

Abstract
Electron transfer dissociation (ETD) has been broadly adopted and is now available on a variety of commercial mass spectrometers. Unlike collisional activation techniques, optimal performance of ETD requires considerable user knowledge and input. ETD reaction duration is one key parameter that can greatly influence spectral quality and overall experiment outcome. We describe a calibration routine that determines the correct number of reagent anions necessary to reach a defined ETD reaction rate. Implementation of this automated calibration routine on two hybrid Orbitrap platforms illustrate considerable advantages, namely, increased product ion yield with concomitant reduction in scan rates netting up to 75% more unique peptide identifications in a shotgun experiment.
Keywords: ETD, Ion/ion, Kinetics, Proteomics, Mass spectrometry, Calibration, Orbitrap
Graphical Abstract
Introduction
We are pleased to offer this article in celebration of John R. Yates III for his seminal 1994 contribution of automated tandem mass spectral interpretation [1]. Now, two decades removed from this landmark publication, his methodology remains at the core of any proteomic analysis, including the one presented here. Though John’s software tools were developed for interpretation of collision-based peptide fragmentation (CAD), the ability to process thousands of tandem mass spectra revealed the limitations of these methods and spawned innovation in the area of peptide dissociation. One of these developments was electron transfer dissociation (ETD), described in 2004 by Hunt and co-workers [2, 3].
Now, one decade since its first description, ETD is commercially available on multiple MS platforms by a variety of vendors including: rf 3D quadrupole ion traps [4, 5], radial ejection rf linear (2D) quadrupole ion traps (QLT) [2, 3], hybrid QLT-Orbitraps [6, 7], quadrupole mass filter Fourier transform ion cyclotron resonance (Q-FT-ICR), and quadrupole time-of-flight (Q-TOF) systems [8]. With this rapid expansion in accessibility, there now exist a large population of new ETD users, most with no experience using ion/ion reactions. This, combined with simple substitution of ETD into non-optimal workflows, often fails to capitalize on ETDs potential [9, 10]. A main confounding factor is that recognizing a high quality ETD MS/MS spectrum is not trivial. This is important as current instrument calibration routines require user input to determine reaction settings [11–20]. ETD reactions dissociate multiple protonated precursors following transfer of an electron from a radical reagent anion [2, 3]. Unlike ion trap CAD, the products of ETD are subject to further reaction so that c- and z•-type product ion signal-to-noise (S/N) ratios can be reduced by overexposure to the reagent anions, especially when the entire precursor population is consumed. That is, although the complete removal of precursor is diagnostic of quality CAD, such is not the case for ETD. In addition to sub-optimal product ion S/N, extension of the reaction considerably slows scan rate, reducing the number of identifications achievable during a shotgun experiment [21]. CAD methods, having benefited from decades of development, shield users from the many parameters that require calibration to produce optimal results [22–26]. No such calibration exists for ETD.
The primary driver of this work is to improve ETD functionality and to ensure that users of all levels can achieve the best possible result with the method. First, we developed a calibration routine to determine the amount of reagent anions necessary to achieve a chosen ETD reaction rate (k). We then used this rate constant to calculate charge state-dependent reaction times. This approach produced up to 40% more unique identifications than a static reaction time on QLT-Orbitrap hybrid system [27]. Application of this method on a newer quadrupole-Orbitrap-quadrupole ion trap (Q-OT-qIT) [28] system showed even more benefit—32% more MS/MS scans, 76% more peptide spectral matches (PSMs), and 75% more unique peptides compared with a static reaction time. With an ETD calibration routine based on ion/ion reaction kinetics, we take a key step forward in maturation of ETD, easing the burden on the user while simultaneously improving spectral quality and overall performance.
Experimental
Calibration of ETD Reaction Rate
The routine for calibration of ETD reaction rates was written in the instrument control language to enable automated execution of the algorithm. Briefly, the calibration procedure reacts the triply charged precursor of angiotensin (AGC = 30,000) with various amounts of fluoranthene reagent anions (20,000 to 450,000) for a range of reaction times. Angiotensin I was diluted to 2 pmol/μL in 50% acetonitrile with 0.2% formic acid and directly infused using electrospray ionization.
Yeast Sample Preparation
Saccharomyces cerevisiae strain BY4742 was grown in yeast extract peptone dextrose media (YPD, 1% yeast extract, 2% peptone, 2% dextrose). Cells were allowed to propagate for at least 10 generations to an optical density at 600 nm of 0.6. The cells were harvested by centrifugation at 5000 rpm for 5 min. The cells were washed three times and centrifuged for the final pelleting at 5000 rpm for 10 min. The cell pellet was resuspended in lysis buffer composed of 50 mM Tris pH8, 8 M urea, 75 mM sodium chloride, 100 mM sodium butyrate, protease (Roche, Indianapolis, IN, USA), and phosphatase inhibitor tablet (Roche, Indianapolis, IN, USA). The cells were then lysed by glass bead milling (Retsch, Germany). Briefly, 2 mL of acid washed glass beads were combined with 2.5 mL of resuspended yeast cells in a stainless steel container and shaken 8 times at 30 Hz for 4 min with a 1 min rest in between. The concentration of extracted protein was measured by bicinchoninic acid (BCA) protein assay. Protein was reduced by addition of 5 mM dithiothreitol and incubated for 45 min at 55°C. The mixture was cooled to room temperature, followed by alkylation of free thiols by addition of 15 mM iodoacetamide in the dark for 30 min. The alkylation reaction was quenched with 5 mM dithiothreitol. To create large, highly charged peptides for the large scale analysis of optimal ETD reaction times, proteins were digested for approximately 60 min at ~4°C after the addition of 1 mM CaCl2, 50 mM Tris to decrease urea to 1 M, and incubated with trypsin at an enzyme-to-substrate ratio of 1:250 (Promega, Madison, WI, USA). Endo LysC (Wako, Richmond, VA, USA) was also used with similar conditions except it was incubated overnight, at 4 M urea, at room temperature, and with an enzyme-to-substrate ratio of 1:100. All digests were quenched by the addition of TFA to a final concentration of 0.5% (pH 2), and desalted via solid phase extraction on a 50-mg tC18 SepPak cartridge (Waters, Milford, MA, USA).
