

Abstract
Mapping of DNA methylation is essential in understanding the process of HBV covalently closed circular DNA (cccDNA) transcription. Here, bisulfite sequencing PCR and methylation-specific PCR, two PCR- based approaches used in determining and quantifying the DNA methylation pattern, are described.
Keywords: HBV cccDNA, DNA methylation, Bisulfite sequencing PCR, Methylation-specific PCR
1 Introduction
DNA methylation is one of the epigenetic modification which correlates with alteration in gene expression [1, 2]. The target sequence of methylation in mammalian genome is 5′ cytosine of CpG dinucleotides, and clusters of CpGs are mostly found associated with gene promoters [3]. The 5-methyl-cytosine was recognized by mCpG-binding proteins, including the methyl-CpG-binding domain (MBD), UHRF, and Kaiso protein families, which interact with histone-modifying and chromatin- remodeling enzymes, mediating stable repression of genes [4].
The first detection of virus DNA methylation was been reported in adenovirus in 1980 [5]. The inverse correlation between methylation level of virus DNA segments and their expression provides clues to the involvement of DNA methylation in inactivation of DNA virus genomes. Evidence demonstrates that several DNA virus infections will trigger an epigenetic response, including HIV, BLV, EBV and HBV [6–8].
Hepatitis B virus (HBV) is an enveloped hepatotropic DNA virus, containing a 3.2 kb partially double stranded circular DNA genome [9, 10]. Without interacting with DNA methyltransferase, the newly synthesized HBV DNA within the viral capsid and Dane particle remain unmethylated. However, once in the nucleus of the infected hepatocyte, the relaxed circular DNA genome converts into covalently closed circular DNA (cccDNA), which is further organized into a viral minichromosome by histone and nonhistone proteins. CccDNA has been shown to be susceptible to DNA methylation mediated by DNMTs in the infected hepatocyte nuclei [11]. As the transcription template for the production of all viral mRNA, methylation of cccDNA results in the repressed transcription and consequently low level of viral replication. The HBV genome contains three major CpG islands. Among which, island I overlaps the start site of the small surface (S) gene, island II spans a region that overlaps enhancer I/II and is proximal to the core promoter, and island III covers the start codon of the polymerase (P) gene and upstream region of SP1 promoter. Among which, the CpG island II methylation has been shown to be associated with decreased pgRNA transcription and consequently viral replication [12, 13].
To understand further the biology that HBV cccDNA methylation state changes in HBV infection, here we report two methods used in determining the distribution of 5-methyl-cytosine, the bisulfite sequencing PCR (BSP) and methylation-specific PCR (MSP). Chemical conversion using sodium bisulfite is performed prior to both two approaches. Cytosine undergoes sulfonation, hydrolytic deamination, and alkali desulfonation and converts to uracil (the uracil amplified as thymine in PCR), while 5-methyl- cytosine remains as cytosine. For BSP, the primers are designed to amplify the bisulfite-treated DNA in the target region. The PCR product is cloned and sequenced; the selective reaction helps to discriminate between unmethylated cytosine (T) and methylated cytosine (C) (Fig. 1). For MSP, separate pairs of primers are designed to specifically amplify the methylated DNA or the unmethylated DNA, respectively. Due to the low copy number of cccDNA within infected liver cells, we applied nested PCR to amplify the CpG islands of HBV cccDNA.
Fig. 1.
