

Abstract
Enteropathogenic Escherichia coli (EPEC) induces dramatic remodeling of enterocyte brush borders, a process that includes microvillar effacement and actin pedestal formation. Although the Arp2/3 complex is involved in formation of a branched actin network within pedestals, the fate of parallel actin bundles in microvilli during infection remains unclear. here, we find that in polarized intestinal epithelial cells, EPEC stimulates long-range microvillar dynamics, pulling protrusions toward sites of bacterial attachment in a process mediated by the adhesion molecule protocadherin-24. Additionally, retraction of the EPEC bundle forming pilus stimulates directed elongation of nearby microvilli. These processes lead to coalescence of microvilli and incorporation of the underlying parallel actin bundles into pedestals. Furthermore, stabilization of microvillar actin bundles delays pedestal formation. Together, these results suggest a model where ePec takes advantage of pre-existing actin filaments in microvillar core bundles to facilitate pedestal formation.
Keywords: microvilli, brush border, EPEC, intestine, espin, bundle forming pilus, Caco-2BBE
Introduction
The mammalian intestine is home to ∼1014 bacterial cells, and while many of these are required for physiological homeostasis, pathogenic species originating from the environment can give rise to severe perturbations of gastrointestinal function.1-3 A leading cause of diarrheal disease in the developing world is a group of virulent Escherichia coli strains found in contaminated food or water.4 One prominent strain is Enteropathogenic E. coli (EPEC), which targets enterocytes, nutrient absorbing epithelial cells that line the intestinal tract. The apical surface of each enterocyte is home to a dense array of membrane-enveloped parallel actin bundles called microvilli. Collectively known as the brush border, these protrusions function to increase the surface area available for nutrient absorption and act as a platform for release of vesicles containing host defense machinery.5 As an attaching/effacing (A/E) pathogen, EPEC remains on the extracellular surface of enterocytes and forms characteristic microcolonies, where multiple bacterial cells auto-aggregate in a process called localized adherence.6 The bacteria are drawn into close contact with the host cell surface and other bacteria through the activity of the bundle forming pilus (BFP).7,8 Using a type III secretion system (T3SS), EPEC co-opt host cell machinery to produce an actin-rich pedestal that anchors the bacterium while allowing it to move across the cell surface.9 During this phase of intimate attachment, microvilli surrounding the bacteria elongate and orient toward the pathogen.10 Microvillar-like processes (MLPs), which are the result of microvillar elongation, have long been observed as part of the normal A/E process during EPEC infection.7,11-14 Ultimately, microvilli surrounding the microcolony are destroyed during infection, leaving an area around the bacteria devoid of brush border and giving rise to a cellular morphology referred to as the “A/E lesion.”
The molecular events surrounding pedestal formation have been well characterized. Bacterial effector proteins injected via the T3SS initiate an actin signaling cascade, culminating in activation of the Actin-related protein (Arp) 2/3 complex.15,16 In turn, Arp2/3 assembles a branched actin network underneath the microbe that comprises the pedestal. The signaling cascade leading to Arp2/3 activation during EPEC infection has been thoroughly studied,15 but significant gaps in our understanding of Arp2/3 function in this context still remain. For example, Arp2/3 binds to the sides of F-actin filaments and nucleates new daughter filaments at a 70° angle.17 Therefore, formation of a branched actin network, such as a bacterial pedestal, requires pre-existing F-actin “templates.” In a polarized enterocyte, the brush border contains a massive population of pre-existing filaments in the form of microvillar core actin bundles. However, whether these pre-existing filaments contribute to pedestal formation during EPEC attachment remains unclear.
Other fundamental questions also persist. Seemingly peripheral features of the A/E process, such as microvillar elongation, have been noted as hallmarks of EPEC infection but not studied in the context of their potential contributions to pedestal formation. Additionally, despite what is known about the molecular pathways involved in pedestal formation, the early stages of EPEC attachment to and interaction with the host cell surface are not well characterized. Although scanning electron microscopy (SEM) has provided important insight into the ultrastructure of the early A/E process, the field lacks kinetic information on the morphological changes that the host cell surface must undergo during A/E. Live cell imaging would therefore provide valuable insight on the process of pedestal formation during early EPEC attachment.