Mass Spectrometry and Chromatography
An ETD-enabled hybrid, dual cell-quadrupole ion trap-Orbitrap mass spectrometer (Orbitrap Elite, Thermo Fisher Scientific) was used for the bulk of these studies [6, 7, 27]. When noted, a quadrupole-Orbitrap-quadrupole ion trap (Q-OT-qIT) hybrid mass spectrometer (Orbitrap Fusion, Thermo Fisher Scientific) was utilized [28]. Reversed phase columns for capillary chromatography were prepared in-house. Briefly, a 75 μm-360 μm inner-outer diameter bare-fused silica capillary, with a laser pulled electrospray tip, was packed with 1.7 μm diameter, 130 Å pore size, Bridged Ethylene Hybrid C18 particles (Waters, Milford, MA, USA) to a final length of 35 cm. The column was installed on a nanoAcquity UPLC (Waters, Milford, MA, USA) using a stainless steel ultra-high pressure union formatted for 360 μm outer diameter columns (IDEX) and heated to 60°C for all runs. Mobile phase buffer A was composed of water, 0.2% formic acid, and 5% DMSO. Mobile phase B was composed of acetonitrile, 0.2% formic acid, and 5% DMSO. Samples were loaded onto the column for 12 min at 0.35 μL/min. Mobile phase B increases to 4% in the first 0.1 min, then to 12% B at 32 min, 22% B at 60 min, and 30% B at 70 min, followed by a 5 min wash at 70% B and a 20 min re-equilibration at 0% B.
For the section, Experimental Validation of Optimal ETD Reaction Time, precursors were selected from MS1 scans with an AGC target of 1,000,000 and collected at 60,000 resolving power (at m/z 400). MS/MS events in the ion trap or Orbitrap (15,000 resolving power at m/z 400) used AGC targets of either 30,000 or 100,000, respectively. For the section, Varying ETD Reaction Time Constants (τ) for Optimal Peptide Identifications, MS1 scanning was performed at a resolving power of 60,000, and from these spectra the data-dependent acquisition method selected the 20 most intense precursors for MS/MS. Precursors with a charge greater than 2 were reacted with the calibrated number of reagent anions for the specified multiple of the ETD time constant. The product ions were analyzed in either the Orbitrap (AGC target of 100,000, 15,000 resolving power) or the ion trap (AGC target of 30,000) in separate experiments. For both ion trap and Orbitrap mass analysis, one experiment was performed using a static ETD reaction time of 100 ms for all precursors. In all nanoLC-MS/MS experiments, precursors were isolated at 1.8 Th and an exclusion window of ±10 ppm was constructed around the monoisotopic peak of each selected precursor for 45 s. A quadrupole-Orbitrap-quadrupole ion trap (Q-OT-qIT) hybrid mass spectrometer (Orbitrap Fusion, Thermo Fisher Scientific) was employed for the study described in the section, Implementation of Normalized ETD Reaction Time Method for Global Proteomic Analysis. This system was operated in top speed mode where MS1 scans were performed at least every 5 s and MS/MS events are triggered in a data-dependent fashion (resolving power 60,000 at m/z 200). ETD reactions were conducted using either the “use calibrated ETD times” setting, where the fluoranthene anion population and reaction duration are defined using the kinetic-based approach described in this work, or using a static reaction time of 100 ms with a reagent anion AGC target of 200,000. ETD product ions were analyzed with either the ion trap (AGC target of 30,000) or Orbitrap (AGC target of 100,000, 15,000 resolving power at m/z 200). Precursors were isolated using the quadrupole mass filter with a window of ±0.7 Th. For the data-dependent precursor selection, an exclusion window of ±10 ppm was constructed around the monoisotopic peak of each selected precursor for 45 s.
Database Searching and Data Analysis
Tandem mass spectral data were searched against a database containing canonical protein and protein isoform sequences from Uniprot (6563 total entries) [29]. For data presented in the section, Experimental Validation of Optimal ETD Reaction Time, database searching was performed using a precursor mass tolerance of ±4.5 Th, Orbitrap MS/MS product ion tolerance of ±0.01 Th, and ion trap MS/MS product ion tolerance of ±0.35 Th while considering up to nine missed cleavages. These parameters allowed identification of large, higher charge state entities to improve the number of such species in the calculations. For all other experiments, a precursor mass tolerance of ±150 ppm was used while allowing up to four isotopes and three missed cleavages. Search parameters included carbamidomethylation of cysteine as a fixed modification and oxidation of methionine as a variable modification. In either approach, raw data files were converted to text files and searched using a version of OMSSA (ver. 2.1.8) modified to allow searching of more than three missed cleavages [30]. OMSSA search results were filtered to a 1% FDR within COMPASS software suite [31].