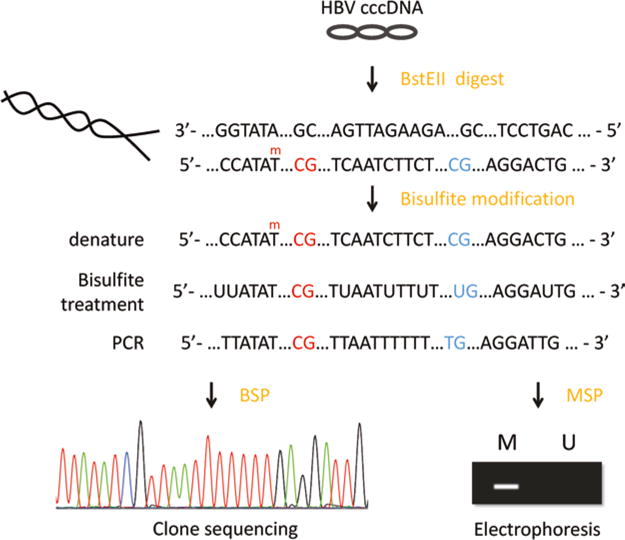
Bisulfite sequencing of hepatitis B virus covalently closed circular DNA. HBV cccDNA are linearized with BstEII digestion prior to the bisulfite conversion, which converts the unmethylated cytosine (blue) to uracil (the uracil amplified as thymine in PCR), while 5-methyl-cytosine remains as cytosine (red). The converted DNA are PCR amplified and cloned into T-vectors. Methylated cytosine is discriminated from the unmethylated cytosine through the selective reaction
2 Materials
2.1 DNA Isolation
Cell lysis buffer: 50 mM Tris–HCl (pH 8.0), 1 mM ethylenediaminetetraacetic acid (EDTA), 0.2 % NP-40, 0.15 M NaCl.
Nuclear lysis buffer: 6 % SDS, 0.1 N NaOH.
Neutralization buffer: 3 M KAc (pH 4.8).
Ethanol (100 %, 70 %).
3 M NaAc (pH 5.5).
10 mg/ml yeast RNA (Ambion).
Phenol/chloroform: phenol/chloroform/isoamyl alcohol (25:24:1), saturated with 10 mM Tris–HCl (pH 8.0) and 1 mM EDTA.
TE buffer: 10 mM Tris–HCl, pH 8.0, and 1 mM EDTA.
2.2 Pretreatment of DNA
Plasmid-Safe DNase with appropriate buffer.
Restriction enzyme with appropriate buffer (Bst EII).
PCR Clean-Up Kit (Axygen).
2.3 Bisulfite Conversion
EpiTect Bisulfite Kit (Qiagen).
Dissolve the required number of aliquots of bisulfite mix with 800 ul RNase-free water.
Prepare buffer BL containing 10 μg/ml carrier RNA.
Ethanol (100 %).
2.4 Primer Design
The primer can be designed manually or using the online software: MethPrimer (http://www.uro-gene.org/methprimer/) and MSP primer (http://www.mspprimer.org/cgi-bin/design.cgi).
2.5 PCR Amplification of Bisulfite- Treated DNA and DNA Sequencing
MightyAmp DNA Polymerase (TAKARA), PCR buffer, MgCl2 solution, and dNTP (10 μM).
Primers (10 μM).
1× TAE buffer: 0.04 M Tris base, 0.04 M glacial acetic acid, 1 mM EDTA, pH 8.2–8.4. Prepare 30× stock solution, and store at room temperature.
Agarose (molecular biology grade).
10× DNA gel loading buffer: 10 mM EDTA (pH 8.0), 50 % (V/V) glycerol, 0.25 % (W/V) bromophenol blue.
Gel Extraction Kit.
T-vector (Takara).
T4 DNA ligase with appropriate buffer.
DH5α Chemically Competent Cell.
Ampicillin (100 mg/ml).
3 Methods
3.1 DNA Isolation
Lyse liver biopsy tissues (2–10 mg) by adding 400 μl pre-chilly cell lysis buffer [50 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.2 % NP-40, 0.15 M NaCl]. Homogenize the tissue with Qiagen Tissue Ruptor on ice until the tissue disruption was efficient (see Note 1).
Centrifuge at 16,000 × g for 10 min at 4 °C. Remove the supernatant. The pellet was treated with 400 μl nuclear lysis buffer (6 % SDS, 0.1 N NaOH), followed by incubation for 30 min at 37 °C, vortex intermittently.
The lysate was then neutralized by 100 ul of neutralization buffer [3 M KAc (pH 4.8)], and centrifuged at 16,000 × g for 15 min at 4 °C.
