

Abstract

Starting from our previous eIF4A3-selective inhibitor 1a, a novel series of (piperazine-1-carbonyl)pyridin-2(1H)-one derivatives was designed, synthesized, and evaluated for identification of orally bioavailable probe molecules. Compounds 1o and 1q showed improved physicochemical and ADMET profiles, while maintaining potent and subtype-selective eIF4A3 inhibitory potency. In accord with their promising PK profiles and results from initial in vivo PD studies, compounds 1o and 1q showed antitumor efficacy with T/C values of 54% and 29%, respectively, without severe body weight loss. Thus, our novel series of compounds represents promising probe molecules for the in vivo pharmacological study of selective eIF4A3 inhibition.
Keywords: Eukaryotic initiation factor 4A3 (eIF4A3) inhibitor, RNA helicase, solubility, metabolic stability, P-gp substrate, pyridin-2(1H)-one derivative
The eukaryotic initiation factor 4A(eIF4A) family is a member of the ATP-dependent RNA helicase protein family and consists of three isoforms, eIF4A1, eIF4A2, and eIF4A3.1−4 While eIF4A1 and eIF4A2 are required for translation initiation,5−7 eIF4A3 controls RNA metabolism including nonsense-mediated mRNA decay (NMD) and RNA localization as a core component of the exon junction complex (EJC).8−15 NMD is a surveillance pathway to eliminate mRNAs that contain premature termination codons (PTCs), preventing the generation of detrimental gain-of-function or dominant-negative proteins.16−18 Cancer cells often harbor a wide variety of mutations introducing PTCs, and cancer cells are considered to be highly dependent on the NMD pathway to avoid the accumulation of aberrant proteins.19−22 Therefore, targeting eIF4A3 inhibition could potentially represent a promising approach for anticancer therapy; however, the detailed physiological functions of eIF4A3 are not clear. While some researchers have reported on the biological function of pan-eIF4A inhibition utilizing known natural products, hippuristanol23−26 and Pateamine A,27 selective eIF4A3 inhibition would be required to enable more definitive biological and pharmacological evaluation both in vitro and in vivo.
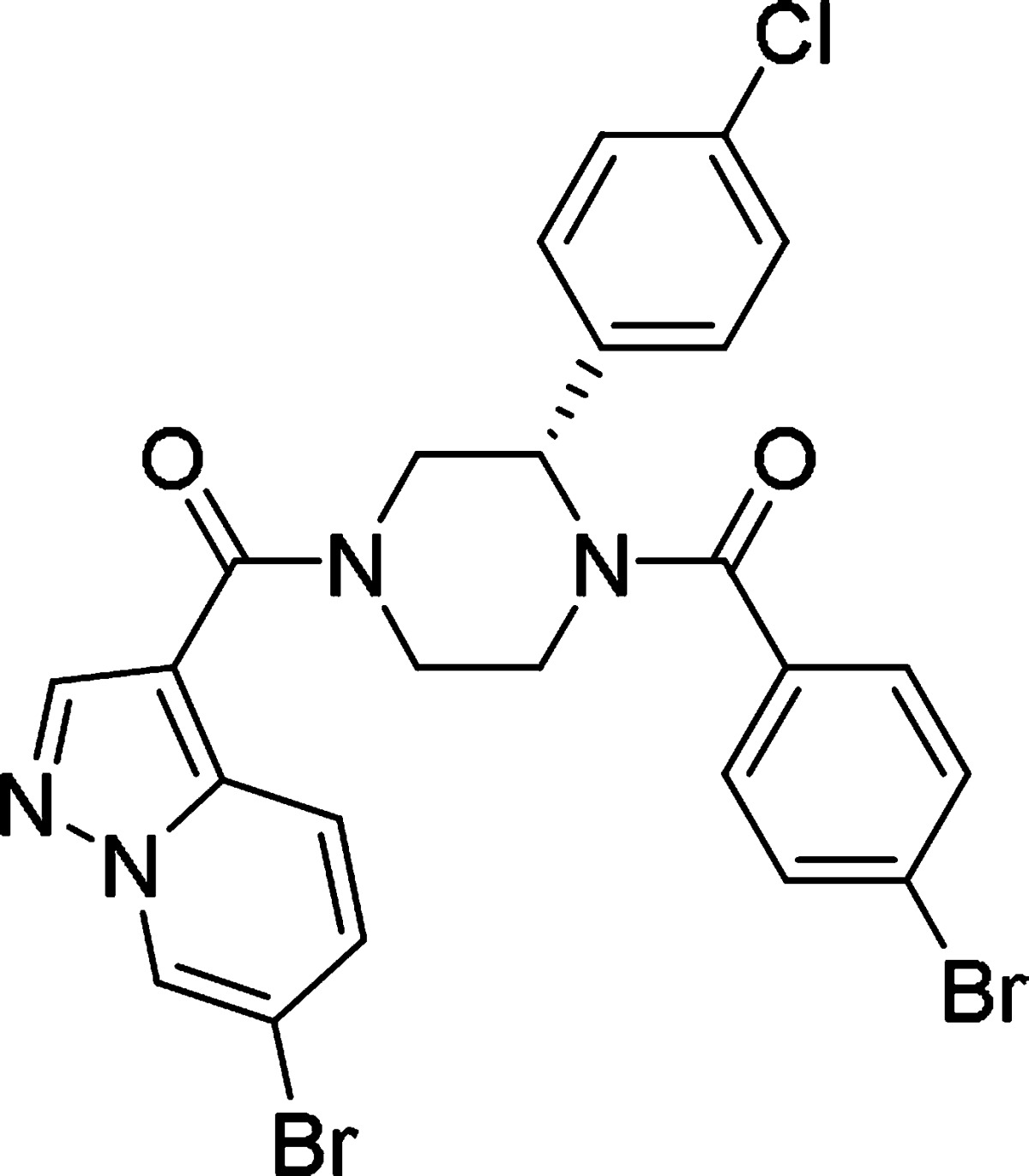
Recently, we discovered several classes of both ATP-competitive and allosteric eIF4A3 inhibitors, which showed potent and selective eIF4A3 inhibition28−30 from hit compounds through HTS of an in-house compound library. Among these, ATP-competitive inhibitors were characterized using ITC and SPR analyses.28 On the other hand, the allosteric eIF4A3 inhibitors exhibited significant NMD inhibition in a cellular reporter assay along with eIF4A3 inhibitory potency29 and were examined for understanding the function of eIF4A3 in RNA homeostasis.30 Although the allosteric inhibitors showed potent enzymatic activity as well as cellular potency, the physicochemical properties such as solubility, metabolic stability, and pharmacokinetics were insufficient for conducting further biological and pharmacological studies. To identify improved probe molecules for the study of eIF4A3 function, we continued our chemistry efforts on allosteric inhibitors using the novel 3-(4-chlorophenyl)-1,4-diacylpiperazine derivative 1a as a lead compound (Table 1).