Results and Discussion
ETD Reaction Kinetics
ETD reactions are governed by many operational parameters including the precursor and reagent ion populations, precursor charge (z), ion/ion reaction vessel characteristics, and reaction duration [21, 32–37]. Samples analyzed with traditional shotgun methodology comprise a diverse pool of precursor peptide cations so that optimal reaction conditions vary considerably from one scan to the next. For collisional activation methods, this variation is accommodated by normalizing collision energy as a function of precursor mass and charge [25, 26]. Ion/ion reactions, however, involve more parameters and normalization is not as straightforward. To approach this challenge, we first consider the pseudo-first order kinetic dependence of ETD. Gas phase ion/ion reactions are well-understood and, provided the reagent ion population is in excess, are modeled as an exponential decay function [32, 33, 38, 39]:
| (1) |
where Np(0) represents the initial precursor population, Np(t) is the amount of precursor remaining at time t, [R] is the average number density of ion cloud overlap, and k is the ion/ion reaction rate constant. This rate constant is defined as:
| (2) |
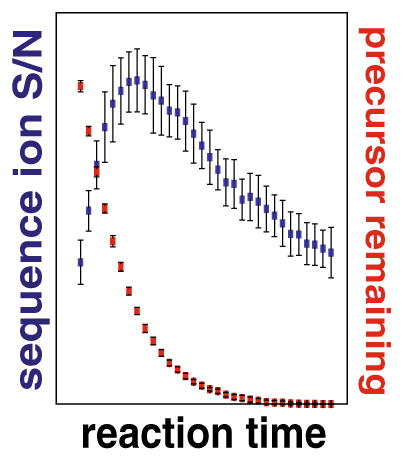
where Zp is precursor charge, mp is precursor mass, Zr is reagent charge, mr is reagent mass, and the quantity c(|v|) is a function of the distribution of the magnitude of differential velocities, |v|, of precursor and reagent ions in the overlapping ion clouds. Holding Np(0) and [R] constant, as a typical MS/MS experiment would, the ion/ion reaction is dependent upon k and t (Equation 1). Thus, optimal reaction duration (t) (i.e., the shortest reaction time to achieve the maximal quality spectra), can be achieved by selecting the appropriate ETD rate constant, k. From Equation 2, k scales with the square of the precursor charge and the number of precursor and reagent ions. Note the contribution of reduced mass to k—for precursors spanning 1000 to 10,000 Da—is negligible relative to precursor charge and ion populations and, thus, is presumed constant. The upshot is that higher charge state precursors react more quickly, requiring reduced reaction times for ideal performance. Besides improving scan speed, shortened reactions limit the occurrence of secondary electron transfer events that erode signal and complicate spectra. Thus, for every selected precursor there is an ETD reaction time that will result in an optimal creation of product ions. Figure 1a illustrates this concept by plotting the c-and z•-type product ion signal-to-noise (S/N) as a function of ion/ion reaction duration for triply protonated cations of angiotensin I (DRVYIHPFHL). In this case, the maximum product ion S/N is achieved using a 40 ms ion/ion reaction, which leaves ~12% of total MS/MS signal attributed to the intact precursor. Continuing the reaction beyond this duration consumes remaining precursor; however, product ion S/N is likewise reduced because of secondary electron transfer events. This is shown in the three example spectra where individual product ion S/N is highest at the 40 ms interval (Figure 1b).
Figure 1.

ETD reaction duration is a key variable for production of quality MS/MS spectra. Panel (a) plots the product ion S/N (blue) and rate of precursor (red, triply protonated angiotensin 1) consumption for reaction times ranging from 5 to 160 ms. From these data, we conclude the maximal product ion S/N is achieved when between 10% and 15% of the precursor remains. Extension of the reaction beyond this point both degrades spectral quality and slows the instrument scan cycle. Single scan ETD MS/MS spectra from the 5, 40, and 100 ms reactions are shown in panel (b). Note that while the precursor is nearly absent in the 100 ms scan, the overall product ion S/N is lower than the optimal 40 ms reaction
From these observations, we conclude that an algorithm to calibrate ETD reaction times, rooted in ion/ion reaction theory, would generate increased quality ETD MS/MS spectra for automated, large-scale analyses. Early implementations of ETD used static reaction conditions, commonly leading to reaction times substantially longer than necessary [21, 40–43]. On late model instrumentation ETD reaction times are dynamically adjusted, but based solely on charge state and user-defined estimates of ideal performance (e.g., ion/ion reaction time for dynamic scaling) [44]. Most users will likely have little experience with the technique and are likely to select reaction durations based on complete precursor consumption rather than peak product ion S/N (i.e., Figure 1). Further, existing algorithms scale reaction times primarily using charge, rather than the ETD reaction rate constant (k), which depends on both charge and reagent ion population (Equation 2). We surmise that an algorithm based on k will enable optimal product ion yield and improved duty cycle for higher performance.
Determination of ETD Reaction Rate Constant (k)
First considering the reagent ion population, we developed a calibration routine to determine the amount of reagent anions necessary to achieve an optimal ETD reaction rate (k). Using triply charged angiotensin, we first determined k by varying reagent anion population (AGC target, 20,000 to 450,000) and reaction durations (5 to 115 ms). From Equation 1, the ETD reaction rate is proportional to ln(A/Ao), where A0 is the precursor intensity with no reaction, and A is the precursor intensity after the ETD reaction. Figure 2a depicts three examples of this, showing the effect of reagent population on k. Here, four spectra per time point were collected, and the resulting data fitted with a linear function where the slope represents -(k). Plotting results from all AGC targets creates a curve with a maxima around k = 50 at a reagent anion AGC target of ~400,000 (Figure 2b). Using Equation 2, we can relate the reaction rate for triply charged precursors (k3) to the reaction rate for any precursor with charge state n, thus describing the charge state-dependent rate constant (kn), where:
| (3) |
Figure 2.

Calibration of reagent anion population to achieve optimal ETD reaction rate constant (k). The ratio of the remaining precursor population (a) to the initial population (A0) is linearly correlated with the magnitude of the reagent anion population (as set by the reagent AGC target, panel (a). Note the negative slope of the linear fit is the ETD reaction rate constant (k). Panel (b) presents the experimentally determined ETD rate constant (k) for reagent AGC target values ranging from 20,000 to 500,000. The maximum k is achieved at a reagent AGC target of 400,000
Commercial ETD systems will have varied performance in reagent anion generation depending on the tuning and cleanliness of the reagent source. Even so, we envision a standard calibration routine, using the method described above, to determine k for any system in an automated fashion, regardless of source conditions. By adjusting the reagent ion population to achieve a desired k, the ETD reaction rate constant is regularly calibrated and will automatically inform the reaction duration calculation on any instrument platform.