Transfer the supernatant to a fresh 2 ml EP tube, add equal volume of phenol to the supernatant, and mix thoroughly by hand shaking for 15 s. Centrifuge at 16,000 × g for 15 min at 4 °C, and transfer the aqueous phase to a fresh 2 ml tube. Add equal volume of phenol–chloroform to the supernatant and mix thoroughly by hand shaking for 15 s. Centrifuge at 16,000 × g for 15 min at 4 °C, and transfer the aqueous phase to a fresh 2 ml tube.
Add 1/10 volumes of 3 M NaAc, 2 volumes of 100 % ethanol, and 2 μl tRNA, and mix thoroughly by pipette. Precipitate DNA at −20 °C overnight.
On the second day, centrifuge the tube at 16,000 × g for 30 min at 4 °C, and discard the supernatant. Add 1 ml 70 % ethanol and gently rotate the tube to wash the DNA pellet. Centrifuge at 16,000 × g for 15 min at 4 °C. Discard the supernatant.
Allow the pellet to air-dry for about 5 min at room temperature. Dissolve the DNA pellet in 50 μl TE buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA).
3.2 Pretreatment of DNA
-
Add DNA solution (<3.33 μg, Vmax = 50 μl) to the following reaction to remove the potential contaminating host genomic DNA by the Plasmid-Safe DNase treatment.
Plasmid-Safe DNase preparation:
Sterile water 42 μl
25 mM ATP 2 μl
10× reaction buffer 5 μl
Plasmid-Safe DNase (10 U) 1 μl
Total reaction volume 50 μl
Incubate at 37 °C for 1 h
Purify the cccDNA using a PCR Clean-Up Kit. Elute the DNA with 30 μl of TE buffer.
-
PCR amplification of GAPDH gene.
To exclude the possible contamination of the integrated HBV DNA, GAPDH (or other control gene) PCR amplification should be carried out before the CpG island amplification. Primer sequences are listed as follows:
Forward primer: 5′- ATTCCACCCATGGCAAATTC-3′.
Reverse primer: 5′-GGATCTCGCTCCTGGAAGATG-3′).
Amplification were performed with initial denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 60 s, with a final extension of 5 min at 72 °C.
The conversion efficiency of plasmid DNA was poor due to the quick reannealing of the single-stranded DNA after the dena-turation during the bisulfite conversion. Therefore, HBV cccDNA was linearized with BstEII (or other restriction endo-nuclease which would not affect the target region) before the bisulfite treatment to maximize the conversion efficiency (see Note 2). Digestion reaction components: Add 2 μl BstEII, 5 μl NEBuffer 3.1, 0.5 μl 10 × BSA, and 12.5 μl sterile water into the cccDNA solution (30 μl), and mix thoroughly. Incubate at 37 °C for 3 h.
Purify the cccDNA using a PCR Clean-Up Kit. Elute the DNA with 40 μl of TE buffer.
3.3 Bisulfite Conversion of HBV cccDNA
Bisulfite treatment of the HBV cccDNA from liver biopsy tissue samples was performed by using Qiagen EpiTect Bisulfite Kit (Qiagen) or EZ DNA Methylation Kit (Zymo Research) or EpiJET Bisulfite Conversion Kit (Invitrogen).
Bisulfite reaction components: Add 85 μl DNA bisulfite mix and 15 μl DNA Protect Buffer into the cccDNA solution (<2 μg, Vmax = 40 μl), and mix thoroughly (see Note 3).
Incubate in a thermocycler with the following conditions: Denaturation at 95 °C for 5 min → Incubation at 60 °C for 25 min → Denaturation at 95 °C for 5 min → Incubation at 60 °C for 85 min → Denaturation at 95 °C for 5 min → Incubation at 60 °C for 175 min → Hold at 20 °C.
Transfer the completed bisulfite reactions to clean 1.5 ml microcentrifuge tubes. Add 560 μl buffer BL (containing 10 μg/ml carrier RNA). Vortex and centrifuge the tube briefly. Transfer the mixture into the spin column. Centrifuge at 16,000 × g for 1 min.