Table 1. Biological and Physicochemical Profile of Lead Compound 1a.
| mouse
cassette PKd |
|||||
|---|---|---|---|---|---|
| eIF4A3 IC50 (μM)a | solubility (μg/mL)b | metabolic stability (μL/min/mg)c | AUCpo (ng h/mL)e | F (%)f | |
| 1a | 0.11 (0.09–0.13) | 0.46 | 227 | 9.0 | 5.7 |
IC50 is presented as the mean of duplicate experiments along with 95% confidence intervals (95% CI) in parentheses.
Solubility in pH 6.8.
Metabolic stability in mice microsomes.
Male ICR mice (n = 3). Dose: i.v. at 0.1 mg/kg; p.o. at 1 mg/kg.
Area under the curve from 0 to 8 h.
Bioavailability.
According to our previous research, we found that the (S)-3-phenyldiacylpiperazine scaffold of 1a was essential for potent eIF4A3 inhibition.29 Substitution or modification of the piperazine ring, including investigations of the corresponding (R)-enantiomer, demonstrated substantially reduced biological activity. In addition, SAR around the pendant 4-chlorophenyl moiety was also found to be narrow, with most modifications resulting in equal or reduced activity, as well. Therefore, we fixed the core scaffold as shown in Figure 1 and thought to modify the two amide moieties, the 4-bromobenzoyl moiety in the East region and the pyrazolo[1,5-a]pyridin-3-yl moiety in the West region, to optimize the biological and physicochemical properties of 1a.
Figure 1.
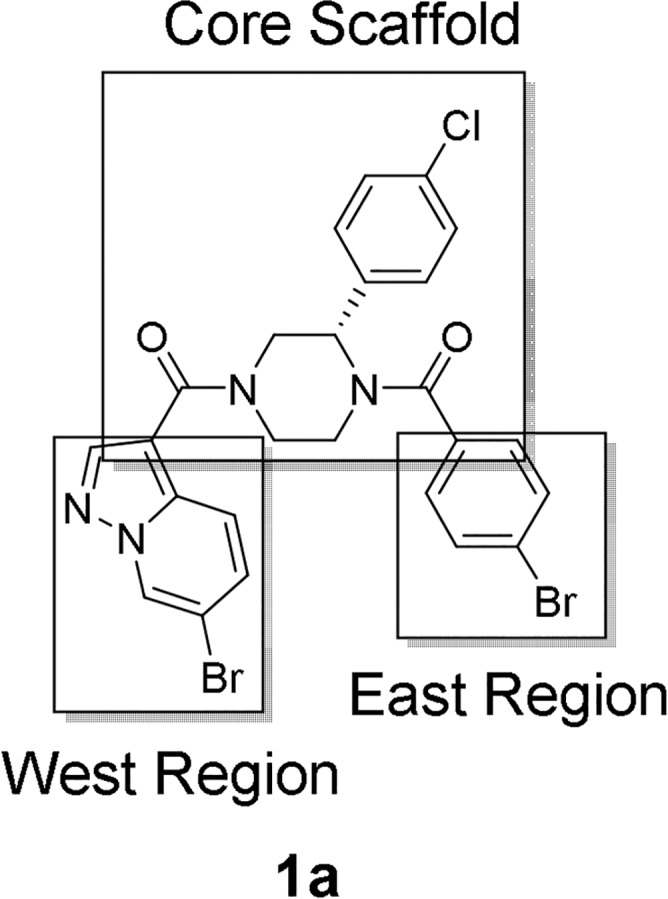
Synthetic strategies for improvement of physicochemical properties and PK profile.