The ETD Time Constant (τ)
From Figure 1 we conclude optimal ETD product ion S/N directly relates to the population of precursor remaining at time t. Therefore, our aim is to determine the reaction time (t) that maximizes the product ion S/N. To simplify the relationship of the calibrated reaction rate k to the actual ETD reaction time used for a given reaction, we measure time in units of the mean lifetime of the precursor (where the initial precursor population is reduced by 1/e), which we term the ETD time constant (τ):
| (4) |
The ETD time constant (τ) is a basis for finding optimal charge state-dependent ETD reaction times. Experimental determination of k3, above, allowed us to use Equations 3 and 4, with z3 = 3 and k3 = 50, to calculate τ3 = 20 ms (time constants for all charge states in Supplementary Table 1). Note from Figure 1 we concluded a 40 ms reaction produced optimal product ion S/N for the triply charged precursor of angiotensin. Relating this optimal reaction time to the ETD time constant (τ3 = 20 ms) we find the optimal reaction time for angiotensin is 2τ. From these results we postulate that maximum product ion yield for a precursor of any charge state would occur at a reaction time equivalent to 2τ, the point at which the precursor population is depleted by 1/e2 (~13.5% its initial abundance).
Experimental Validation of Optimal ETD Reaction Time
Above, we calculate the required ETD reaction time to generate spectra with the maximal product ion S/N using a single peptide precursor. To explore the relationship between the ETD time constant (τ) and maximal ETD product ion yield globally, we performed a large-scale experiment testing a range of reaction times for thousands of precursors. Yeast cells were lysed, partially trypsin-digested to produce a precursor population with charge state diversity, and chromatographed into an ETD-enabled Orbitrap mass spectrometer. During the LC-MS/MS experiments, each selected precursor was dissociated in 11 consecutive MS/MS scans, accounting for nine different times ranging from 1 to 4τ—we term this spectral collection a reaction set. Product ions were analyzed in either the ion trap or Orbitrap. The time required to sequentially sample a single precursor for 11 tandem mass spectral events was typically 3 to 4 s, meaning precursor signal could vary over the course of the reaction set. To address this, failure to reach the precursor AGC target value for any MS/MS scan within the reaction set (i.e., peptide eluted prior to completion of the eleven tandem mass spectra), resulted in dismissal of the entire reaction set from subsequent data analysis. To mitigate systematic error in data collection, the order in which multiples of τ were collected was randomized.
Raw data files were converted to text files, searched using OMSSA [30], and the results filtered to a 1% FDR [31]. Altogether, we collected 10,987 reaction sets of which 4194 were mapped to sequence and also met the precursor AGC target value criteria described above. These sets comprised 46,134 tandem mass spectra and permitted detailed analysis of ETD reactions for charge states ranging from +2 to +6 (Figure 3a). Using in-house algorithms, we calculated the theoretical c- and z•-type product ions for each peptide sequence and then extracted the observed product ion intensities (15 ppm tolerance for Orbitrap and 0.5 Da for ion trap) from corresponding spectra. For precursors with charge greater than three, multiply charged product ions were considered. Within each reaction set, total product ion intensities were summed for each spectrum and then normalized to the maximum summed ion intensity of that set, resulting in a score ranging from 0 to 100 for each τ multiple for a given precursor. Figure 3 presents these results for spectra analyzed within the ion trap, showing the distribution of normalized fragment intensities of all precursors for each multiple of τ. Supplemental Figure 1 displays the same data, but with Orbitrap mass analysis. With these results, we extend the single peptide infusion studies presented above to include thousands of peptides and tens of thousands of tandem mass spectra, confirming that maximum product ion yield for a precursor of any charge state occurs within a range of 1.4 to 2τ.
Figure 3.

Experimental validation of optimal ETD reaction time. We performed large-scale experiments testing a range of reaction times for thousands of precursors with charge states ranging from +2 to +6. Panel (a) displays the number of reaction sets (i.e., peptide sequences) that were used to generate the data shown in panels (b)–(f). For each charge state, the distribution of normalized product ion intensities are plotted as a function of reaction duration, as measured in either τ or ms. These data were collected using the ion trap mass analyzer; similar data showing Orbitrap mass analysis are presented in Supplementary Figure 1
Varying ETD Reaction Time Constants (τ) for Optimal Peptide Identifications
For large-scale studies, the ultimate goal is to generate the most proteomic depth (i.e., greatest numbers of peptide identifications) in the shortest timeframe possible [45]. In this context, optimal product ion yield must be balanced with instrument scan speed. Having analyzed peptides individually using a series of ETD time constant (τ) multiples to obtain the maximum product ion S/N, we now explore how multiples of τ affect the rate of peptide identifications.
A complex mixture of yeast peptides, resulting from digestion with endo LysC, was analyzed using nano LC-MS/MS experiments (ion trap product ion analysis) with each analysis using a designated multiple of the ETD time constant (i.e., 0 to 20τ). To provide a comparison to a common implementation of ETD, we conducted an additional analysis with a static ETD reaction time of 100 ms for all charge states. Depending upon the ETD time constant used, the only variable in this study, the experiments generated between 9000 and 21,000 tandem mass spectra. This broad range exemplifies the balance that one must strike when tuning the time spent on an ETD reaction, as long reaction times will decrease the overall number of MS/MS events and reduce proteomic depth (Figure 4). Likewise, too short of a reaction and success rates will plummet. Figure 4b presents the tradeoff between success rates and unique peptide identifications. The highest rate of success (30.6%) was achieved with a 5.2τ; not surprisingly, however, this setting does not produce the optimal number of identifications.
Figure 4.