Discard the filtrate, add 500 μl buffer BW, and centrifuge at 16,000 × g for 1 min. Discard the filtrate, add 500 μl buffer BD, and incubate at room temperature for 15 min.
Wash the spin column with 500 μl buffer BW twice. Centrifuge at 16,000 × g for 1 min and discard the filtrate.
Place the spin columns into a clean 1.5 ml EP tube, and elute the DNA with 30–40 μl of TE buffer. The bisulfite-converted DNA was stored at −80 °C, which should be used for PCR as soon as possible.
3.4 Design of BSP and MSP Primers
MethPrimer can be used to predict CpG islands within the HBV whole genome. The CpG islands were defined based on the following criteria: (1) a GC content of ≥50 %, (2) an observed-to- expected CpG dinucleotide ratio ≥0.60. and (3) a sequence window longer than 100 bp. As mentioned above, bisulfite modification converts the unmethylated “C” to “T,” while mC remains “C.” The modified DNA turns into two DNA strands which are not complementary. The primers can be designed according to either strand. We designed the primers according to the negative strand (Table 1).
Table 1.
Primers used in the bisulfite-converted cccDNA amplification
|
BSP primers:
| |||
|---|---|---|---|
| Target | Primers, 5′-3′ | Position (bp) | |
| CpG1 | 53–283 | ||
| Nested PCR options: ① f2 + down f2 + r2 ② f2 + down 1′ f2 + r2 ③ ViveF + down1′ viveF + viveR | |||
| CpG1-BSP-f2 | TATTTTTTTGTTGGTGGTTTTAGTTT | 54–77 | |
| CpG1-BSP-r2 | TAAAAAATTAAAAAAAATCCACCA | 286–266 | |
| CpG1-BSP-seminested downstream | ACAAAAAAATAAAACATAACAACAAAATA | 439–411 | |
| CpG1-BSP-semidown 1′ | AACAACATACCTTAATAATCCAA | 470–448 | |
| Primers from reference [16]: | |||
| Vivekanandan-CpG1-F | TTTGTTGGTGGTTTTAGTTTAGGA | 58–81 | |
| Vivekanandan-CpG1- R | TCCCCCTAAAAAATTAAAAAAAATC | 288–264 | |
|
| |||
| CpG2 | 1195–1666/1389–1666 | ||
| Nested PCR options: ① f2 + down f2 + r2 ② f2 + down CF2 + CR2 ③ CF2 + CR2 f3 + r2 | |||
| CpG2-BSP-f2 | GTAATTTTTATTGGWTGGGGTTTGGT | 1048–1069 | |
| CpG2-BSP-r2 | ATCCTCTTATAYAAAACCTTAAACAA | 1680–1655 | |
| CpG2-BSP-seminested downstream | TTATACCTACAACCTCCTAATACAAAA | 1794–1768 | |
| CpG2-CF2 | TTTTATGGTTGTTAGGDTGTGTTGTT | 1052–1077 | |
| CpG2-CR2 | TAACCTAAHCTCCTCCCCCAAC | 1762–1741 | |
| CpG2-BSP-f3 | TGTGTTGTTAATTGGATTTTG | 1389–1409 | |
| Primers from reference [13, 16, 17]: | |||
| Vivekanandan-CpG2- F | GTAATTTTTATTGGTTGGGGTTTG | 1194–1217 | |
| Vivekanandan-CpG2- R | TCCAATTAACAACACAHCCTAACAAC | 1404–1379 | |
| Kim-CpG2-F | GGGATTGATAATTTTGTTGTTTTTTT | 1329–1354 | |
| Kim-CpG2-R | TCCAAAAATCCTCTTATATAAAACCTTAA | 1672–1644 | |
| Guo-CpG2-F | GTTTTTTGTTTATGTTTTGTTGG | 1418–1440 | |
| Guo-CpG2-R | AAATAAAACAAAATACACACAATCCG | 1598–1573 | |
|
| |||
| CpG3 | 2206–2461 | ||
| Nested PCR: up + r2 f2 + r2 | |||
| CpG3-BSP-f2 | GTGGTTTTATATTTTTTGTTTTAT | 2208–2228 | |
