMed] [Google Scholar]
- König W.; Geiger R. New method for the synthesis of peptides: activation of the carboxyl group with dicyclohexylcarbodiimide by using 1-hydroxybenzotriazoles as additives. Chem. Ber. 1970, 103, 788–798. 10.1002/cber.19701030319. [DOI] [PubMed] [Google Scholar]
- Shulman E.; Belakhov V.; Wei G.; Kendall A.; Meyron-Holtz E. G.; Ben-Shachar D.; Schacht J.; Baasov T. Designer aminoglycosides that selectively inhibit cytoplasmic rather than mitochondrial ribosomes show decreased ototoxicity: a strategy for the treatment of genetic diseases. J. Biol. Chem. 2014, 289, 2318–2330. 10.1074/jbc.M113.533588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.