Effect of the ETD reaction time constant (τ) for shotgun proteomics. Using a complex mixture of yeast peptides, we conducted a set of nanoLC-MS/MS experiments using a designated multiple of the ETD time constant (i.e., 0 to 20τ) for each. The summary of MS/MS scans, PSMs, and unique peptides resulting from each analysis are presented in panel (a). Panel (b) explores the tradeoff between success rates (ratio PSMs to MS/MS events) and unique peptide identifications. ETD time constant multiples of 1 to 2 produce the highest number of both peptide spectral matches (PSMs) and unique peptides. To correlate the quality of the ETD MS/MS spectrum with the number of peptides identified, we calculated the median peptide sequence coverage for all identified spectra; 2τ yields the highest median sequence coverage and delivers high numbers of unique identifications [panel (c)]
ETD time constant multiples of 1 to 2 produce the highest number of both peptide spectral matches (PSMs) and unique peptides (Figure 4). Specifically, 1 and 2τ produce 19% (4344) and 16% (4243) more unique identifications than the 100 ms static reaction (3655). As calculated above, a reaction lasting 1.4 to 2τ produced the highest product ion intensity (Figure 3); however, in this experiment, 1τ produced slightly more unique identifications (Figure 4). This is most likely a result of additional MS/MS events performed. To better correlate the quality of the ETD MS/MS spectrum with the number of peptides identified, we calculated the median peptide sequence coverage for all identified spectra. In this calculation, a tandem mass spectrum that provides evidence for cleavage of 9 out of 10 backbone bonds has a 90% sequence coverage (Figure 4c); 2τ yields the highest median sequence coverage of all reaction times tested, corroborating results from our experiments above. Identical experiments performed with Orbitrap product ion analysis also produced a maximum number of peptide identifications using 1–2τ (Supplementary Figure 3). When using the Orbitrap, reaction times of 1–2τ produced ~40% more unique identifications than the 100 ms static reaction. These results indicate that using optimal ETD reaction times are particularly important when analyzing product ions in the Orbitrap.
Implementation of Normalized ETD Reaction Time Method for Global Proteomic Analysis
Above, we provide a theoretical and experimental strategy to dynamically adjust ETD reaction times based on calibrated ion/ion rate constants. The intent is to implement a standard approach so that any user can generate optimal ETD tandem mass spectra, regardless of instrument platform or level of expertise in ion/ion kinetics. As such, the calibration of ETD reaction times, as we describe herein, is a standard feature on the newest generation quadrupole-Orbitrap-quadrupole ion trap (Q-OT-qIT) mass spectrometer (Orbitrap Fusion) [28]. Within the method editor, an option to “use calibrated ETD times” sets charge state-dependent reaction times to ~2τ. When used to analyze a complex mixture of peptides (endo LysC digested yeast proteins), employment of calibrated ETD times afforded 38% more MS/MS scans (29,457 versus 21,261), 50% more PSMs (9145 versus 6076), and 52% more unique peptides (7915 versus 5210, Figure 5a). Representative MS/MS spectra of the triply charged peptide VSIAGRIHAK demonstrate lower product ion yield in the 100 ms static reaction time (Figure 5b, top) compared with the calibrated ETD reaction time (~2τ or 46.5 ms, Figure 5b, bottom). When product ions are analyzed in the Orbitrap, the benefit of calibrated ETD times is even greater with 32% more MS/MS scans (26,461 versus 20,017), 76% more PSMs (6116 versus 3466), and 75% more unique peptides (5418 versus 3090) compared with a 100 ms static reaction time (Supplementary Figure 3A). These results support our conclusion that optimal product ion yield is especially critical when analyzing ETD reactions in the Orbitrap.
Figure 5.

Implementation of calibrated ETD reaction time method for global proteomic analysis on a Q-OT-qIT hybrid. Results following the analysis of complex peptide mixtures using either calibrated or static ETD reaction times panel (a). Panel (b) displays representative spectra (VSIAGRIHAK, +3) demonstrating lower product ion yield in the 100 ms static reaction time (top) compared with the calibrated ETD reaction time (~2τ or 46.5 ms, bottom)
Conclusion
By integrating an ion/ion reaction kinetic model, we present an automated ETD calibration routine that maximizes performance and does not require expert knowledge. In doing so, we define a new concept, ETD reaction time constant (τ), which scales charge-state dependent ETD reaction times based on the experimentally determined rate constant k. These results provide a template for extending this approach to a variety of ETD-enabled systems, as exemplified by its implementation on the Orbitrap Fusion platform. This approach considerably outperforms early implementations of ETD, where static reaction times slowed duty cycle and generated lower quality spectra [21, 40–43]. Our kinetic model also provides advantages over more recent methods that scale ETD reaction times based on charge state by eliminating the need for user input, a major source of performance variability [44]. Finally, these existing algorithms scale reaction times primarily using charge rather than the ETD reaction rate constant (k), which depends on both charge and reagent ion population.
Implementation of the ETD calibration algorithm in standard data-dependent experiments revealed that ETD reaction times scaled to 1–2 multiples of the ETD time constant (τ) maximized peptide identifications and spectral quality, improving identifications by nearly 20% for the ion trap and 40% for the Orbitrap analyses, compared with a static reaction time. Use of this procedure on the newer, faster scanning Orbitrap Fusion system produced even stronger results. Specifically, calibrated ETD times afforded 38% more MS/MS scans, 50% more PSMs, and 52% more unique peptides when using ion trap product ion analysis. For MS/MS with the Orbitrap, the benefit of calibrated ETD times is even greater: 75% more unique peptides, compared with a 100 ms static reaction time. These results highlight transferability of the routine as the Fusion platform utilizes an entirely different approach for reagent anion generation and introduction [46]. We conclude that as instrument scan speed continues to increase, so too will the importance of the ETD calibration algorithm.