| CpG3-BSP-r2 | AAAATACTAACATTAAAATTCCCAAA | 2460–2435 | |
| CpG3-BSP-seminested upstream | GTTATGTTAATGTTAATATGGGT | 2162–2184 | |
|
| |||
| MSP primers (primers from reference [15]): | |||
| CpG1-MSP-F | ACGTGTTTTGGTTAAAATTCGTAGTTTTTA | 292–322 | 95 °C 5 min (95 °C 10s, 55 °C 30s, 72 °C 10s) × 50 cycles |
| CpG1-MSP-R | AATATAATAAAACGCCGCAAACACATC | 376–402 | |
| Probe: [6FAM] GTTTTTTAATTTGTTTTGGTTATCGTTGGATG [BHQ1] | 348–379 | ||
| CpG2-MSP-F | TGTCGTTTCGGTCGATTAC | 1502–1520 | 95 °C 5 min (95 °C 10s, 52 °C 30s, 72 °C 10s) × 45 cycles, Melting curve, 40 °C 30s |
| CpG2-MSP-R | CACGATCCGACAAATAAAAA | 1560–1579 | |
| SYBR Green | |||
| CpG3-MSP-F | GTGTGGATTCGTATTTTTTTC | 2270–2290 | 95 °C 10 min (95 °C 10s, 53 °C 30s, 72 °C 10s) × 45 cycles |
| CpG3-MSP-R | GACGATTAAAACCTTCGTCT | 2393–2412 | |
| Probe: [6FAM]AACCTACCTCGTCGTCTAACAACAAT[BHQ1] | 2339–2364 | ||
BSP: (1) PCR products should be less than 500 bp; (2) primers should not contain CpG dinucleotides; (3) and the non-CpG “C” can be included at 3′ of the primers ensure that complete converted DNA being amplified (see Note 4).
MSP: (1) PCR products should be less than 200 bp; (2) primers should contain at least one CpG site at the most 3′ end, and primer sequence should span more than one CpG site; and (3) primers amplifying methylated or unmethylated DNA should contain the same number of CpG sites [14].
3.5 PCR Amplification of Bisulfite- Converted HBV cccDNA
BSP:
-
Set up a PCR in a total of 50 μl, as follows (or using other DNA polymerase such as AmpliTaq Gold 360 Master Mix or ZymoTaq DNA Polymerase)
MightyAmp DNA Polymerase 1 μl
PCR mix 25 μl
Forward primer (10 μM) 1.5 μl
Reverse primer (10 μM) 1.5 μl
Bisulfite-converted DNA 5–10 μl
Total reaction volume 50 μl
Amplification condition: initial denaturation at 98 °C for 3 min, followed by 30 cycles of 98 °C for 15 s, 55 °C for 20 s, and 68 °C for 45 s, with a final extension of 10 min at 68 °C. 1 μl of the ten times diluted PCR product was then subjected to a second round of amplification: 98 °C for 3 min, followed by 35 cycles of 98 °C for 15 s, 55 °C for 20 s, and 68 °C for 45 s, and a final extension of 10 min at 68 °C (see Note 5).
Separate the PCR products on a 1.5 % agarose gel. Purify the PCR products with a Gel Extraction Kit. Elute the DNA with 25 μl of TE buffer.
The PCR products of the three CpG islands were cloned into T-vector (mol ratio, 3 ~ 10:1) and subject to sequencing to study the methylation status of each CpG island. More than ten clones need to be analyzed for each island to get a reliable conclusion.
MSP (Protocols from Reference)
Amplification condition: 95 °C 10 min (95 °C 10s, 53 °C 30s, 72 °C 10s) × 45 cycles.
The quantitative analysis of HBV DNA methylation is carried out by calculating the ratio methylated HBV copies/3000copies of BS-actin DNA [15].