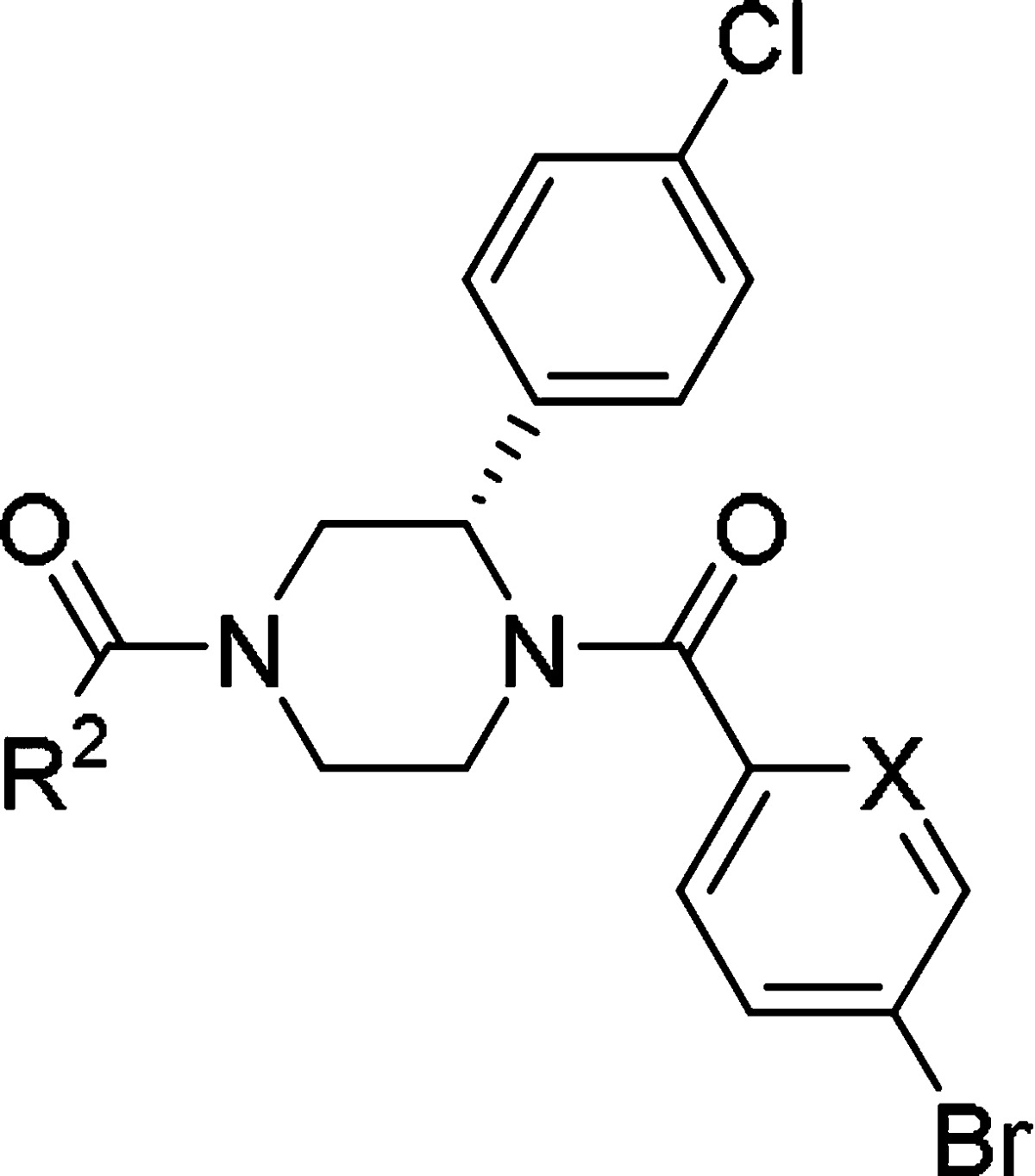
During the chemical modification of the East region, it was found that replacement of the 4-bromobenzoyl moiety with a 5-bromo-2-picolinoyl moiety generally improved solubility while maintaining eIF4A3 inhibitory potency (1b eIF4A3 IC50: 0.28 μM), although broad investigation of other substituents in the East region,29 acyl and otherwise, generally showed substantially reduced biological activity. On the other hand, an initial SAR campaign suggested that modification of the West region was tolerated for a wide variety of substituents to modulate both potency and physicochemical properties. Thus, we focused on the chemical modification of this region for further investigation. During derivative synthesis around this amide moiety, quinoline derivative 1c and isoquinoline derivative 1d were identified as promising bicyclic analogues. Further investigation of bicyclic 6–6 fused ring systems led us to find isoquinolin-1(2H)-one derivative 1e as a potent lead compound with improved solubility and metabolic stability while still maintaining eIF4A3 inhibitory potency. Additionally, 1e showed a slightly improved PK profile compared to 1a, along with improved physicochemical properties as shown in Table 2.
Table 2. Initial SAR of Diacyl Moieties: All Compounds Are (S)-Enantiomers.

IC50 is presented as the mean of duplicate experiments along with 95% confidence intervals (95% CI) in parentheses.
Solubility in pH 6.8.
Metabolic stability in mice microsomes.
Male ICR mice (n = 3). Dose: i.v. at 0.1 mg/kg; p.o. at 1 mg/kg.
Area under the curve from 0 to 8 h.
Bioavailability.
As the start of compound optimization for 1e, we examined the replacement of bicyclic ring of 1f, the racemate of 1e, into a biaryl moiety in order to facilitate synthetic accessibility to new analogues during our synthesis campaign aimed at efficient SAR data collection. The 3-phenylpyridin-2(1H)-one derivative 1g thus synthesized showed potent eIF4A3 inhibition (1g eIF4A3 IC50: 0.41 μM). Therefore, we chose 1g as a new lead compound and conducted further optimization of related 3-arylpyridin-2(1H)-one derivatives. In order to collect SAR information rapidly for these new pyridin-2(1H)-one derivatives, we examined substituent effects on the phenyl ring of pyridin-2(1H)-one utilizing parallel synthesis. Assay results for representative compounds are listed in Table 3.
Table 3. Representative Compounds in Parallel Synthesis for 3-Phenylpyridin-2(1H)-one Derivatives.

| R | eIF4A3 IC50 (μM)a | R | eIF4A3 IC50 (μM)a | ||
|---|---|---|---|---|---|
| 1h | 2-Cl | 0.42 (0.33–0.53) | 1k | 3-CN | 0.19 (0.12–0.29) |
| 1i | 3-Cl | 0.29 (0.25–0.34) | 1l | 3-CF3 | 0.7 (0.5–0.9) |
| 1j | 4-Cl | 1.1 (0.8–1.5) | 1m | 3-SO2Me | 0.49 (0.35–0.69) |
IC50 is presented as the mean of duplicate experiments along with 95% confidence intervals (95% CI) in parentheses.