Here we demonstrate an algorithm for calibration of the ETD reaction primarily in the context of shotgun proteomics. ETD is doubtless a useful technique for large-scale peptide discovery and is especially valuable for applications mapping labile post-translational modifications, including glycosylation, phosphorylation, and others [11, 16, 42, 47–57]. Beyond these applications, there are biomolecule classes where traditional collisional activation methods yield inadequate or less than satisfactory results. Peptides having abundant basicity (e.g., histone tails) [44, 58, 59], lacking basic C-terminal residues (e.g., antigens), and containing crosslinks (e.g., disulfides) [60] are a few examples. Finally, intact protein sequencing methods (i.e., top-down) routinely leverage ETD for extensive backbone cleavage and whole protein characterization [36, 61–66]. We note all of these applications will benefit from the ETD calibration routine presented here as our kinetic modeling will accommodate a broad range of precursor charge states.
A primary driver of this work is to improve ETD functionality and ensure that users of all levels can achieve the best possible result with the method. A comparable evolution transpired in the decades following introduction of ion trap CAD in 1986 [67, 68]. In modern commercial implementations of ion trap CAD, most of the critical parameter choices are made automatically by the instrument control software according to a number of calibrations. The user is near-completely shielded from the many parameters that require calibration for reliable and successful resonant excitation [25, 26]. Of course in early commercial implementations of ETD, the instrument control software shielded the user from many experimental parameters; however, those that remained demanded a high level of understanding to balance production of quality spectra and maintain scan speed. With an ETD calibration routine based on ion/ion reaction kinetics, we take a key step forward in maturation of ETD, easing the burden on the user while simultaneously improving spectral quality and overall performance.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support from Thermo Fisher Scientific and NIH grant R01 GM080148. C.M.R was funded by an NSF Graduate Research Fellowship and NIH Traineeship (T32GM008505). N.M.R. was funded through an NSF Graduate Research Fellowship (DGE-1256259). A.E.M. was supported by an NIH post-doctoral traineeship (5T15LM007359). The authors thank John Syka for helpful discussions, experimental suggestions, and critical reading of the manuscript.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-015-1183-1) contains supplementary material, which is available to authorized users.
References
- 1.Eng JK, Mccormack AL, Yates JR. An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 2.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF. Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. Int J Mass Spectrom. 2004;236:33–42. [Google Scholar]
- 4.Stephenson JL, McLuckey SA. Adaptation of the Paul trap for study of the reaction of multiply charged cations with singly charged anions. Int J Mass Spectrom Ion Process. 1997;162:89–106. [Google Scholar]
- 5.Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA. Electron transfer ion/ion reactions in a three-dimensional quadrupole ion trap: reactions of doubly and triply protonated peptides with SO2*- Anal Chem. 2005;77:1831–1839. doi: 10.1021/ac0483872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McAlister GC, Phanstiel D, Good DM, Berggren WT, Coon JJ. Implementation of electron-transfer dissociation on a hybrid linear ion trap-Orbitrap mass spectrometer. Anal Chem. 2007;79:3525–3534. doi: 10.1021/ac070020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAlister GC, Berggren WT, Griep-Raming J, Horning S, Makarov A, Phanstiel D, Stafford G, Swaney DL, Syka JEP, Zabrouskov V, Coon JJ. A proteomics grade electron transfer dissociation-enabled hybrid linear ion trap-Orbitrap mass spectrometer. J Proteome Res. 2008;7:3127–3136. doi: 10.1021/pr800264t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Y, Chrisman PA, Erickson DE, Liu J, Liang XR, Londry FA, Yang MJ, McLuckey SA. Implementation of ion/ion reactions in a quadrupole/time-of-flight tandem mass spectrometer. Anal Chem. 2006;78:4146–4154. doi: 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Good DM, Coon JJ. Advancing proteomics with ion/ion chemistry. Biotechniques. 2006;40:783–789. doi: 10.2144/000112194. [DOI] [PubMed] [Google Scholar]
- 10.Coon JJ. Collisions or electrons? Protein sequence analysis in the 21st century. Anal Chem. 2009;81:3208–3215. doi: 10.1021/ac802330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, McAlister GC, Thomson JA, Coon JJ. Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:4093–4098. doi: 10.1073/pnas.0710515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phanstiel D, Zhang Y, Marto JA, Coon JJ. Peptide and protein quantification using iTRAQ with electron transfer dissociation. J Am Soc Mass Spectrom. 2008;19:1255–1262. doi: 10.1016/j.jasms.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Good DM, Wenger CD, McAlister GC, Bai DL, Hunt DF, Coon JJ. Post-acquisition ETD spectral processing for increased peptide identifications. J Am Soc Mass Spectrom. 2009;20:1435–1440. doi: 10.1016/j.jasms.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phanstiel D, Unwin R, McAlister GC, Coon JJ. Peptide quantification using 8-Plex isobaric tags and electron transfer dissociation tandem mass spectrometry. Anal Chem. 2009;81:1693–1698. doi: 10.1021/ac8019202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadygov RG, Good DM, Swaney DL, Coon JJ. A new probabilistic database search algorithm for ETD spectra. J Proteome Res. 2009;8:3198–3205. doi: 10.1021/pr900153b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimsrud PA, den Os D, Wenger CD, Swaney DL, Schwartz D, Sussman MR, Ane JM, Coon JJ. Large-Scale phosphoprotein analysis in Medicago truncatula roots provides insight into in vivo kinase activity in legumes. Plant Physiol. 