3.6 Data Processing
The methylation status can be obtained by comparing the original HBV CpG island sequence and the sequencing data of the BSP products. For CHB patients in China, we use the consensus sequence of B or C genotype HBV as the template sequence for each patient. The analysis can be conducted manually by using software such as VectorNTI, or using the methylation analyzing software such as BiQ Analyzer, which can transform the methylation status into a visualized graph:
Import the original HBV CpG island sequence (unconverted sequence) and then all the sequencing files.
Perform the sequence alignment; the conversion rate of each sequence will be calculated by the software, and the sequence with conversion rate lower than 90 % will be excluded. The inverted sequence needs the reverse and complementary transformation (see Notes 6 and 7).
The results of analysis will be presented in two ways: (A) Lollipop diagram: The black and white circles represent methylated and unmethylated CpG dinucleotides, respectively. The vertical line indicates dinucleotides other than CpG at the corresponding site. (B) Box diagram: The vertical box indicates all HBV DNA clones from patients at corresponding CpG position. The blue and yellow regions represent the proportion of unmethylated and methylated clones, respectively. The gray color refers to the absence of CG dinucleotide due to single nucleotide polymorphism. The number of unmethylated and methylated clones is listed under the corresponding dinucleotides (Fig. 2).
Fig. 2.
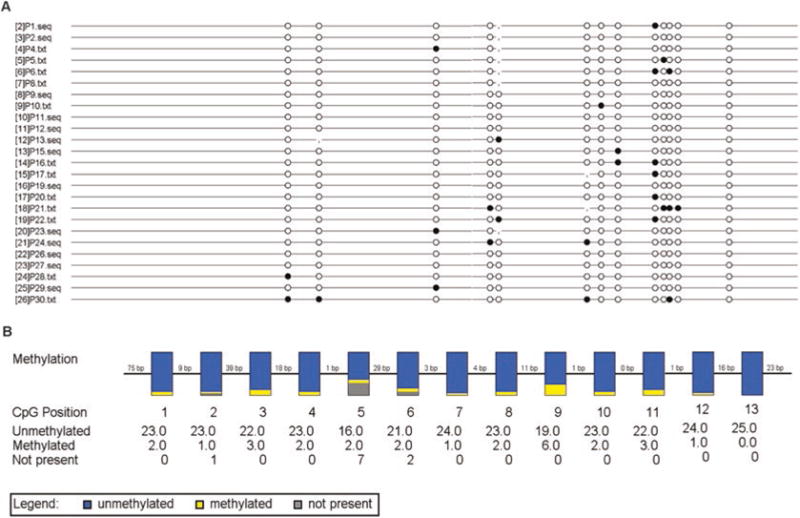
BiQ Analyzer output of the methylation status of HBV cccDNA CpG island III. (a) Lollipop diagram: The black and white circles represent methylated and unmethylated CpG dinucleotides, respectively. (b) Box diagram: The vertical box indicates all HBV DNA clones from patients at corresponding CpG position. The blue and yellow regions represent the proportion of unmethylated and methylated clones, respectively. The gray color refers to the absence of CG dinucleotide due to single nucleotide polymorphism. The number of unmethylated and methylated clones is listed under the corresponding dinucleotides
Footnotes
Make sure the tissue ruptor is placed below the liquid surface of cell lysis buffer to avoid the production of foam, which will cause sample loss and make it difficult in adding the following reagent.
To maximize the bisulfite conversion rate of DNA samples, it is critical to linearize cccDNA, which is a supercoid double- stranded DNA before starting the bisulfite treatment. Or during the subsequent PCR, the unconverted cccDNA may also be amplified, and the presence of unconverted “C” makes it difficult for data analysis.
The bisulfite mix needs to be freshly prepared, and the bisulfite- converted DNA samples need to be aliquoted in the thin-wall plastic PCR tubes and stored at −80 °C. Avoid multiple freeze–thaw cycles.
The restriction endonuclease used in the linearization of cccDNA should not be chosen in the region of CpG islands.
Choosing DNA polymerase which can perform well in the AT- rich or GC-rich DNA sample amplification. The annealing temperature for each island amplification and the amount of the DNA template used in the second round of PCR need to be tested and optimized.
