

Abstract
T cells recognize antigens at the two-dimensional (2D) interface with antigen-presenting cells (APCs), which trigger T-cell effector functions. T-cell functional outcomes correlate with 2D kinetics of membrane-embedded T-cell receptors (TCRs) binding to surface-tethered peptide-major histocompatibility complex molecules (pMHCs). However, most studies have measured TCR–pMHC kinetics for recombinant TCRs in 3D by surface plasmon resonance, which differs drastically from 2D measurements. Here, we compared pMHC dissociation from native TCR on the T-cell surface to recombinant TCR immobilized on glass surface or in solution. Force on TCR–pMHC bonds regulated their lifetimes differently for native than recombinant TCRs. Perturbing the cellular environment suppressed 2D on-rates but had no effect on 2D off-rate regardless of whether force was applied. In contrast, for the TCR interacting with its monoclonal antibody, the 2D on-rate was insensitive to cellular perturbations and the force-dependent off-rates were indistinguishable for native and recombinant TCRs. These data present novel features of TCR–pMHC kinetics that are regulated by the cellular environment, underscoring the limitations of 3D kinetics in predicting T-cell functions and calling for further elucidation of the underlying molecular and cellular mechanisms that regulate 2D kinetics in physiological settings.
Keywords: Adhesion, Cellular immunology, Molecular immunology, TCRs, T cells
Introduction
The interaction between T-cell receptors (TCRs) and peptide-major histocompatibility complex molecules (pMHCs) is the focus of a large body of literature due to its central role in a broad range of T-cell functions [1–5]. Kinetics of the TCR–pMHC interaction is commonly analyzed by surface plasmon resonance (SPR) using recombinant TCR or pMHC tetramer decay from the T-cell surface [3, 6, 7]. In both cases, one of the interacting molecules is soluble in fluid phase dispersed in a three-dimensional (3D) space. Despite the remarkable sensitivity of T cells for extremely low densities of antigenic pMHC on the surface of antigen-presenting cells (APCs), 3D kinetic measurements generally show weak binding (dissociation constant Kd in the 1–100 μM range and half-life t½ on the order of seconds) [1, 8]. 3D kinetic parameters have been widely used as the foundation for various T-cell activation models [1, 2, 9, 10]. Recently, two sets of novel methods—one mechanical-based [4] and the other fluorescent-based [5, 11, 12]—have been developed to make direct in situ measurements of TCR–pMHC kinetics on live T cells interacting with pMHC anchored on a two-dimensional (2D) surface. Some in situ 2D kinetic measurements dramatically differ from their 3D counterparts [4–6, 11]. In 2D, the TCR binds agonist pMHC class I with a high affinity comparable to the high affinity of leukocyte function-associated antigen binding to intercellular adhesion molecule [4, 6]. In terms of discriminating antigens of varying biological activities, the 2D affinities and on-rates display far broader dynamic ranges than 3D parameters [4, 6]. Importantly, T-cell responses correspond better to 2D than 3D kinetics [4–6, 13–15].
The drastic differences between the reported 2D and 3D TCR–pMHC kinetic parameters have generated considerable interest and debates regarding the underlying mechanisms. It has been argued that SPR measurements capture intrinsic kinetic properties of the physical chemistry local to the TCR–pMHC binding interface whereas measurements on the T-cell membrane are influenced by cellular regulations [4, 5, 15]. Despite that some elements of the differences between binding in 2D versus 3D may be accounted for by physical considerations [16] and mod-eled mathematically [17, 18], the complex cellular environment (e.g., lipid rafts, protein islands, TCR clustering, cytoskeleton, and active transport) may regulate molecular interactions across intercellular junctions biologically, making 2D measures apparent rather than intrinsic parameters [19, 20]. Thus, to relate 2D and 3D parameters requires an understanding of the nature, characteristics, and mechanisms of the hypothetical biological regulations. However, little is known about these regulations, and many questions remain unanswered. For example, among a panel of pMHC ligands specific for the 42F3 TCR, complexes with the peptide p3A1 had the highest 3D affinity by SPR but the lowest 2D affinity by micropipette adhesion frequency assay [13]. This is at odds with the notion that the cellular environment generally increases 2D affinity by boosting on-rate [4–6]. Also, some in situ measurements reported faster pMHC dissociation from the native TCR (i.e., naturally expressed on the T-cell surface) than the recombinant TCR [4, 5]. An extreme case is that 2D off-rates of the OT1 TCR measured on naïve T cells using the micropipette adhesion frequency assay [21] and the thermal fluctuation assay [22] was more than 300 times (at room temperature) or 8000 times (at 37°C) faster than the 3D value previously measured for agonist OVA:H2-Kb [4, 23]. No satisfactory explanation is yet available for such a large difference between 2D and 3D off-rates.
Most previous studies focused on kinetic measurements under stress-free conditions. Recently, kinetics of TCR–pMHC dissociation under mechanical force has received growing attention [24–28]. Using a biomembrane force probe (BFP) to apply pico-Newton (pN) forces to individual TCR–pMHC bonds on live T cells, force was found to prolong rather than shorten lifetimes of TCR bonds with agonist pMHC—a counterintuitive phenomenon called catch bond [26, 28]. By comparison, the TCR forms slip bonds with less biologically active pMHC ligands where bond lifetimes are shortened by force. In effect, this peptide-dependent regulation of bond dissociation by force reversed the zero-force lifetime ranking [4] such that the more biologically active the ligand, the longer the bond lifetime—a trend that is shared with SPR measurements [29]. However, for the same ligands, their 2D bonds with the native OT1 TCR under force still far shorter-lived than their 3D bonds with the recombinant OT1 TCR. By comparison, neither discrepancies between 2D and 3D off-rates nor catch bonds were found when recombinant 1G4 TCR was analyzed by a flow chamber [27].
To investigate the discrepant results of TCR–pMHC kinetics, we interrogated several TCRs from three perspectives. We asked whether the differences were due to physical distinction between 2D and 3D measurements, biological dissimilarity between the recombinant proteins produced by Escherichia coli and the native proteins expressed on cell surface, or biomechanical regulation by force. Our new SPR measurements (3D, recombinant, zero force) revealed much faster off-rates than previously reported [29, 30]. This is consistent with our 2D measurements with BFP for both the recombinant and the native TCRs at zero force. Under tensile forces, both the recombinant and the native TCRs formed catch bonds with their agonist pMHCs, but lifetimes for the native TCRs were much longer than those for the recombinant TCRs. Perturbation of T cells with pharmaceutical agents severely suppressed 2D affinity and on-rate, but not off-rates under either zero or tensile forces, of native TCR bonds with pMHC. In contrast, the force-dependent 2D off-rates of an anti-TCR antibody dissociation from the recombinant TCR and the native TCR were the same, and the 2D affinity of native TCR–antibody interaction was not affected by pharmaceutical treatments. These data suggest that the on-rate of pMHC association to native TCR is regulated by the T cell but the off-rate is insensitive to such regulation, and that force applied via an engaged pMHC may induce different bonding conformations on native TCR from recombinant TCR.
Results
Agonist dissociates fast from both the recombinant and the native TCR in 3D
We first attempted to measure dissociation of soluble OVA:H2-Kbα3A2 from the native OT1 TCR on live T cells using a real-time flow cytometry assay [31]. Unlike the tetramer decay assay that fixes cells at various time points and tests them in separate runs [6], real-time flow cytometry collects data continuously. We reasoned that the improved temporal resolution (~10 s) might allow us to measure the 3D off-rate of OT1 TCR–OVA:H2-Kbα3A2 dissociation, should it be as slow as previously reported (with a half-life of 30 s) [23, 29, 30]. We observed a well-behaved dissociation curve for the OVA:H2-Kbα3A2 tetramer from OT1 T cells (Fig. 1A), enabling a reliable evaluation of an apparent half-life of 105 s. In sharp contrast, the dissociation curve of the OVA:H2-Kbα3A2 monomer from the OT1 T cells were much faster—the mean fluorescent intensity (MFI) immediately dropped to background level at the first measurable time point (Fig. 1B), showing that the corresponding half-life is much shorter than the temporal resolution of the assay.