2010;152:19–28. doi: 10.1104/pp.109.149625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Good DM, Wenger CD, Coon JJ. The effect of interfering ions on search algorithm performance for electron-transfer dissociation data. Proteomics. 2010;10:164–167. doi: 10.1002/pmic.200900570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia QW, Lee MV, Rose CM, Marsh AJ, Hubler SL, Wenger CD, Coon JJ. Characterization and diagnostic value of amino acid side chain neutral losses following electron-transfer dissociation. J Am Soc Mass Spectrom. 2011;22:255–264. doi: 10.1007/s13361-010-0029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey DJ, Rose CM, McAlister GC, Brumbaugh J, Yu PZ, Wenger CD, Westphall MS, Thomson JA, Coon JJ. Instant spectral assignment for advanced decision tree-driven mass spectrometry. Proc Natl Acad Sci U S A. 2012;109:8411–8416. doi: 10.1073/pnas.1205292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards AL, Vincent CE, Guthals A, Rose CM, Westphall MS, Bandeira N, Coon JJ. Neutron-encoded signatures enable product ion annotation from tandem mass spectra. Mol Cell Proteomics. 2013;12:3812–3823. doi: 10.1074/mcp.M113.028951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Compton PD, Strukl JV, Bai DL, Shabanowitz J, Hunt DF. Optimization of electron transfer dissociation via informed selection of reagents and operating parameters. Anal Chem. 2012;84:1781–1785. doi: 10.1021/ac202807h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunt DF, Buko AM, Ballard JM, Shabanowitz J, … Giordani AB. Sequence-analysis of polypeptides by collision activated dissociation on a triple quadrupole mass-spectrometer. Biomed Mass Spectrom. 1981;8:397–408. doi: 10.1002/bms.1200080909. [DOI] [PubMed] [Google Scholar]
- 23.Mcluckey SA. Principles of collisional activation in analytical mass-spectrometry. J Am Soc Mass Spectrom. 1992;3:599–614. doi: 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- 24.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Ficarro SB, Li SJ, Marto JA. Optimized Orbitrap HCD for quantitative analysis of phosphopeptides. J Am Soc Mass Spectrom. 2009;20:1425–1434. doi: 10.1016/j.jasms.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 26.McAlister GC, Phanstiel DH, Brumbaugh J, Westphall MS, Coon JJ. Higher-energy collision-activated dissociation without a dedicated collision cell. Mol Cell Proteom. 2011;10(5):O111.009456. doi: 10.1074/mcp.O111.009456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Muller M, Viner R, Schwartz J, Remes P, Belford M, Dunyach JJ, Cox J, Horning S, Mann M, Makarov A. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteom. 2012:11. doi: 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senko MW, Remes PM, Canterbury JD, Mathur R, Song QY, Eliuk SM, Mullen C, Earley L, Hardman M, Blethrow JD, Bui H, Specht A, Lange O, Denisov E, Makarov A, Horning S, Zabrouskov V. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal Chem. 2013;85:11710–11714. doi: 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- 29.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 30.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang XY, Shi WY, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 31.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11:1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLuckey SA, Stephenson JL, Asano KG. Ion/ion proton-transfer kinetics: implications for analysis of ions derived from electrospray of protein mixtures. Anal Chem. 1998;70:1198–1202. doi: 10.1021/ac9710137. [DOI] [PubMed] [Google Scholar]
- 33.Tolmachev AV, Udseth HR, Smith RD. Modeling the ion density distribution in collisional cooling rf multipole ion guides. Int J Mass Spectrom. 2003;222:155–174. [Google Scholar]
- 34.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Chalkley RJ, Medzihradszky KF, Lynn AJ, Baker PR, Burlingame AL. Statistical analysis of peptide electron transfer dissociation fragmentation mass spectrometry. Anal Chem. 2010;82:579–584. doi: 10.1021/ac9018582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose CM, Russell JD, Ledvina AR, McAlister GC, Westphall MS, Griep-Raming J, Schwartz JC, Coon JJ, Syka JEP. Multipurpose dissociation cell for enhanced ETD of intact protein species. J Am Soc Mass Spectrom. 2013;24:816–827. doi: 10.1007/s13361-013-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frese CK, Nolting D, Altelaar AFM, Griep-Raming J, Mohammed S, Heck AJR. Characterization of electron transfer dissociation in the Orbitrap velos HCD cell. J Am Soc Mass Spectrom. 2013;24:1663–1670. doi: 10.1007/s13361-013-0618-9. [DOI] [PubMed] [Google Scholar]
- 38.Stephenson JL, McLuckey SA. Ion/ion reactions in the gas phase: proton transfer reactions involving multiply-charged proteins. J Am Chem Soc. 1996;118:7390–7397. [Google Scholar]
- 39.McLuckey SA, Stephenson JL. Ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom Rev. 1998;17:369–407. doi: 10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 40.Williams DK, McAlister GC, Good DM, Coon JJ, Muddiman DC. Dual electrospray ion source for electron-transfer dissociation on a hybrid linear ion trap-Orbitrap mass spectrometer. Anal Chem. 2007;79:7916–7919. doi: 10.1021/ac071444h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose CM, Venkateshwaran M, Volkening JD, Grimsrud PA, Maeda J, Bailey DJ, Park K, Howes-Podoll M, den Os D, Yeun LH, Westphall MS, Sussman MR, Ane JM, Coon JJ. Rapid phosphoproteomic and transcriptomic changes in the rhizobia-legume symbiosis. Mol Cell Proteomics. 2012;11:724–744. doi: 10.1074/mcp.M112.019208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myers SA, Daou S, Affar EB, Burlingame A. Electron transfer dissociation (ETD): the mass spectrometric breakthrough essential for O-GlcNAc protein site assignments—a study of the O-GlcNAcylated protein host cell factor C1. Proteomics. 2013;13:982–991. doi: 10.1002/pmic.201200332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frey BL, Ladror DT, Sondalle SB, Krusemark CJ, Jue AL, Coon JJ, Smith LM. Chemical derivatization of peptide carboxyl groups for highly efficient electron transfer dissociation. J Am Soc Mass Spectrom. 