In order to address these gaps, we utilized live cell time-lapse deconvolution imaging, structured illumination microscopy (SIM), laser scanning confocal, SEM, and transmission electron microscopy (TEM) to visualize EPEC attachment and the associated ultrastructural changes that take place on the surface of polarized human intestinal epithelial cells during A/E. Our results reveal significant dynamics of the apical surface that, to our knowledge, have not been reported previously. We find that the elongation of microvilli surrounding surface-attached EPEC requires retraction of the bundle forming pilus (BFP) and is independent of factors injected by the T3SS. In addition to extensive microvillar elongation, we observe directed flow of distal microvilli toward sites of EPEC attachment. Microvillar flow requires intermicrovillar adhesion, which is driven by tip-localized protocadherin-24 (PCDH24). The temporal coincidence of microvillar flow and pedestal formation suggests that microvillar actin is directly incorporated into nascent pedestals, a result supported by time-lapse imaging and SEM. Additionally, stabilization of F-actin bundles inhibits pedestal formation, further suggesting that microvillar actin provides a source for the actin networks supporting A/E lesions. In combination, our data lead us to propose that microvillar elongation and flow are two mechanisms that EPEC uses to sequester pre-existing microvillar actin filaments during pedestal assembly. These filaments could then serve as templates for Arp2/3-mediated polymerization, or be bundled together as part of a complementary process.
Results
EPEC induces extensive microvillar dynamics across the apical surface
Because the temporal progression of pedestal formation is poorly characterized, we turned to live cell imaging to capture microvillar dynamics induced by EPEC infection. We transiently transfected Caco-2BBE cells with a fluorescent protein-tagged version of the utrophin calponin homology domain (mCherry-UtrCH), which binds to F-actin without disrupting actin dynamics.18 Additionally, we transfected the cells with a fluorescently labeled version of the intermicrovillar adhesion molecule protocadherin-24 (PCDH24-GFP) both to label membrane and to promote brush border maturation.19 EPEC was added to Caco-2BBE cells on glass-bottom dishes and imaged at 10–20 s intervals using deconvolution microscopy. F-actin began to accumulate around bacteria ∼5–10 min after initial attachment, and by ∼20–25 min, fully developed pedestals labeled with highly enriched mCherry-UtrCH signal had formed beneath attached bacteria (Fig. 1A and B; Video 1).
Figure 1.
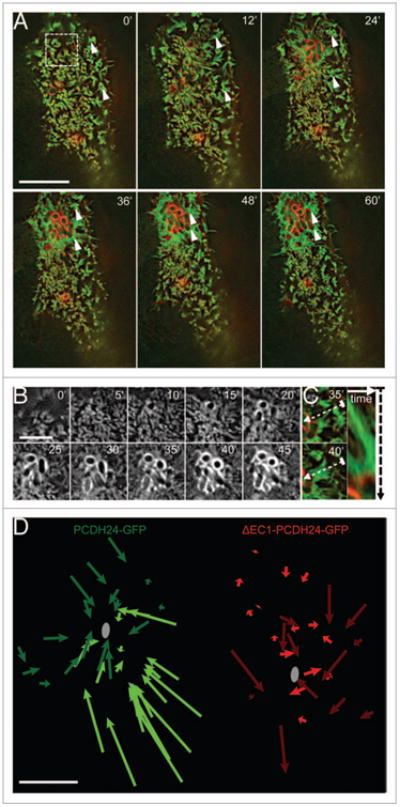
Brush border microvilli flow toward sites of EPEC attachment. (A) Time-lapse images of EPEC infected caco-2BBE cell expressing PCDH24-GFP (green) and mcherry-Utrch (red). Microvilli cluster and flow (arrowheads) toward the site of bacterial attachment; scale bar = 10 μm. (B) Time-lapse images of actin (mcherry-Utrch) accumulation and pedestal formation in cell shown in (A); scale bar = 5 μm. (C) Example of clustered microvilli on cell shown in (A) flowing toward developing pedestal. Right: Kymograph of microvillar cluster along dashed line between frames shown at Left. (D) Plots of microvillar flow in Caco-2BBE cells expressing GFP-tagged full-length PCDH24 (green) or the dominant negative ΔEC1-PCDH24 (red). Shades indicate different cells; gray circles represent site of bacterial attachment closest to each microvillus tracked. Scale bar = 5 μm.
In addition to stereotypical pedestal formation, we observed that the dynamics of microvilli at sites distal to EPEC attachment were significantly affected within 5–10 min of initial contact (Video 1). In cells with well-organized brush borders, clusters of microvilli appeared to flow toward the bacterium during attachment and microcolony formation (Fig. 1A, arrowheads; Fig. 1C; Video 1). Over the course of 1 h, as the A/E lesion grew due to division and attachment of additional bacteria, this directed flow expanded across the entire surface of the cell. Microvillar clusters distal to the attachment site moved toward the microcolony and, subsequently, appeared to be incorporated into the pedestal (Fig. 1A, arrowheads). As a result, the periphery of the lesion was surrounded by an expanding ring of microvillar membrane that enveloped F-actin within the growing pedestal (Fig. 1A). From these time-lapse data, we conclude that EPEC attachment and brush border effacement is not simply a matter of localized microvillar destruction. Rather, it is a dynamic process that engages the entire cell surface, with microvilli flowing to and coalescing at sites of attachment. The net effect of such flow is the enrichment of pre-existing actin filaments (in the form of microvillar core bundles) immediately adjacent to growing pedestals. Long-range microvillar dynamics may, therefore, represent one mechanism that EPEC uses to sequester host cell F-actin.