Figure 1.
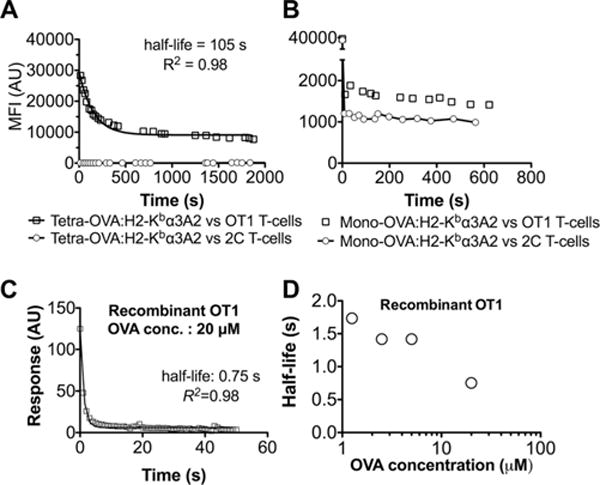
Agonist OVA:H2-Kb dissociates rapidly from both recombinant and native OT1 TCR at the force-free condition. (A, B) Dissociation of (A) tetrameric or (B) monomeric OVA:H2-Kbα3A2 (a H2-Kb mutant that does not bind CD8) from naïve OT1 (square) or 2C (circle, serving as control) T-cells measured by a real-time flow cytometry. MFI denotes mean fluorescence intensity. The dissociation curve of OVA:H2-Kbα3A2 tetramer from OT1 cells was fitted by a first-order dissociation model (curve) with the indicated half-life. The dissociation of OVA:H2-Kbα3A2 monomer from OT1 cells was too fast to fit the model. Shown is one of two replicates. (C) Representative SPR dissociation sensorgram of recombinant OT1 TCR (at 20 μM) dissociating from OVA:H2-Kb immobilized on sensor chip. Data (points) were fitted by a first-order dissociation model (curve) with the indicated half-life. (D) Plot of half-life versus pMHC concentration used to obtain the dissociation sensorgram. (A–D) Data shown are representative of two independent experiments.
Having demonstrated fast dissociation of OT1 TCR–agonist interaction on live T cells, we wanted to reproduce the previous SPR measurement of the same interaction using the recombinant OT1 TCR. We immobilized the TCR on a BIAcore sensor chip and applied a range of concentrations of OVA:H2-Kb as the soluble analyte. Surprisingly, the dissociation was faster than our BIA-core machine can reliably resolve. This is evident from the fact that we were only able to record a few data points in the initial unbinding phase (Fig. 1C). Nevertheless, fitting the data with a single exponential decay yielded a nominal half-life value of ~1 s (Fig. 1C and D). Therefore, the new SPR measurement is consistent with our real-time flow cytometry data, both showing much faster dissociation than previous SPR measurements [23, 29, 30].
2D lifetimes under tensile forces are much longer for native than recombinant TCRs
We recently reported that low forces prolong TCR–agonist bond lifetime on live OT1 and 2C T cells, that is, the counterintuitive catch bond behavior. Beyond 10 pN, further increase of force shortens lifetimes of these bonds, that is, the ordinary slip bond behavior. This is in contrast to bonds of the same native TCRs with antagonists, which exhibit slip bond behavior only, where force shortens lifetimes over the entire force range. By comparison, only slip bonds were observed for recombinant 1G4 TCR interactions with both agonists and antagonists in a flow chamber study [27]. To determine whether and how force might impact pMHC dissociation from recombinant and native TCRs differently, we used a BFP to measure force-dependent lifetimes of agonist pMHC bonds with two native TCRs and their recombinant counterparts. Both forms of OT1 TCRs formed catch-slip bonds with OVA:H2-Kbα3A2 (Fig. 2A). Although qualitatively similar, the catch bond for the native OT1 TCR is much more pronounced than that for the recombinant protein, demonstrating greater mechanical strength for the native TCR bonds. As a result, force amplified the 40% small difference of bond lifetime at zero force (0.10 s for the recombinant TCR versus 0.14 s for the native TCR), reaching maximal effect of >10 folds at ~10 pN (0.08 s for the recombinant TCR versus 0.83 s for the native TCR). The marked difference in bond lifetimes of the two forms of OT1 TCR under force was not an isolated observation as similar results were also obtained for two forms of 2C TCR interacting with QL9:H2-Ld (m31, Fig. 2B).
Figure 2.
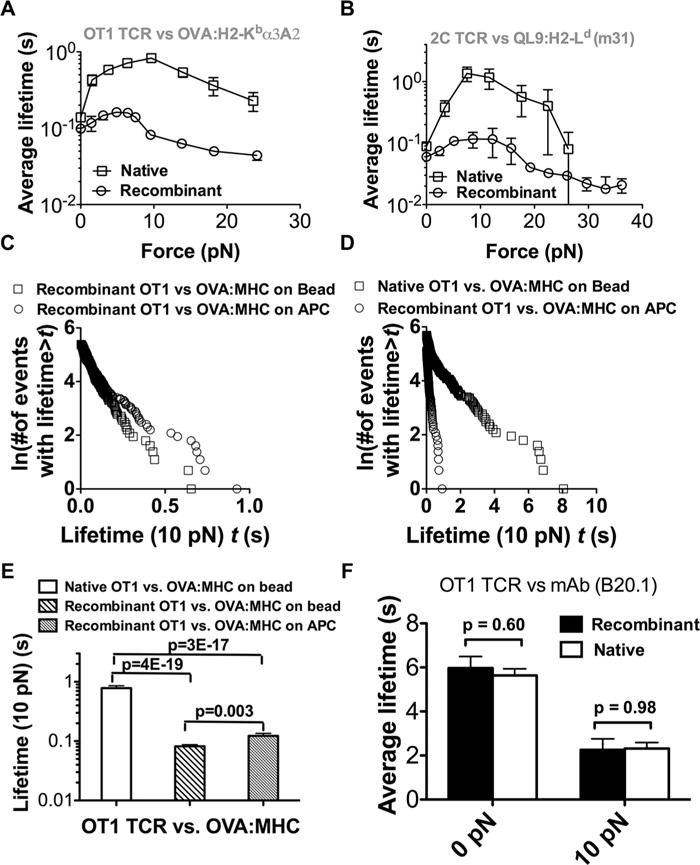
Substantially different force-dependent 2D off-rates between the native and recombinant TCRs but not native and recombinant pMHC. (A, B) Force-dependent average lifetimes of native (square) or recombinant (circle) OT1 (A) or 2C (B) TCR dissociating from OVA:H2-Kbα3A2 or QL9:H2-Ld (a minimal α1α2 variant of H2-Ld termed m31 presenting covalently linked peptides). (C) Comparison between lifetimes at 10 pN of recombinant OT1 TCR bonds with recombinant OVA:H2-Kbα3A2 (square) or APC-expressed H2-Kb loaded with the OVA peptide (circle). (A–C) Each force corresponds to >50 measurements of lifetime events. (D) Comparison between lifetimes at 10 pN of native OT1 TCR bonds with recombinant OVA:H2-Kbα3A2 (square) and of recombinant OT1 TCR bonds with APC-expressed H2-Kb loaded with the OVA peptide (circle). (E) Mean + SEM of >100 bond lifetimes under 10 pN for the three cases in (C, D). (F) Mean + SEM of >50 Bond lifetime of antibody binding to the recombinant OT1 TCR was the same as that to the native OT1 TCR at both 0 and 10 pN force. (A and B) Data are shown as mean ± SEM and (A–F) are pooled from one to five independent experiments.