Investigation of substituents on the benzene ring (compounds 1h–1j) suggested that substitution at the C3 position was favorable for obtaining potent eIF4A3 inhibitory activity (1i eIF4A3 IC50: 0.29 μM). Therefore, we focused our examination of substituent effects at the benzene C3-position. Replacement of the chloro group with a cyano group slightly improved potency (1k eIF4A3 IC50: 0.19 μM) compared to 1i. We postulated that an electron-withdrawing group (EWG) could be effective at enhancing eIF4A3 inhibition. However, further investigation of EWGs such as a trifluoromethyl group (1l) or a methanesulfonyl group (1m) at the C3-position did not yield improvements relative to the 3-cyano group. As a result, we concluded the 3-cyanophenyl group to be the most potent substituent for this series. Next, we synthesized the optically active (S)-pyridin-2(1H)-one derivative with a 3-cyanophenyl group possessing a 5-bromo-2-picolinoyl moiety in the East region. As expected, compound 1n showed highly potent eIF4A3 inhibitory activity (1n eIF4A3 IC50: 0.05 μM) with acceptable solubility (2.4 μg/mL at pH 6.8); however, it exhibited poor metabolic stability in mouse microsomes (113 μL/min/mg) (Table 4). We hypothesized that its instability in mouse microsomes originated from the acidity of the pyridin-2(1H)-one moiety. Based on this hypothesis, we designed and synthesized N-methyl compound 1o to reduce the acidity of the compound, which showed dramatically improved solubility and metabolic stability while maintaining eIF4A3 inhibitory potency, as expected. However, 1o was found to act as a P-gp substrate, with a high efflux ratio of 25.0. It is well-known that P-gp mediated efflux can have a negative impact a compound’s PK profile.31 Thus, we conducted further modification of these pyridin-2(1H)-one derivatives to address this issue.
Table 4. Compound Optimization of Pyridin-2(1H)-one Derivatives: All Compounds Are (S)-Enantiomers.
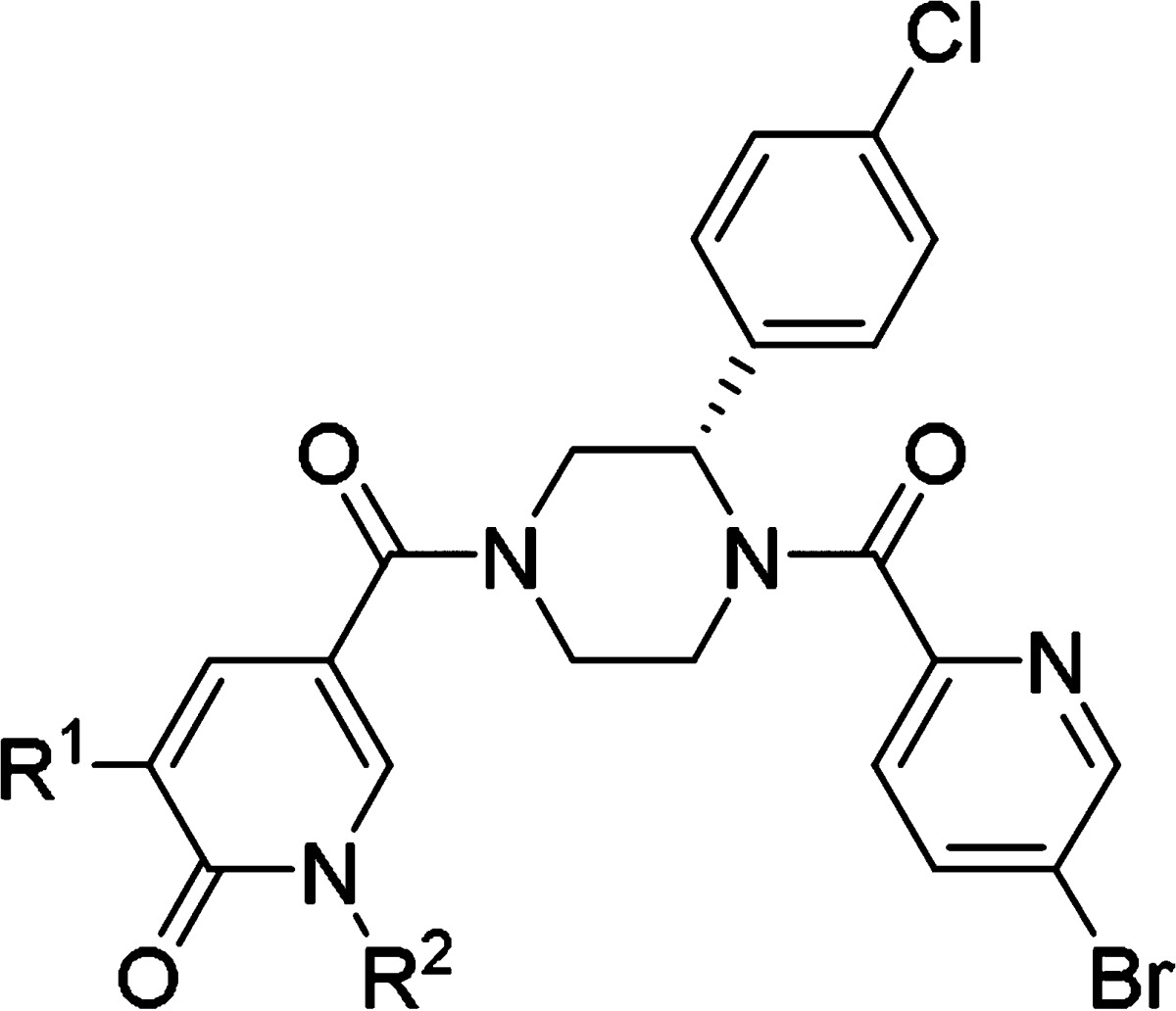
| R1 | R2 | eIF4A3 IC50 (μM)a | solubility (μg/mL)b | metabolic stability (μL/min/mg)c | efflux ratiod | tPSA (Å2)e | |
|---|---|---|---|---|---|---|---|
| 1n | 3-CNPh | H | 0.05 (0.04–0.05) | 2.4 | 113 | 35.0 | 110.2 |
| 1o | 3-CNPh | Me | 0.1 (0.06–0.15) | 12 | 6 | 25.0 | 99.3 |
| 1p | CF3 | Me | 0.75 (0.52–1.1) | 32 | 0 | 7.4 | 75.5 |
| 1q | CF3 | Ph | 0.14 (0.09–0.22) | 5.9 | 0 | 0.8 | 75.5 |
IC50 is presented as the mean of duplicate experiments along with 95% confidence intervals (95% CI) in parentheses.