The software BiQ Analyzer will exclude the sequence with high nucleotide sequence homology (sequences are equal in all of the genomic sequence’s aligned C positions) to make sure that the data are not the repeat sequencing results from the same clone. However, we choose to do the clone sequencing which have ensured that each sequencing result comes from one separate clone, so each sequencing result needs to be involved for a reliable analysis.
The most accurate alignment should include the original CpG island sequences, at least from the dominant stain of the patient. Aligning using the original sequence as template can minimize the impact of the sequence polymorphism on the methylation analysis. Such as the nucleotide in template is “AG,” while the sequencing shows a “CG” in the corresponding position, the methylated “C” will be missed, which will lead to the false negativity.
References
- 1.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19(2):187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293(5532):1068–1070. doi: 10.1126/science.10638523. [DOI] [PubMed] [Google Scholar]
- 3.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321(6067):209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 4.Rottach A, Leonhardt H, Spada F. DNA methylation-mediated epigenetic control. J Cell Biochem. 2009;108(1):43–51. doi: 10.1002/jcb.22253. [DOI] [PubMed] [Google Scholar]
- 5.Sutter D, Doerfler W. Methylation of integrated adenovirus type 12 DNA sequences in transformed cells is inversely correlated with viral gene expression. Proc Natl Acad Sci U S A. 1980;77(1):253–256. doi: 10.1073/pnas.77.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki K, Shijuuku T, Fukamachi T, Zaunders J, Guillemin G, Cooper D, Kelleher A. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J RNAi Gene Silencing. 2005;1(2):66–78. [PMC free article] [PubMed] [Google Scholar]
- 7.Bergbauer M, Kalla M, Schmeinck A, Gobel C, Rothbauer U, Eck S, Benet-Pages A, Strom TM, Hammerschmidt W. CpG- methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog. 2010;6(9):e1001114. doi: 10.1371/journal.ppat.1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vivekanandan P, Daniel HD, Kannangai R, Martinez-Murillo F, Torbenson M. Hepatitis B virus replication induces methylation of both host and viral DNA. J Virol. 2010;84(9):4321–4329. doi: 10.1128/JVI.02280-099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Summers J, O’Connell A, Millman I. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from dane particles. Proc Natl Acad Sci U S A. 1975;72(11):4597–4601. doi: 10.1073/pnas.72.11.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64(1):51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. The covalently closed duplex form of the hepadna-virus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol. 1995;69(6):3350–3357. doi: 10.1128/jvi.69.6.3350-3357.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Mao R, Yan R, Cai D, Zhu H, Kang Y, Liu H, Wang J, Qin Y, Huang Y, Guo H, Zhang J. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS One. 2014;9(10):e110442. doi: 10.1371/journal.pone.0110442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JW, Lee SH, Park YS, Hwang JH, Jeong SH, Kim N, Lee DH. Replicative activity of hepatitis B virus is negatively associated with methylation of covalently closed circular DNA in advanced hepatitis B virus infection. Intervirology. 2011;54(6):316–325. doi: 10.1159/00032145014. [DOI] [PubMed] [Google Scholar]
- 14.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 15.Jain S, Chang TT, Chen S, Boldbaatar B, Clemens A, Lin SY, Yan R, Hu CT, Guo H, Block TM, Song W, Su YH. Comprehensive DNA methylation analysis of hepatitis B virus genome in infected liver tissues. Sci Rep. 2015;5:10478. doi: 10.1038/srep1047816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vivekanandan P, Thomas D, Torbenson M. Hepatitis B viral DNA is methylated in liver tissues. J Viral Hepat. 2008;15(2):103–107. doi: 10.1111/j.1365-2893.2007.00905.x. [DOI] [PubMed] [Google Scholar]
- 17.Guo Y, Li Y, Mu S, Zhang J, Yan Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol. 2009;81(7):1177–1183. doi: 10.1002/jmv.21525. [DOI] [PubMed] [Google Scholar]