Similar to TCR [32], pMHC has been observed to form microclusters on APCs that modulates TCR’s recognition sensitivity [33, 34], suggesting a possible cellular regulation of TCR–pMHC kinetics by the APC. Measured at 10 pN, lifetimes of recombinant TCR bonds with native and recombinant pMHCs were similar, but lifetimes of recombinant pMHC bonds were much longer with native than recombinant TCR (Fig. 2C–E). Interestingly, pulling the OT1 TCR with a monoclonal antibody (mAb) did not yield different lifetimes between the native and the recombinant TCRs (Fig. 2F). Collectively, these data suggest that (i) recombinant TCRs can also form catch bonds with agonists; (ii) significant difference exists between force-dependent lifetime of pMHC bonds with the two forms of TCR but not with the two forms of pMHC; and (iii) such difference is pMHC specific.
The antibody data suggest that the force-dependent lifetime disparity for the two forms of TCR could not be solely due to the differential organizations of the native TCR on the T-cell surface and the recombinant TCR on the bead. To further test this idea, we looked at clustering effects on TCR–pMHC dissociation by measuring lifetimes of pMHC captured by either tetravalent or monovalent streptavidin [35] on the BFP bead interacting with recombinant TCR captured in the same manner on the target bead. We previously showed that force-regulated lifetime of P-selectin bonds with P-selectin glycoprotein ligand-1 was longer for multimeric than monomeric interaction [36, 37]. The same differential bond lifetime patterns were produced by using tetravalent or monovalent streptavidin-captured 2GSP6 (a short glycolsulfate peptide mimicking the ligand-binding site of P-selectin glycoprotein ligand-1) to prompt dimeric or monomeric bond formation with an L-selectin construct captured by the dimeric HPC4 mAb (Fig. 3A and B). However, under both the force-free condition and at 10 pN, indistinguishable lifetimes were observed between tetrameric and monomeric pMHC interacting, respectively, with tetrameric and monomeric recombinant TCR (Fig. 3A, C, and D). TCR and/or pMHC clustering mediated by the multivalent streptavidin–biotin interaction did not have significant effect on the lifetime of pMHC bonds with recombinant TCR. In addition, indistinguishable bond lifetimes were observed between tetrameric and monomeric pMHC-interacting native TCR under both 0 and 10 pN (Fig. 3A, E, and F), suggesting that native TCR on live T-cell surface could not support multimeric interaction with tetrameric pMHC any better than with monomeric pMHC. These data do not support TCR and/or pMHC clustering as a possible explanation for the different bond lifetimes observed for the two forms of TCRs.
Figure 3.
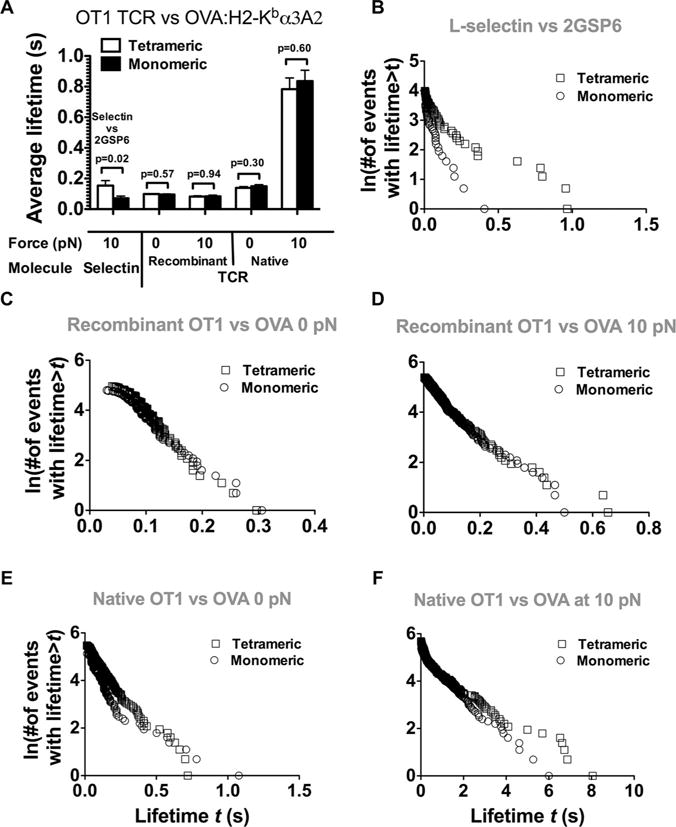
Effect of binding valence on bond lifetime. (A) Receptor and/or ligand multimericity or clustering does not affect bond lifetime between OT1 TCR (either recombinant or native) and OVA:H2-Kbα3A2. For the recombinant OT1 TCR, tetrameric and monomeric binding was measured, respectively, using tetravalent and monovalent streptavidin (SA) to capture both the TCR and the pMHC. For the native OT1 TCR, only valence of pMHC was manipulated using tetravalent and monovalent SA coupling, respectively. Divalent L-selectin was used as a control. p-Values were indicated for interactions between tetrameric and monomeric receptor(s) and ligand(s). Data are shown as mean + SEM of >50 lifetime events and are pooled from one to five independent experiments. (B–F) Lifetime distributions corresponding to panel (A).
Differences between native and recombinant TCRs affect affinity and on-rate
In a previous study [13], we observed that the 2D affinities of the 42F3 TCR for a panel of peptide ligands measured on live T cells correlates well with their biological activity whereas the 3D affinities by SPR using the recombinant TCR yielded a different affinity ranking that does not correlate with function (Fig. 4A). Of particular interest is the “outlier” peptide p3A1, which had the highest 3D affinity but the lowest 2D affinity and exhibited the lowest activity to stimulate T cells. To test whether the difference in affinity trend was due to the different 2D and 3D measurement methods used, we further measured 2D affinities (Fig. 4B) and off-rates (Fig. 4C) of the four pMHCs with the recombinant 42F3 TCR. The resulting 2D affinities followed the same trend as that from 3D SPR measurements (Fig. 4B) but different trend from the 2D affinities of the native TCR for the same panel of pMHCs, thus supporting the assertion that the affinity difference seen in Figure 4A was due to differences between the native and the recombinant TCRs rather than differences between binding in 2D and 3D. The resulting 2D zero-force off-rates of the recombinant 42F3 TCR are comparable to those of its native counterpart for the same panel of pMHCs (Fig. 4C). These data isolate the effect of differential TCR forms on 2D affinity to 2D on-rate and stipulate such effect to be peptide dependent.
Figure 4.