Solubility in pH 6.8.
Metabolic stability in mice microsomes.
Efflux ratio in vitro MDR1 overexpressing cell line.
Calculated by Daylight software.
To reduce the potential for P-gp recognition, our medicinal chemistry strategy focused on the introduction of less polar substituents into the pyridin-2(1H)-one moiety, using the calculated total polar surface area (tPSA) value for reference (1o has a high tPSA value of 99.3 Å2).32 Replacement of the 3-cyanophenyl group with a trifluoromethyl group reduced the tPSA value to 75.5 Å2. Since eIF4A3 inhibitory potency was decreased by this modification (1p eIF4A3 IC50: 0.75 μM), we hypothesized that the biaryl scaffold was important for obtaining high potency. Based on this hypothesis, we investigated the introduction of an aryl substituent into the pyridin-2(1H)-one moiety while maintaining a tPSA value comparable to 1p. Accordingly, the introduction of an N-phenyl group instead of an N-methyl group into the pyridin-2(1H)-one moiety improved eIF4A3 inhibitory potency (1q IC50: 0.14 μM) comparable to 1o (1o IC50: 0.10 μM) while maintaining a low tPSA value of 75.5 Å2. During this modification, 1q showed lower P-gp efflux (efflux ratio of 0.8) compared to 1o (efflux ratio of 25.0). The PK profile of pyridin-2(1H)-one derivatives 1o and 1q are shown in Table 5. Expectantly, their PK profiles were dramatically improved relative to 1a.
Table 5. PK Profile of 1o and 1q in Mouse Cassettea.
Male ICR mice (n = 3). Dose: i.v. at 0.1 mg/kg; p.o. at 1 mg/kg.
Area under the curve from 0 to 8 h.
Bioavailability.
Given the discovery of potent eIF4A3 inhibitors 1o and 1q, which showed acceptable PK profiles, we examined their biochemical selectivity. As shown in Table 6, compounds 1o and 1q were both found to show highly selective eIF4A3 inhibitory activity over not only other eIF4A family proteins (eIF4A1 and eIF4A2) but also against other ATP-dependent RNA helicases such as BRR2 and DHX29, which is consistent with the activity found for compound 1a. Accordingly, we advanced these potent and selective eIF4A3 inhibitors further in vitro and in vivo biological evaluation.
Table 6. Biochemical Selectivity of 1o and 1q.
| eIF4A1 IC50 (μM) | eIF4A2 IC50 (μM) | eIF4A3 IC50 (μM)a | BRR2 IC50 (μM) | DHX29 IC50 (μM) | |
|---|---|---|---|---|---|
| 1a | >100 | >100 | 0.11 (0.09–0.13) | >100 | >100 |
| 1o | >100 | >100 | 0.1 (0.06–0.15) | >100 | >100 |
| 1q | >100 | >100 | 0.14 (0.09–0.22) | >100 | >100 |
IC50 is presented as the mean of duplicate experiments along with 95% confidence intervals (95% CI) in parentheses.
In a separate report, we developed a luciferase-based cellular NMD reporter assay to evaluate the cellular potency of our eIF4A3 inhibitors.30 However, this assay method could not be applied to in vivo pharmacodynamics (PD) evaluation. On the other hand, serine/arginine-rich splicing factor 2 (SC35) 1.6 and 1.7 kb have been known as substrates for NMD33 and are measurable in vivo. Thus, their mRNA expression levels could be a potential PD marker for NMD inhibition in vivo. To evaluate cellular potency, we chose SC35 1.6 kb as a PD marker in consideration of its applicability in vivo studies. Six hour treatment of 1o and 1q upregulated SC35 1.6 kb mRNA expression by ca. 8-fold at 3 μM and ca. 20-fold at 10 μM in colorectal carcinoma HCT-116 cells, whereas 1a showed 2.59-fold upregulation at 3 μM and 12.88-fold at 10 μM at 7 h, respectively. Consistent with these results, both 1o and 1q exhibited more potent growth inhibitory activity in HCT-116 cancer cells compared to 1a, as shown in Table 7. According to these results, ca. 2-fold upregulation of SC35 1.6 kb mRNA expression appeared to correlate with a meaningful change in GI50 value.