2013;24:1710–1721. doi: 10.1007/s13361-013-0701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eliuk SM, Maltby D, Panning B, Burlingame AL. High resolution electron transfer dissociation studies of unfractionated intact histones from murine embryonic stem cells using on-line capillary LC separation determination of abundant histone isoforms and post-translational modifications. Mol Cell Proteomics. 2010;9:824–837. doi: 10.1074/mcp.M900569-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. The 1-hour yeast proteome. Mol Cell Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Earley L, Anderson LC, Bai DNL, Mullen C, Syka JEP, English AM, Dunyach JJ, Stafford GC, Shabanowitz J, Hunt DF, Compton PD. Front-end electron transfer dissociation: a new ionization source. Anal Chem. 2013;85:8385–8390. doi: 10.1021/ac401783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. Complementary structural information from a tryptic N-linked glycopeptide via electron transfer ion/ion reactions and collision-induced dissociation. J Proteome Res. 2005;4:628–632. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JEP, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taouatas N, Drugan MM, Heck AJR, Mohammed S. Straightforward ladder sequencing of peptides using a Lys-N metalloendopeptidase. Nat Methods. 2008;5:405–407. doi: 10.1038/nmeth.1204. [DOI] [PubMed] [Google Scholar]
- 51.Swaney DL, Wenger CD, Thomson JA, Coon JJ. Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2009;106:995–1000. doi: 10.1073/pnas.0811964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou WD, Ross MM, Tessitore A, Ornstein D, VanMeter A, Liotta LA, Petricoin EF. An initial characterization of the serum phosphoproteome. J Proteome Res. 2009;8:5523–5531. doi: 10.1021/pr900603n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swaney DL, Wenger CD, Coon JJ. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J Proteome Res. 2010;9:1323–1329. doi: 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darula Z, Chalkley RJ, Baker P, Burlingame AL, Medzihradszky KF. Mass spectrometric analysis, automated identification and complete annotation of O-linked glycopeptides. Eur J Mass Spectrom. 2010;16:421–428. doi: 10.1255/ejms.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frese CK, Altelaar AFM, Hennrich ML, Nolting D, Zeller M, Griep-Raming J, Heck AJR, Mohammed S. Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J Proteome Res. 2011;10:2377–2388. doi: 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- 56.Kim YC, Udeshi ND, Balsbaugh JL, Shabanowitz J, Hunt DF, Olszewski NE. O-GlcNAcylation of the Plum pox virus capsid protein catalyzed by SECRET AGENT: characterization of O-GlcNAc sites by electron transfer dissociation mass spectrometry. Amino Acids. 2011;40:869–876. doi: 10.1007/s00726-010-0706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frese CK, Zhou HJ, Taus T, Altelaar AFM, Mechter K, Heck AJR, Mohammed S. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD) J Proteome Res. 2013;12:1520–1525. doi: 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rose KL, Li A, Zalenskaya I, Zhang Y, Unni E, Hodgson KC, Yu YP, Shabanowitz J, Meistrich ML, Hunt DF, Ausio J. C-terminal phosphorylation of murine testis-specific histone H1t in elongating spermatids. J Proteome Res. 2008;7:4070–4078. doi: 10.1021/pr8003908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jung HR, Pasini D, Helin K, Jensen ON. Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol Cell Proteomics. 2010;9:838–850. doi: 10.1074/mcp.M900489-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu SL, Jiang HT, Hancock WS, Karger BL. Identification of the unpaired cysteine status and complete mapping of the 17 disulfides of recombinant tissue plasminogen activator using LC-MS with electron transfer dissociation/collision induced dissociation. Anal Chem. 2010;82:5296–5303. doi: 10.1021/ac100766r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc Natl Acad Sci U S A. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsybin YO, Fornelli L, Stoermer C, Luebeck M, Parra J, Nallet S, Wurm FM, Hartmer R. Structural analysis of intact monoclonal antibodies by electron transfer dissociation mass spectrometry. Anal Chem. 2011;83:8919–8927. doi: 10.1021/ac201293m. [DOI] [PubMed] [Google Scholar]
- 63.Fornelli L, Damoc E, Thomas PM, Kelleher NL, Aizikov K, Denisov E, Makarov A, Tsybin YO. Analysis of intact monoclonal antibody IgG1 by electron transfer dissociation Orbitrap FTMS. Mol Cell Proteomics. 2012;11:1758–1767. doi: 10.1074/mcp.M112.019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fornelli L, Parra J, Hartmer R, Stoermer C, Lubeck M, Tsybin YO. Top-down analysis of 30–80 kDa proteins by electron transfer dissociation time-of-flight mass spectrometry. Anal Bioanal Chem. 2013;405:8505–8514. doi: 10.1007/s00216-013-7267-5. [DOI] [PubMed] [Google Scholar]
- 65.Rhoads TW, Rose CM, Bailey DJ, Riley NM, Molden RC, Nestler AJ, Merrill AE, Smith LM, Hebert AS, Westphall MS, Pagliarini DJ, Garcia BA, Coon JJ. Neutron-encoded mass signatures for quantitative top-down proteomics. Anal Chem. 2014;86:2314–2319. doi: 10.1021/ac403579s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fornelli L, Ayoub D, Aizikov K, Beck A, Tsybin YO. Middle-down analysis of monoclonal antibodies with electron transfer dissociation Orbitrap Fourier transform mass spectrometry. Anal Chem. 2014;86:3005–3012. doi: 10.1021/ac4036857. [DOI] [PubMed] [Google Scholar]
- 67.Louris JN, Cooks RG, Syka JEP, Kelley PE, Stafford GC, Todd JFJ. Instrumentation, applications, and energy deposition in quadrupole ion-trap tandem mass-spectrometry. Anal Chem. 1987;59:1677–1685. [Google Scholar]
- 68.Louris JN, Brodbeltlustig JS, Cooks RG, Glish GL, Vanberkel GJ, Mcluckey SA. Ion isolation and sequential stages of mass-spectrometry in a quadrupole ion trap mass-spectrometer. Int J Mass Spectrom Ion Process. 1990;96:117–137. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.