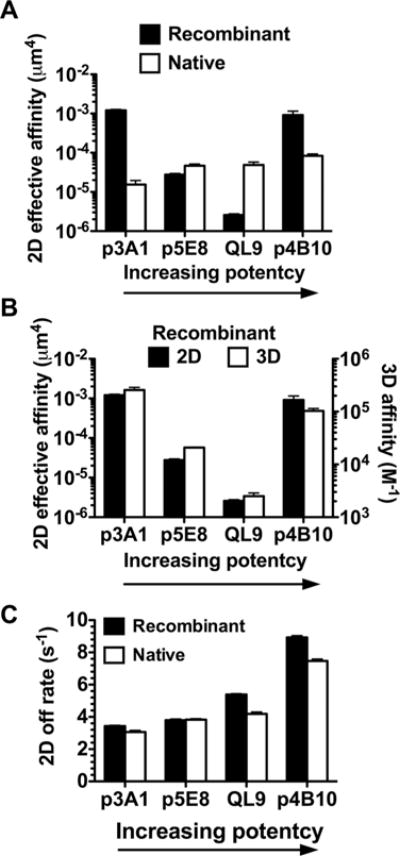
Comparison of affinity and off-rate dependencies on a panel of pMHC between native and recombinant 42F3 TCRs in 2D and 3D. (A) The peptide-dependent 2D affinity pattern of recombinant TCR (black bar, left ordinate) differs from that of the native TCR (white bar, right ordinate). Only the latter correlates with the biological activity of the ligand (indicated by arrow). Note that the exact values of 2D affinity for recombinant and native TCRs cannot be directly compared as they were presented on very different surfaces (RBCs for the recombinant 42F3 TCR and hybridoma cells for the native 42F3 TCR) [38]. The TCR and pMHC site densities are listed in Supporting Information Table 1A. (B) The peptide-dependent affinity patterns of recombinant TCR are indistinguishable in 2D (black bar, left ordinate) and 3D (white bar, right ordinate). (A and B) 2D affinity was measured with adhesion frequency assay at contact time of 2 s from two to five pairs of cell pairs; error bars represent uncertainty calculated by error propagation from adhesion frequency measurements. 3D affinity was from [13]. (C) The peptide-dependent zero-force 2D off-rate patterns are indistinguishable for recombinant (black bar) and native (white bar) TCRs. Data are shown as mean + SEM of >100 lifetime events and are pooled from one to five independent experiments.
Perturbing the cellular environment alters 2D affinity and on-rate but not off-rate
We used the OT1 system to investigate potential impact of the cellular environment on the 2D kinetics of native TCR. We reasoned that, should the cellular environment play a role in regulating in situ TCR–pMHC binding kinetics, its perturbation would change the measured parameters. We first examined the off-rate/lifetime using BFP thermal fluctuation and force clamp assays. Treating cells with latrunculin A (LA) to disrupt cytoskeleton or cholesterol oxidase (CO) to deplete lipid rafts did not significantly affect lifetimes of OT1 TCR bonds with OVA:H2-Kbα3A2 and G4:H2-Kbα3A2 at either 0 or 10 pN force (Fig. 5). This indifference of off-rate to cellular perturbations is consistent with the insensitivity of whether the off-rate was measured using native or recombinant TCRs.
Figure 5.
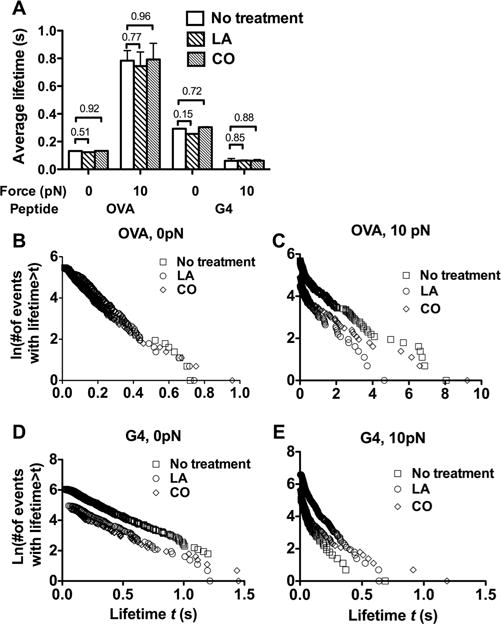
Pharmaceutical treatments of native T cells did not affect 2D TCR–pMHC off-rate. (A) Lack of influence of perturbing cellular environment on lifetimes of OT1 TCR bonds with OVA:H2-Kbα3A2 and G4:H2-Kbα3A2 at 0 and 10 pN. Data are shown as mean + SEM of >100 lifetime events and are pooled from one to five independent experiments. (B–E) Lifetime distributions corresponding to panel (A).
In sharp contrast to the lack of effect on off-rates, both LA and CO treatments greatly reduced the effective 2D affinity of the native TCR–OVA:H2-Kbα3A2 binding by 20- and 25-folds, respectively, as measured by the adhesion frequency assay (Fig. 6A). Since these treatments had no effect on off-rates (Fig. 5), the affinity changes were due to changes in effective 2D on-rates of association of the native OT1 TCR with OVA:H2-Kbα3A2 (Fig. 6B). No effect was observed in control experiments using recombinant TCR (Fig. 6), confirming that the pharmacological treatments exerted their effects on the cell rather than on the TCR molecule. These treatments did not alter kinetic properties of the native OT1 TCR binding to a mAb that blocks pMHC binding (Fig. 7), indicating that the pMHC-specific drug effects on affinity and on-rate is not caused by potential changes in cell surface microtopology and/or stiffness [38, 39].
Figure 6.
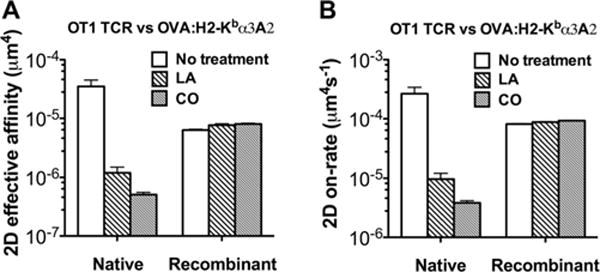
Pharmaceutical treatments greatly altered 2D affinity and on-rates of pMHC binding to the native but not the recombinant TCR. Treatment of either 1 μM LA or 1 U/mL CO suppresses effective 2D affinity (A) and on-rates (B) of OVA:H2-Kbα3A2 interactions with native (left group) but not recombinant (right group) TCRs. The TCR and pMHC site densities are listed in Supporting Information Table 1B. Two-dimensional affinity was measured with adhesion frequency assay at contact time of 2 s; 2D on-rate was calculated from 2D affinity and off-rate. Error bars represent uncertainty calculated by error propagation. (A and B) Data shown are pooled from one to five independent experiments.
Figure 7.
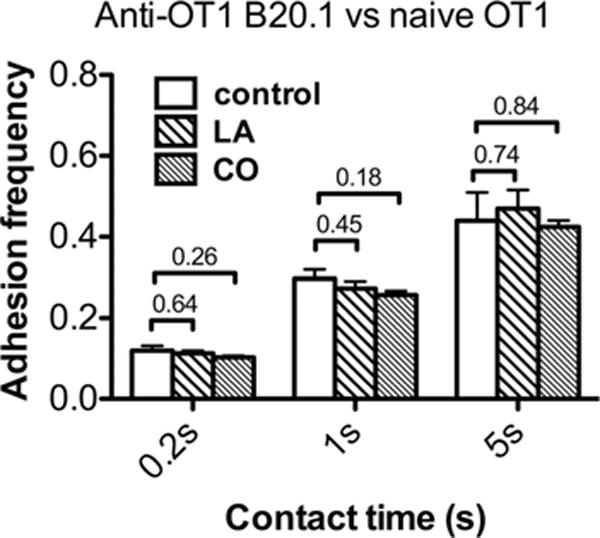
The cellular environment did not regulate 2D kinetics of anti-TCR (B20.1) binding to TCR. At each condition for each contact time, adhesion frequency was calculated from two to five cell pairs. The TCR and pMHC site densities are listed in Supporting Information Table 1C. Data are shown as mean + SEM of n = 2–5 samples.