Table 7. Cellular Potency of eIF4A3 Inhibitorsa.
| SC35
1.6kb (fold of change)b |
||||
|---|---|---|---|---|
| 1 μM | 3 μM | 10 μM | HCT-116 GI50 (μM)c | |
| 1a | 0.9d | 2.59d | 12.88d | 3.06 |
| 1o | 2.17 | 8.41 | 19.08 | 1.34 |
| 1q | 1.79 | 8.31 | 21.76 | 1.18 |
Data are representative of three independent experiments.
Fold change relative to control for mRNA expression of SC35 1.6 kb after 6 h treatment.
Intracellular ATP amount measured 72 h after compound treatment. GI50 calculated using GraphPad Prism5.
Fold change relative to control of mRNA expression after 7 h treatment.
After confirming cellular potency of 1o and 1q in HCT-116 cells, we designed in vivo studies for further assessment of our eIF4A3 inhibitors. In order to improve the exposure of 1o and 1q effectively for in vivo evaluation, we examined solubilized formulations for both compounds. As a result of our investigations, the compounds were dissolved in 10% DMSO/10% Cremophor EL/30% PEG400/20% propylene glycol (PG)/30% 0.4 mol/L citrate solution. Next, we conducted in vivo PD studies in an HCT-116 xenograft model. The in vivo PD profiles of 1o and 1q are shown in Figure 2. Expression of SC35 1.6 kb in tumors was measured at 4, 8, and 24 h after oral administration of 1o and 1q at 100 mg/kg in the above solubilizing formulation in HCT-116 xenograft model mice. As a result, PD responses were confirmed by single oral administration of 1o and 1q. Remarkably, 1o showed 4-fold upregulation of SC35 1.6 kb mRNA expression after 4 h as a rapid-acting profile, while 1q displayed ca. 2-fold upregulation of SC35 1.6 kb mRNA expression after 24 h as a long-acting profile. Thus, we examined both compounds for an antitumor efficacy study to assess the differing profiles of these eIF4A3 inhibitors.
Figure 2.

Effects of compounds 1o and 1q on expression of SC35 1.6kb in HCT-116 xenograft tumors. *P < 0.05 versus vehicle group by Dunnett’s test.
The in vivo antitumor activity of 1o and 1q was examined in an HCT-116 xenograft mouse model. As shown in Figure 3, oral administration of both compounds in our solubilizing formulation (100 mg/kg, bid) for 14 days exhibited antitumor efficacy with T/C values of 54% and 29%, respectively. The compounds were tolerated during the course of the study, showing less than 5% decrease in body weight. These data suggested that a long-acting profile in PD studies might be favorable for antitumor efficacy. Accordingly, compound 1q is an orally bioavailable eIF4A3-selective inhibitor with in vivo antitumor efficacy, representing a useful new probe molecule for assessing the pharmaceutical potential of eIF4A3 inhibition in cancer.
Figure 3.

Antitumor efficacy of compounds 1o and 1q in HCT-116 xenograft model mice. **P < 0.001 versus vehicle group by Dunnett’s test.
We have described the identification of a novel series of orally bioavailable eIF4A3-selective inhibitors as chemical probes for oncology research and potential use in the development of anticancer agents. Our initial lead 1a was optimized to improve its physicochemical properties and PK profile and converted to 3-(3-cyanophenyl)pyridin-2(1H)-one derivative 1o. Compound 1o showed improved solubility and metabolic stability relative to compound 1a, while maintaining eIF4A3 inhibitory potency; however, further testing suggested the need to reduce P-gp efflux. To address this need, a 3-trifluoropyridin-2(1H)-one derivative was prepared using the tPSA value as a guide for reducing polarity, which lead us to the discovery of N-phenyl-3-trifluoropyridin-2(1H)-one derivative 1q. Compounds 1o and 1q showed improved physicochemical and ADMET profiles, while maintaining potent and subtype-selective eIF4A3 inhibitory potency. Based on their promising PK profiles and results from initial in vivo PD studies, both compounds demonstrated antitumor efficacy in a xenograft model. As a result, compounds 1o and 1q showed antitumor efficacy with T/C values of 54% and 29%, respectively, without severe body weight loss. Accordingly, compound 1q was judged to be a useful in vivo probe molecule to further study the fundamental biology and therapeutic potential of selective eIF4A3 inhibition in cancer.
Acknowledgments
We sincerely appreciate Drs. Hiroshi Miyake and Tomoyasu Ishikawa for intellectual support during this work. We would like to express our appreciation to Masahiro Ito, Toshio Tanaka, and Hironobu Maezaki for useful discussion in medicinal chemistry; Shinsuke Araki, Shoichi Nakao, Momoko Ohori, and Atsushi Nakanishi for helpful support in biological evaluation of compounds; Mitsuo Yamamoto for supporting the solubilizing formulation studies; Nao Morishita and Satoshi Endo for computational chemistry support; Natsumi Fujii, Tomoko Izukawa, Chie Kushibe, and Katsuhiko Miwa for chiral HPLC analysis of compounds; and Atsuko Ochida, Mika Inoue, Kouichi Iwanaga, and Izumi Nomura for conducting parallel synthesis. We extend gratitude to Prof. Samuel Aparicio for his dedicated backup and support of the splicing projects.
Glossary
ABBREVIATIONS USED
- ATP
adenosine triphosphate
- HATU
O-(7-aza-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- (A-taPhos)2PdCl2
(bis(ditert-butyl(4-dimethylaminophenyl)phosphine)dichloropalladium(II))
- DME
1,2-dimethoxyethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
- TMSCl
trimethylsilyl chloride
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00283.
Synthetic procedures, analytical data, and assay protocol (PDF)
Author Present Address
† Ono Pharmaceutical Co., Ltd., Mishima-gun, Osaka 618-8585, Japan.
Author Present Address
‡ Astellas Pharma Inc., Tsukuba-shi, Ibaraki 305-8585, Japan.