Discussion
Two sets of recent studies on 2D TCR–pMHC interactions showed variable results in comparison to prior 3D SPR measurements [4–6, 11–13, 26, 27]. These 2D results were obtained for both native and recombinant TCRs using two distinct methodologies. One set of studies used mechanical-based assays. We used micropipette adhesion frequency assay [21, 40] and thermal fluctuation assay [22, 40] to measure force-free 2D affinity and off-rate of native TCRs on naive OT1 T cells [4] or on T-cell lines expressing 42F3 TCR [13] or melanoma antigen gp100-specific TCRs [6]. Robert et al. used a flow chamber [41] to measure force-dependent off-rates of recombinant 1G4 TCR [27]. The other set of studies used fluorescence-based methods. Huppa et al. used single-molecule FRET imaging [5] and Axmann et al. used diffusion analysis [11] to monitor dissociation of lipid bilayer anchored pMHC from in vitro activated 2B4 and 5c.c7 T blasts. O’Donoghue et al. analyzed single-molecule diffusion of pMHC reconstituted on supported lipid bilayer of the same TCR systems [12]. However, several caveats make it difficult to directly compare the discrepant results. The first caveat is the potential effect of force. We measured zero-force 2D off-rates [4, 6, 13], whereas measurements in the FRET-based method might involve mechanical force generated by the cell. Indeed, the faster 2D off-rates of the two MHC class II-restricted TCRs than their 3D counterparts were explained by possible active cellular force on TCR–pMHC bonds that might accelerate dissociation [2, 5]. However, this could not explain the much faster 2D off-rate for the OT1 TCR than its 3D counterpart [4] because (i) the 2D off-rates were measured at zero force [21, 40], (ii) force decelerates rather than accelerate OT1 TCR–OVA:H2-Kb dissociation [26], and (iii) even the slowest 2D off-rate at optimal force (10 pN) is still orders of magnitude faster [26] than the reported 3D off-rate [23].
Another caveat is the reliability of some of the previous data. One telling aspect of the previous SPR measurements for the OT1 TCR–OVA:H2-Kb kinetics is that the half-life at 37°C was much longer (t½ ~ 500 s) than that at room temperature (t½ ~ 30 s) [23]. Although the authors explained this observation using a TCR dimerization model at the higher temperature, it is also possible that it was caused by protein aggregation. In our 2D analysis, dimeric interaction was not observed between tetravalent TCR and tetravalent pMHC. Also, the present SPR measurement yielded an estimated half-life for OT1 TCR–OVA:H2-Kb dissociation at room temperature ~40-folds faster (t½ = 0.75 s) than the previous result. Since this time scale is beyond the temporal resolution of the BiaCore 2000 instrument, the actual dissociation could be even faster, potentially bringing it even closer to 2D lifetime for the native TCR [4]. This is consistent with a recent study showing that dissociation of OVA:H2-Kb from recombinant OT1 TCR was too fast to be reliably measured by SPR [42]. Similarly, fast off-rates of 2C TCR and 42F3 TCR have been observed in other SPR studies [13, 43, 44]. Furthermore, the fast off-rate of the TCR–pMHC bond has been supported by real-time flow cytometry data of dissociation of the fluorescent monomeric OVA:H2-Kbα3A2 from live naïve OT1 T cells. Moreover, the 2D lifetimes at zero force were comparable between the recombinant and the native TCRs. Therefore, our data collectively demonstrated fast TCR–pMHC dissociation under zero force condition; however, we could not ascertain that the dissociation rate was numerically equal between the recombinant and the native TCR.
There may also be differences between native and recombinant TCRs. The a priori assumption that the in situ ligand-binding properties of native TCR can be faithfully recapitulated by recombinant TCR is based on the belief that such properties are solely determined by the ligand-binding domain. This assumption is implied in most published work but has not been tested experimentally. The 2D kinetics data in this paper challenges its validity. Disrupting either the cytoskeleton or lipid rafts by pharmaceutical treatments severely suppressed 2D on-rate and affinity but not off-rate, highlighting the regulation by the T cell. The impact of cellular environment on 2D on-rate could be due to cell surface mechanical properties and topology [38, 45], orientation and flexibility of TCRs [46], confinement of TCRs within microclusters or protein islands [47–49], or possible TCR association with actin cytoskeleton or lipid rafts [50–52]. However, such TCR-extrinsic factors could not have been the primary reason underlying the on-rate/affinity regulation, otherwise binding of TCR to its antibody should also be similarly affected, but this was not observed. The cellular regulation of TCR–pMHC on-rate is also highlighted by data from the 42F3 TCR system. For the same peptide p3A1, on-rate and affinity were the highest for the recombinant TCR but the lowest for the native TCR. This would not be possible if differences between native and recombinant TCRs were merely due to TCR-extrinsic factors, which should not cause change of ranking of affinity and on-rate. Together, these data suggest that there could be fundamental difference between native and recombinant TCRs at the single molecule level that results in distinct pMHC-binding kinetics. This is despite that the two forms of TCRs have identical ligand-binding domains, which exhibit the same binding kinetics for an anti-TCR mAb.
Our data on force-regulated TCR–pMHC bond lifetime further support the above notion. Force elicited a much more pronounced catch bond to result in a much longer pMHC bond lifetime with the native TCR than the recombinant TCR. The longer lifetime could be due to possible TCR clustering on the cell surface. As a first step toward examining this effect, we used monovalent and multivalent streptavidin to capture recombinant TCR but found indistinguishable bond lifetimes. These data do not exclude the possibility that TCR–pMHC binding kinetics may be regulated by TCR clustering not recapitulated by streptavidin-mediated multivalency. However, for this to be the case, such effect should not be disrupted by LA or CO because neither treatment changes lifetime of recombinant or native TCR bond with pMHC. Furthermore, the hypothetical TCR clustering effect is inconsistent with our anti-TCR experiment that yielded the same bond lifetime under zero or tensile force conditions irrespective of whether the TCR is recombinant or native. Moreover, similar lifetimes were observed for recombinant TCR bonds with either recombinant or native pMHC. Therefore, our single-bond lifetime measurements are more compatible to an intrinsic difference between the recombinant and the native TCR.
As a first step toward understanding our results, we speculate that the TCR functions as a mechanosensor the pMHC recognition of which requires the intact TCR residing in its native membrane environment [53]. Compromising this mechanosensory machinery may adversely affect the TCR’s ability to interpret the information embedded in the pMHC, manifested as changing the in situ TCR–pMHC kinetics but not TCR–antibody kinetics. Compared with the recombinant TCR, the native TCR is glycosylated and interacts with chains of CD3 dimers (γδ, γε, and ζζ). Increasing glycosylation of TCR and coreceptor CD8 has been shown to dramatically decrease binding affinity/avidity and accelerate dissociation from pMHC [54–56]. Therefore, TCR glycosylation does not seem to be a plausible mechanism for the longer lifetime. Combined with the single-bond nature of bond lifetime measurements [4, 6, 22, 26, 57], our data collectively suggest that interaction with components of the CD3 complexed with the αβTCR, which was absent in the experiments with recombinant TCRs, might play an important role in regulating TCR–pMHC binding kinetics.
As function is dictated by structure, our TCR–pMHC kinetics data suggest that the known X-ray structures of the recombinant TCR, particularly at the pMHC-binding interface, may not faithfully represent the native, dynamic TCR, especially under tensile forces. We postulate that the αβTCR-associated CD3 complex may impose specific constraints on the TCR structure via extracellular interactions [58] to regulate the TCR–pMHC binding. Under tensile forces, such regulation may strengthen the TCR–pMHC bond to facilitate channeling of the force to the CD3 tails to initiate sig-naling. Such hypothesis can be tested by measuring in situ binding kinetics of pMHC with cell surface TCR mutated to perturb interactions between αβTCR and CD3.