Author Present Address
§ Axcelead Drug Discovery Partners Inc. Fujisawa-shi, Kanagawa 251-8585, Japan.
Author Present Address
∥ Senjyu Pharmaceutical Co. Ltd., Osaka-shi, Osaka 541-0046, Japan.
Author Present Address
⊥ PeptiDream Inc., Meguro-ku, Tokyo 153-8904, Japan.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. R.M., S.S., K.T., K.F., M.K., S.S., R.A., and D.R.C. contributed to the design and synthesis of compounds; D.N., Y.S., D.M., S.S., and M.I. conducted in vitro and in vivo studies; Y.K. and M.K. administrated PK analyses; J.T. performed solubilized formulation; Y.I. supervised this project.
The authors declare no competing financial interest.
Supplementary Material
References
- Linder P.; Jankowsky E. From Unwinding to Clamping — the DEAD Box RNA Helicase Family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- Tanner N. K.; Linder P. DExD/H box RNA Helicases: From Generic Motors to Specific Dissociation Functions. Mol. Cell 2001, 8, 251–262. 10.1016/S1097-2765(01)00329-X. [DOI] [PubMed] [Google Scholar]
- Jankowsky E. RNA Helicases at Work: Binding and Rearranging. Trends Biochem. Sci. 2011, 36, 19–29. 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W. T.; Wilczynska A.; Smith E.; Bushell M. The Diverse Roles of the eIF4A Family: You Are the Company You Keep. Biochem. Soc. Trans. 2014, 42, 166–172. 10.1042/BST20130161. [DOI] [PubMed] [Google Scholar]
- Parsyan A.; Svitkin Y.; Shahbazian D.; Gkogkas C.; Lasko P.; Merrick W. C.; Sonenberg N. mRNA Helicases: The Tacticians of Translational Control. Nat. Rev. Mol. Cell Biol. 2011, 12, 235–245. 10.1038/nrm3083. [DOI] [PubMed] [Google Scholar]
- Blum S.; Schmid S. R.; Pause A.; Buser P.; Linder P.; Sonenberg N.; Trachsel H. ATP Hydrolysis by Initiation Factor 4A is Required for Translation Initiation in Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 7664–7668. 10.1073/pnas.89.16.7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin Y. V.; Pause A.; Haghighat A.; Pyronnet S.; Witherell G.; Belsham G. J.; Sonenberg N. The Requirement for Eukaryotic Initiation Factor 4A (elF4A) in Translation Is in Direct Proportion to The Degree of mRNA 5′ Secondary Structure. RNA 2001, 7, 382–394. 10.1017/S135583820100108X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H.; Andersen G. R. Structural Insights into the Exon Junction Complex. Curr. Opin. Struct. Biol. 2008, 18, 112–119. 10.1016/j.sbi.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Palacios I. M.; Gatfield D.; St Johnston D.; Izaurralde E. An eIF4AIII-containing Complex Required for mRNA Localization and Nonsense-mediated mRNA Decay. Nature 2004, 427, 753–757. 10.1038/nature02351. [DOI] [PubMed] [Google Scholar]
- Ferraiuolo M. A.; Lee C.-S.; Ler L. W.; Hsu J. L.; Costa-Mattioli M.; Luo M.-J.; Reed R.; Sonenberg N. A Nuclear Translation-like Factor eIF4AIII is Recruited to the mRNA During Splicing and Functions in Nonsense-mediated Decay. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4118–4123. 10.1073/pnas.0400933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya T.; Tange T. O.; Sonenberg N.; Moore M. J. eIF4AIII Binds Spliced mRNA in the Exon Junction Complex and is Essential for Nonsense-mediated Decay. Nat. Struct. Mol. Biol. 2004, 11, 346–351. 10.1038/nsmb750. [DOI] [PubMed] [Google Scholar]
- Le Hir H.; Séraphin B. EJCs at the Heart of Translational Control. Cell 2008, 133, 213–216. 10.1016/j.cell.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Reed R.; Hurt E. A Conserved mRNA Export Machinery Coupled to Pre-mRNA Splicing. Cell 2002, 108, 523–531. 10.1016/S0092-8674(02)00627-X. [DOI] [PubMed] [Google Scholar]
- Michelle L.; Cloutier A.; Toutant J.; Shkreta L.; Thibault P.; Durand M.; Garneau D.; Gendron D.; Lapointe E.; Couture S.; Le Hir H.; Klinck R.; Elela S. A.; Prinos P.; Chabot B. Proteins Associated with the Exon Junction Complex Also Control the Alternative Splicing of Apoptotic Regulators. Mol. Cell. Biol. 2012, 32, 954–67. 10.1128/MCB.06130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Murigneux V.; Le Hir H. Transcriptome-wide Modulation of Splicing by the Exon Junction Complex. Genome Biol. 2014, 15, 551. 10.1186/s13059-014-0551-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K. E.; Parker R. Nonsense-mediated mRNA Decay: Terminating Erroneous Gene Expression. Curr. Opin. Cell Biol. 2004, 16, 293–299. 10.1016/j.ceb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Trcek T.; Sato H.; Singer R. H.; Maquat L. E. Temporal and Spatial Characterization of Nonsense-mediated mRNA Decay. Genes Dev. 2013, 27, 541–551. 10.1101/gad.209635.