Materials and methods
Cell and proteins
Human red blood cells (RBCs) were isolated from whole blood freshly drawn from healthy volunteers according to a protocol approved by the IRB of Georgia Institute of Technology to serve as a force transducer [26]. Naïve CD8+ T cells were purified from spleens of OT1 or 2C transgenic mice kindly provided by Brian Evavold according to a protocol approved by the IACUC of Emory University. 42F3 T hybridoma cells were kind gifts from David Kranz of University of Illinois at Urbana-Champaign. The TAP-deficient RAM-S T cell lymphoma cell line [59, 60] was provided by Brian Evavold for preparing OVA peptides presented by bona fide APCs. Before loading OVA peptide, RAM-S cells were cultured for 24 h at room temperature (~25°C) to promote expression of empty H2-Kb molecules. 200,000 cells at 1 M/mL in culture medium were then incubated in the presence of 10 μM OVA peptide at 25°C for 2 h, followed by 1-h incubation at 37°C to facilitate internalization of empty H2-Kb. C-terminally biotin-tagged recombinant 2C and 42F3 TCRs as well as H2-Ld presenting covalently linked peptides QL9 (QLSPFPFDL), p3A1 (SPLDSLWWI), p4B10 (QLSDVPMDL), or p5E8 (FLSPFWFDI) [13] were generous gifts from Christopher Garcia of Stanford University. Single biotin-tagged OT1 TCR was produced by bacterial expression and in vitro refolding. The OT1 TCR consisted of the extracellular portions of the α and β chains, terminating immediately preceding the membrane proximal cysteines. Two modifications of the TCR were introduced to improve yields and enhance stability: (i) an interchain disulfide bond was introduced between the Cα and Cβ domains by replacing Cα Thr48 and Cβ Ser55 with cysteines and (ii) the unpaired Cys71 in the Cβ domain was mutated to alanine [61]. TCRα and β were expressed separately as inclusion bodies, solubilized in 6 M guanidine-HCl/0.1 mM DTT, mixed together and added to refolding buffer consisting of 0.4 M arginine-HCl, 3 mM reduced glutathione, 0.3 mM oxidized glutathione, 0.1 M TRIS pH8, 2 mM EDTA at 4°C. After 4 days, the refolding mixture was dialyzed against 25 mM TRIS pH8 and 150 mM NaCl, concentrated and properly folded and disulfide-linked αβ heterodimer was purified by size exclusion chromatography on Superdex 75 (GE Healthcare) and ion exchange chromatography on mono Q (GE Healthcare). BirA-tagged OT1 TCR was prepared by adding the BirA recognition sequence LHHILDAQKMVWNHR at the C-terminus of the TCR β chain. Biotinylation was carried out following manufacturer’s instructions (Avidity LLC, Aurora, CO, USA). Monomeric OVA (SIINFEKL) and G4 (SIIGFEKL) bound to H2-Kb was produced by the NIH Tetramer Core Facility at Emory University [4] as previously described for H2-Dd [62] but with a single biotin at the C-terminus. To prevent CD8 binding, a mutant form of H2-Kb (termed H2-Kbα3A2, with the α3 domain switched to HLA-A2 α3 domain) was used. The H2-Ld used was a minimal α1α2 variant termed m31, which also abolish CD8 binding. L-selectin construct containing the lectin domain and epidermal growth factor domain fused to a C-terminal HPC4-tag and 2GSP6, a synthetic ligand for both P- and L-selectin with a C-terminal biotin tag [63] was kind gifts from Rodger P. McEver of Oklahoma Medical Research Foundation.
Protein coating on RBCs and beads
Detailed procedures have been described previously [26]. In brief, freshly isolated human RBCs were biotinylated, conjugated with streptavidin, and incubated with biotinylated pMHC, TCR, or anti-TCR mAb (clone B20.1, eBioscience, San Diego, CA) for adhesion frequency assay with a micropipette. For BFP experiments, biotinylated RBCs were loaded with nystatin (Sigma-Aldrich, Saint Louis, MO, USA) in N2 buffer. To link biotinylated pMHC, TCR, anti-TCR, or 2GSP6 onto glass beads, bead surfaces were silanized first and then covalently linked to streptavidin–maleimide (Sigma-Aldrich). Subsaturating biotinylated pMHC, TCR, anti-TCR, or 2GSP6 was then conjugated onto streptavidinated beads. Wild-type (WT) tetravalent streptavidin was used unless specified otherwise. HPC4-tagged L-selectin was captured by divalent HPC4 mAb (clone HPC4, Roche Life Science, Indianapolis, IN, USA) covalently coupled on beads.
Measurement of molecular densities on the surfaces of T cells and beads
Detailed procedures have been described previously [26]. Briefly, proteins were stained with PE-conjugated antibodies. The antibodies used for OT1 TCR, OVA:MHC, anti-mouse TCR Vα2 (clone B20.1, eBioscience), and 42F3 TCR were anti-mouse TCR Vα2, anti-mouse OVA:H2-Kb (clone 25-D1.16, eBioscience), anti-rat IgG2a (clone: r2a-21B2), and anti-mouse TCR Vβ8 (clone F23.1, BD Bioscience), respectively. Due to lack of appropriate antibodies, the pMHC densities for H2-Ld presenting QL9, p3A1, p4B10, p5E8 were estimated by measuring OVA:H2-Kb densities on the same batch of streptavdinylated RBCs incubated with the same concentrations of the different pMHCs. Fluorescent intensities of PE-stained T cells or beads were measured by a BD LSR flow cytometer (BD Biosciences), calibrated using standard beads (BD Quantibrite PE Beads, BD), and converted to site densities using known radii of T cells or beads.
Thermal fluctuation assay and force-clamp assay
Thermal fluctuation and force-clamped assays were used to measure 2D bond lifetimes of receptor–ligand dissociation at zero force and under constant tensile forces, respectively [22, 26]. Briefly, the thermal fluctuation assay identifies bond dissociation and association at zero-force from the respective reduction and resumption of the BFP bead thermal fluctuation. Bond lifetime is the period between consecutive association and dissociation events. The force-clamp assay loads a bond to a preset force and holds the force constant until bond rupture to measure a lifetime under that force. Measurements were pooled and binned into several force levels. Bond lifetimes at each force level (including zero force) are visualized as ln(number of events with a lifetime > t) versus lifetime t and presented as arithmetic mean ± SEM.
Effective 2D affinity and on-rate measurements by micropipette adhesion assay
The adhesion frequency assay has been described [21]. Briefly, the effective 2D affinity (AcKa) is calculated by
| (1) |
where mr and ml are the respective surface densities of TCR and pMHC and Pa is adhesion frequency determined from the ratio of the number of adhesions to the total number of repeated 2 s contacts. The effective 2D on-rate is calculated from Ackon = AcKa × koff, where koff is zero-force off-rate measured by thermal fluctuation assay.
Real-time flow cytometry assay
This assay has been described [31]. Briefly, PE-labeled ligand (at a monomer concentration of 10 μg/mL) was incubated with T-cells to allow association with TCR, followed by rapid centrifugation and resuspension of the cells in ligand-free medium supplemented with blocking antibody (clone B20.1). Immediately after resuspension, the sample was subject to flow cytometry analysis. Multiple 2000-event histograms were successively collected to monitor ligand dissociation. As acquiring the first 2000-event histogram took ~10 s, the temporal resolution of the method is 10 s. Between successive collections of individual histograms, the sample was briefly vortexed to ensure sufficient mixing. The average intensity at each time point was derived from each 2000-event histogram and plotted against time.
Pharmacological agent treatments
To examine the impact of pharmacological agents on TCR–pMHC kinetics, T cells were incubated either with LA (Calbiochem) at 1 μM for 30 min at 25 °C or CO (Sigma-Aldrich) at 1 U/mL for 1 h at 37°C before adding to the experiment chamber. Experiments were performed in the continuous presence of the same concentration of the pharmacological agent at room temperature.
3D kinetics measurement by SPR
Binding was performed on a BiaCore 2000. OT1 TCR was immobilized at a density of 4500 resonance units on the dextran surface of a CM-5 biosensor chip using standard NHS/EDC coupling chemistry. All experiments were carried out at 25°C in 10 mM HEPES pH7.2, 150 mM NaCl, 3 mM EDTA, 0.005% Tween-20. Graded concentrations of OVA:H2-Kb ranging from 1.25 to 20 μM were injected over the TCR surface at a flow rate of 10 μL/min at 25°C for 2 min followed by dissocation phase with buffer alone. Data points were acquired at 1 s intervals. Binding to a mock coupled surface was subtracted to establish baselines. The dissociation phase was analyzed with a single exponential decay.
Statistical test
Standard Student’s t-test was performed to test the statistical difference of average bond lifetime and adhesion frequency between paired conditions. p-Values below 0.05 were deemed statistically significant.
Supplementary Material
Acknowledgments
We thank the NIH Tetramer Core Facility at Emory University for providing the H2-Kb monomers, Brian D. Evavold for providing the OT1 and 2C transgenic mice and the RAM-S cell line, K. Christopher Garcia for providing the H2-Ld monomers and the recombinant 2C and 42F3 TCRs, David Kranz for providing the 42F3 hybridoma cells, John Altman for providing the H2-Kbα3A2 construct, Rodger P. McEver for providing L-selectin and 2GSP6, and Larissa Doudy for purifying T cells. This work was supported by NIH grants AI38282 and GM096187 (to C.Z.). K.N. and D.H.M are supported by the Intramural Research Program of the NIAID, NIH.
Abbreviations
- BFP
biomembrane force probe
- LA
latrunculin A
- mAb
monoclonal antibody
- pMHCs
peptide-major histocompatibility complex molecules
- pN
pico-Newton
- SPR
surface plasmon resonance
Footnotes
Additional supporting information may be found in the online version of this article at the publisher’s web-site
Conflict of interest: The authors declare no financial or commercial conflict of interest
References
- 1.Gascoigne NR, Zal T, Alam SM. T-cell receptor binding kinetics in T-cell development and activation. Expert Rev Mol Med. 2001;2001:1–17. doi: 10.1017/S1462399401002502. [DOI] [PubMed] [Google Scholar]
- 2.vander Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11:47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- 3.Aleksic M, Dushek O, Zhang H, Shenderov E, Chen JL, Cerundolo V, Coombs D, et al. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity. 2010;32:163–174. doi: 10.1016/j.immuni.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu BY, Zhong S, Malecek K, Johnson LA, Rosenberg SA, Zhu C, Krogsgaard M. 2D TCR-pMHC-CD8 kinetics determines T-cell responses in a self-antigen-specific TCR system. Eur J Immunol. 2014;44:239–250. doi: 10.1002/eji.201343774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong S, Malecek K, Johnson LA, Yu ZY, deMiera EVS, Darvishian F, McGary K, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunother-apy. Proc Natl Acad Sci U S A. 2013;110:6973–6978. doi: 10.1073/pnas.1221609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, et al. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- 9.Dushek O, Das R, Coombs D. A role for rebinding in rapid and reliable T cell responses to antigen. PloS Comput Biol. 2009;5:e1000578. doi: 10.1371/journal.pcbi.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci U S A. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Axmann M, Huppa JB, Davis MM, Schutz GJ. Determination of interaction kinetics between the T cell receptor and peptide-loaded MHC Class II via single-molecule diffusion measurements. Biophys J. 2012;103:L17–L19. doi: 10.1016/j.bpj.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Donoghue GP, Pielak RM, Smoligovets AA, Lin JJ, Groves JT. Direct single molecule measurement of TCR triggering by agonist pMHC in living primary T cells. Elife. 2013;2:e00778. doi: 10.7554/eLife.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bower-man NA, Chen W, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity. 2011;35:681–693. doi: 10.1016/j.immuni.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabatino J, Huang J, Zhu C, Evavold B. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med. 2011;208:81–90. doi: 10.1084/jem.20101574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang N, Huang J, Edwards LJ, Liu B, Zhang Y, Beal CD, Evavold BD, et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity. 2011;34:13–23. doi: 10.1016/j.immuni.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dustin ML, Bromley SK, Davis MM, Zhu C. Identification of self through two-dimensional chemistry and synapses. Annu Rev Cell Dev Biol. 2001;17:133–157. doi: 10.1146/annurev.cellbio.17.1.133. [DOI] [PubMed] [Google Scholar]
- 17.Wu YH, Vendome J, Shapiro L, Ben-Shaul A, Honig B. Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature. 2011;475:510–513. doi: 10.1038/nature10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell GI. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 19.Zarnitsyna V, Zhu C. T cell triggering: insights from 2D kinetics analysis of molecular interactions. Phys Biol. 2012;9:045005. doi: 10.1088/1478-3975/9/4/045005. [DOI] [PubMed] [Google Scholar]
- 20.Zhu C, Jiang N, Huang J, Zarnitsyna VI, Evavold BD. Insights from in situ analysis of TCR–pMHC recognition: response of an interaction network. Immunol Rev. 2013;251:49–64. doi: 10.1111/imr.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chesla SE, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Evans EA, McEver RP, Zhu C. Monitoring receptor-ligand interactions between surfaces by thermal fluctuations. Biophys J. 2008;94:694–701. doi: 10.1529/biophysj.107.117895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alam SM, Davies GM, Lin CM, Zal T, Nasholds W, Jameson SC, Hogquist KA, et al. Qualitative and quantitative differences in T cell receptor binding of agonist and antagonist ligands. Immunity. 1999;10:227–237. doi: 10.1016/s1074-7613(00)80023-0. [DOI] [PubMed] [Google Scholar]
- 24.Allard JF, Dushek O, Coombs D, vander Merwe PA. Mechanical modulation of receptor-ligand interactions at cell-cell interfaces. Biophys J. 2012;102:1265–1273. doi: 10.1016/j.bpj.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klotzsch E, Schutz GJ. Improved ligand discrimination by force-induced unbinding of the T cell receptor from peptide-MHC. Biophys J. 2013;104:1670–1675. doi: 10.1016/j.bpj.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Chen W, Evavold BD, Zhu C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell. 2014;157:357–368. doi: 10.1016/j.cell.2014.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robert P, Aleksic M, Dushek O, Cerundolo V, Bongrand P, vander Merwe PA. Kinetics and mechanics of two-dimensional interactions between T cell receptors and different activating ligands. Biophys J. 2012;102:248–257. doi: 10.1016/j.bpj.2011.11.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das DK, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, Brady SK, et al. Force-dependent transition in the T-cell receptor beta-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc Natl Acad Sci U S A. 2015;112:1517–1522. doi: 10.1073/pnas.1424829112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NRJ. T-cell-receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 30.Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, Gascoigne NRJ, et al. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity. 2001;15:59–70. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 31.Li P, Jiang N, Nagarajan S, Wohlhueter R, Selvaraj P, Zhu C. Affinity and kinetic analysis of Fcγ receptor IIIa (CD16a) binding to IgG ligands. J Biol Chem. 2007;282:6210–6221. doi: 10.1074/jbc.M609064200. [DOI] [PubMed] [Google Scholar]