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N.; Longman D.; Cáceres J. F. Mechanism and Regulation of the Nonsense-mediated Decay Pathway. Nucleic Acids Res. 2016, 44, 1483–1495. 10.1093/nar/gkw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K.; Sanada M.; Shiraishi Y.; Nowak D.; Nagata Y.; Yamamoto R.; Sato Y.; Sato-Otsubo A.; Kon A.; Nagasaki M.; Chalkidis G.; Suzuki Y.; Shiosaka M.; Kawahata R.; Yamaguchi T.; Otsu M.; Obara N.; Sakata-Yanagimoto M.; Ishiyama K.; Mori H.; Nolte F.; Hofmann W. K.; Miyawaki S.; Sugano S.; Haferlach C.; Koeffler H. P.; Shih L. Y.; Haferlach T.; Chiba S.; Nakauchi H.; Miyano S.; Ogawa S. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- Martin L.; Grigoryan A.; Wang D.; Wang J.; Breda L.; Rivella S.; Cardozo T.; Gardner L. B. Identification and Characterization of Small Molecules That Inhibit Nonsense-mediated RNA Decay and Suppress Nonsense p53 Mutations. Cancer Res. 2014, 74, 3104–3113. 10.1158/0008-5472.CAN-13-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa E. Expression of New Antigens on Tumor Cells by Inhibiting Nonsense-mediated mRNA Decay. Immunol. Res. 2013, 57, 44–51. 10.1007/s12026-013-8442-7. [DOI] [PubMed] [Google Scholar]
- Jia J.; Furlan A.; Gonzalez-Hilarion S.; Leroy C.; Gruenert D. C.; Tulasne D.; Lejeune F. Caspases Shutdown Nonsense-mediated mRNA Decay During Apoptosis. Cell Death Differ. 2015, 22, 1754–1763. 10.1038/cdd.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau M. E.; Mori A.; Oberer M.; Lindqvist L.; Chard L. S.; Higa T.; Belsham G. J.; Wagner G.; Tanaka J.; Pelletier J. Functional Characterization of IRESes by an Inhibitor of the RNA Helicase eIF4A. Nat. Chem. Biol. 2006, 2, 213–20. 10.1038/nchembio776. [DOI] [PubMed] [Google Scholar]
- Lindqvist L.; Oberer M.; Reibarkh M.; Cencic R.; Bordeleau M. E.; Vogt E.; Marintchev A.; Tanaka J.; Fagotto F.; Altmann M.; Wagner G.; Pelletier J. Selective Pharmacological Targeting of a DEAD Box RNA Helicase. PLoS One 2008, 3, e1583. 10.1371/journal.pone.0001583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa T.; Tanaka J.; Tsukitani Y.; Kikuchi H. Hippuristanols, Cytotoxic Polyoxygenated Steroids from the Gorgonian Isis Hippuris. Chem. Lett. 1981, 10, 1647–1650. 10.1246/cl.1981.1647. [DOI] [Google Scholar]
- Cencic R.; Pelletier J. Hippuristanol – A Potent Steroid Inhibitor of Eukaryotic Initiation Factor 4A. Translation 2016, 4, e1137381. 10.1080/21690731.2015.1137381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low W. K.; Li J.; Zhu M.; Kommaraju S. S.; Shah-Mittal J.; Hull K.; Liu J. O.; Romo D. Second-generation Derivatives of the Eukaryotic Translation Initiation Inhibitor Pateamine A Targeting eIF4A as Potential Anticancer Agents. Bioorg. Med. Chem. 2014, 22, 116–125. 10.1016/j.bmc.2013.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M.; Iwatani M.; Kamada Y.; Sogabe S.; Nakao S.; Tanaka T.; Kawamoto T.; Aparicio S.; Nakanishi A.; Imaeda Y. Discovery of Selective ATP-competitive eIF4A3 Inhibitors. Bioorg. Med. Chem. 2017, 25, 2200–2209. 10.1016/j.bmc.2017.02.035. [DOI] [PubMed] [Google Scholar]
- Ito M.; Tanaka T.; Cary D. R.; Iwatani-Yoshihara M.; Kamada Y.; Kawamoto T.; Aparicio S.; Nakanishi A.; Imaeda Y. Discovery of Novel 1,4-Diacylpiperazines as Selective and Call-Active eIF4A3 Inhibitors. J. Med. Chem. 2017, 60, 3335–3351. 10.1021/acs.jmedchem.6b01904. [DOI] [PubMed] [Google Scholar]
- Iwatani-Yoshihara M.; Ito M.; Ishibashi Y.; Oki H.; Tanaka T.; Morishita D.; Ito T.; Kimura H.; Imaeda Y.; Aparicio S.; Nakanishi A.; Kawamoto T. Discovery and Characterization of a Eukaryotic Initiation Factor 4A-3-Selective Inhibitor That Suppresses Nonsense-Mediated mRNA Decay. ACS Chem. Biol. 2017, 12, 1760–1768. 10.1021/acschembio.7b00041. [DOI] [PubMed] [Google Scholar]
- Abel S.; Beaumont K. C.; Crespi C. L.; Eva M. D.; Fox L.; Hyland R.; Jones B. C.; Muirhead G. J.; Smith D. A.; Venn R. F.; Walker D. K. Potential Role for P-glycoprotein in the Nonproportional Pharmacokinetics of UK-343,664 in Man. Xenobiotica 2001, 31, 665–676. 10.1080/00498250110052779. [DOI] [PubMed] [Google Scholar]
- Lunn G.; Mowbray C. E.; Liu W. L. S.; Joynson V. M.; Hay T.; Yeadon M. The Discovery and Profile of PF-0868087, a CNS-sparing Histamine H3 Receptor Antagonist for the Treatment of Allergic Rhinitis. MedChemComm 2012, 3, 339–343. 10.1039/C2MD00276K. [DOI] [Google Scholar]
- Sureau A.; Gattoni R.; Dooghe Y.; Stévenin J.; Soret J. SC35 Autoregulates Its Expression by Promoting Splicing Events That Destabilize Its mRNAs. EMBO J. 2001, 20, 1785–1796. 10.1093/emboj/20.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.