- 32.Molnar E, Deswal S, Schamel WW. Pre-clustered TCR complexes. FEBS Lett. 2010;584:4832–4837. doi: 10.1016/j.febslet.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Bosch B, Heipertz EL, Drake JR, Roche PA. Major histocompatibility complex (MHC) class II-peptide complexes arrive at the plasma membrane in cholesterol-rich microclusters. J Biol Chem. 2013;288:13236–13242. doi: 10.1074/jbc.M112.442640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fooksman DR, Gronvall GK, Tang Q, Edidin M. Clustering class I MHC modulates sensitivity of T cell recognition. J Immunol. 2006;176:6673–6680. doi: 10.4049/jimmunol.176.11.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howarth M, Chinnapen DJ, Gerrow K, Dorrestein PC, Grandy MR, Kelleher NL, El-Husseini A, et al. A monovalent streptavidin with a single femtomolar biotin binding site. Nat Methods. 2006;3:267–273. doi: 10.1038/NMETHXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 37.Ramachandran V, Yago T, Epperson TK, Kobzdej MM, Nollert MU, Cummings RD, Zhu C, et al. Dimerization of a selectin and its ligand stabilizes cell rolling and enhances tether strength in shear flow. Proc Natl Acad Sci U S A. 2001;98:10166–10171. doi: 10.1073/pnas.171248098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams TE, Nagarajan S, Selvaraj P, Zhu C. Quantifying the impact of membrane microtopology on effective two-dimensional affinity. J Biol Chem. 2001;276:13283–13288. doi: 10.1074/jbc.M010427200. [DOI] [PubMed] [Google Scholar]
- 39.Wu L, Xiao BT, Jia XL, Zhang Y, Lu SQ, Chen J, Long M. Impact of carrier stiffness and microtopology on two-dimensional kinetics of P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) interactions. J Biol Chem. 2007;282:9846–9854. doi: 10.1074/jbc.M609219200. [DOI] [PubMed] [Google Scholar]
- 40.Chen W, Zarnitsyna VI, Sarangapani KK, Huang J, Zhu C. Measuring receptor-ligand binding kinetics on cell surfaces: from adhesion frequency to thermal fluctuation methods. Cell Mol Bioeng. 2008;1:276–288. doi: 10.1007/s12195-008-0024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pierres A, Touchard D, Benoliel AM, Bongrand P. Dissecting streptavidin-biotin interaction with a laminar flow chamber. Biophys J. 2002;82:3214–3223. doi: 10.1016/S0006-3495(02)75664-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stepanek O, Prabhakar AS, Osswald C, King CG, Bulek A, Naeher D, Beaufils-Hugot M, et al. Coreceptor scanning by the T cell receptor provides a mechanism for T cell tolerance. Cell. 2014;159:333–345. doi: 10.1016/j.cell.2014.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chervin AS, Stone JD, Holler PD, Bai A, Chen J, Eisen HN, Kranz DM. The impact of TCR-binding properties and antigen presentation format on T cell responsiveness. J Immunol. 2009;183:1166–1178. doi: 10.4049/jimmunol.0900054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Degano M, Garcia KC, Apostolopoulos V, Rudolph MG, Teyton L, Wilson IA. A functional hot spot for antigen recognition in a superagonist TCR/MHC complex. Immunity. 2000;12:251–261. doi: 10.1016/s1074-7613(00)80178-8. [DOI] [PubMed] [Google Scholar]
- 45.Wu L, Xiao B, Jia X, Zhang Y, Lü S, Chen J, Long M. Impact of carrier stiffness and microtopology on two-dimensional kinetics of P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) interactions. J Biol Chem. 2007;282:9846–9854. doi: 10.1074/jbc.M609219200. [DOI] [PubMed] [Google Scholar]
- 46.Huang J, Chen J, Chesla SE, Yago T, Mehta P, McEver RP, Zhu C, et al. Quantifying the effects of molecular orientation and length on two-dimensional receptor-ligand binding kinetics. J Biol Chem. 2004;279:44915–44923. doi: 10.1074/jbc.M407039200. [DOI] [PubMed] [Google Scholar]
- 47.Kumar R, Ferez M, Swamy M, Arechaga I, Rejas MT, Valpuesta JM, Schamel WWA, et al. Increased sensitivity of antigen-experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity. 2011;35:375–387. doi: 10.1016/j.immuni.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 48.Crites TJ, Padhan K, Muller J, Krogsgaard M, Gudla PR, Lockett SJ, Varma R. TCR microclusters pre-exist and contain molecules necessary for TCR signal transduction. J Immunol. 2014;193:56–67. doi: 10.4049/jimmunol.1400315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lillemeier BF, Mortelmaier MA, Forstner MB, Huppa JB, Groves JT, Davis MM. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rozdzial MM, Malissen B, Finkel TH. Tyrosine-phosphorylated T cell receptor zeta chain associates with the actin cytoskeleton upon activation of mature T lymphocytes. Immunity. 1995;3:623–633. doi: 10.1016/1074-7613(95)90133-7. [DOI] [PubMed] [Google Scholar]
- 51.Rozdzial MM, Pleiman CM, Cambier JC, Finkel TH. pp56Lck mediates TCR zeta-chain binding to the microfilament cytoskeleton. J Immunol. 1998;161:5491–5499. [PubMed] [Google Scholar]
- 52.Kabouridis PS. Lipid rafts in T cell receptor signalling. Mol Membr Biol. 2006;23:49–57. doi: 10.1080/09687860500453673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim ST, Takeuchi K, Sun ZYJ, Touma M, Castro CE, Fahmy A, Lang MJ, et al. The alphabeta T cell receptor is an anisotropic mechanosensor. J Biol Chem. 2009;284:31028–31037. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daniels MA, Levine L, Miller JD, Moser JM, Lukacher AE, Altman JD, Kavathas P, et al. CD8 binding to MHC class I molecules is influenced by maturation and T cell glycosylation. Immunity. 2001;15:1051–1061. doi: 10.1016/s1074-7613(01)00252-7. [DOI] [PubMed] [Google Scholar]
- 55.Kuball J, Hauptrock B, Malina V, Antunes E, Voss RH, Wolfl M, Strong R, et al. Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J Exp Med. 2009;206:463–475. doi: 10.1084/jem.20082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moody AM, Chui D, Reche PA, Priatel JJ, Marth JD, Reinherz EL. Developmentally regulated glycosylation of the CD8alphabeta coreceptor stalk modulates ligand binding. Cell. 2001;107:501–512. doi: 10.1016/s0092-8674(01)00577-3. [DOI] [PubMed] [Google Scholar]
- 57.Chen W, Lou J, Zhu C. Forcing switch from short- to intermediate-and long-lived states of the alphaA domain generates LFA-1/ICAM-1 catch bonds. J Biol Chem. 2010;285:35967–35978. doi: 10.1074/jbc.M110.155770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuhns MS, Davis MM. Disruption of extracellular interactions impairs T cell receptor-CD3 complex stability and signaling. Immunity. 2007;26:357–369. doi: 10.1016/j.immuni.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 59.Ortiz-Navarrete V, Hammerling GJ. Surface appearance and instability of empty H-2 class I molecules under physiological conditions. Proc Natl Acad Sci U S A. 1991;88:3594–3597. doi: 10.1073/pnas.88.9.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hansen T, Myers N. Peptide induction of surface expression of class I MHC. Curr Protoc Immunol. 2003:11. doi: 10.1002/0471142735.im1811s57. Chapter 18: Unit 18. [DOI] [PubMed] [Google Scholar]
- 61.Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, Jakobsen BK. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–711. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 62.Li HM, Natarajan K, Malchiodi EL, Margulies DH, Mariuzza RA. Three-dimensional structure of H-2D(d) complexed with an immunodominant peptide from human immunodeficiency virus envelope glycoprotein 120. J Mol Biol. 1998;283:179–191. doi: 10.1006/jmbi.1998.2091. [DOI] [PubMed] [Google Scholar]
- 63.Klopocki AG, Yago T, Mehta P, Yang J, Wu T, Leppanen A, Bovin NV, et al. Replacing a lectin domain residue in L-selectin enhances binding to P-selectin glycoprotein ligand-1 but not to 6-sulfo-sialyl Lewisx. J Biol Chem. 2008;283:11493–11500. doi: 10.1074/jbc.M709785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.