PCDH24 is required for the long-range microvillar dynamics induced by EPEC
In order for newly attached EPEC to stimulate long-range microvillar flow, they must be able to use either biochemical or mechanical pathways to signal to distal sites on the apical surface that in some cases are several microns away. Recent work from our laboratory revealed that microvillar organization and packing density are driven by intermicrovillar adhesion, which in turn is mediated by heterophilic interactions between PCDH24 and mucin-like protocadherin (MLPCDH).19 Thus, we sought to test the possibility that long-range microvillar flow induced by EPEC attachment requires the physical connections between microvilli that are provided by intermicrovillar adhesion. To this end, we overexpressed a dominant-negative PCDH24-GFP construct (ΔEC1-PCDH24-GFP) in which deletion of the N-terminal extracellular cadherin (EC) repeat prevents formation of the PCDH24-MLPCDH heterophilic complex.19 Blocking inter-microvillar adhesion in this way slows maturation of the brush border and reduces microvillar density.19 In cells expressing ΔEC1-PCDH24-GFP, the observed flow of microvilli following EPEC attachment was significantly reduced (Fig. 1D, red arrows; Video 2) compared with PCDH24-GFP expressing cells (Fig. 1D, green arrows). Although microvilli immediately surrounding sites of attachment on ΔEC1-PCDH24-GFP expressing cells displayed localized dynamics, the rest of the cell surface remained relatively unperturbed (Video 2). PCDH24-GFP and ΔEC1-PCDH24-GFP expressing cells did not exhibit microvillar flow in the absence of EPEC or when exposed to inert silica beads (Fig. S1; Videos 3–6). Thus, EPEC takes advantage of host cell intermicrovillar adhesion to affect the flow of microvilli over the apical surface toward sites of developing microcolonies.
Microvilli exhibit directed elongation during EPEC attachment
Consistent with previous work describing MLPs,7,11-14 we observed numerous examples of elongated protrusions on the surface of EPEC-infected Caco-2BBE cells at both early and late time points post-confluency (Fig. 2A and B). SEM of fixed samples revealed clear examples of elongated protrusions that were often >3-fold longer than normal microvilli and arranged in a radial pattern around the attached bacterium (Fig. 2A and B). These are most likely elongated versions of pre-existing microvilli, as SIM imaging of phalloidin-stained Caco-2BBE cells showed that these protrusions are supported by F-actin (Fig. 2C).
Figure 2.
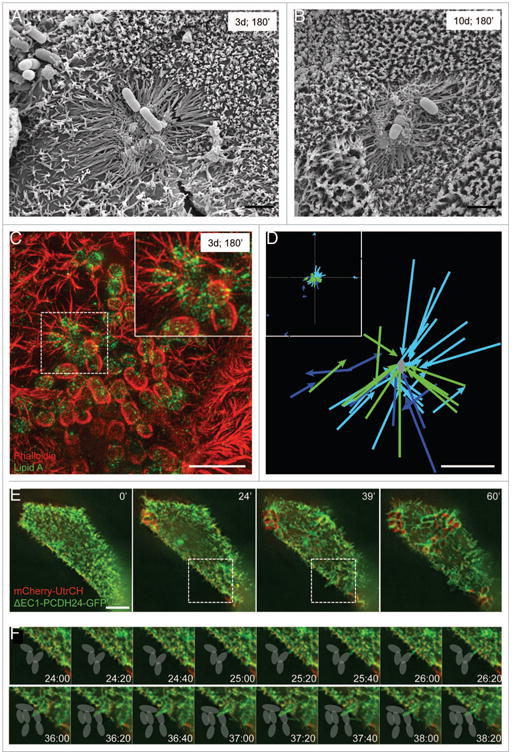
For figure legend see page 508. EPEC stimulates directed microvillar elongation. (A, B) SEM images of microvillar elongation surrounding developing microcolonies 3 d (A) and 10 d (B) post-confluency, following a 3 h incubation with EPEC. (C) SIM image of microvilli elongating toward an attaching EPEC prior to pedestal formation. Dashed square outlines the area magnified in the inset. (D) Plot of elongating microvilli relative to nearest EPEC. Inset: lower magnification of entire field shows random examples of microvilli elongating independent of bacterial stimulation. Each color represents an individual cell. Microvillar tips were tracked until the protrusion reached its max length. (E) Frames from time-lapse deconvolution of EPEC-infected cell expressing ΔEC1-PCDH24-GFP (green) and mcherry-Utrch (red). (F) Individual frames of region indicated by box in (E, middle images), showing microvillar elongation toward bacteria attached to a neighboring cell. Gray ovals indicate location of bacteria. Scale bars = 2 μm (A, B, D), 5 μm (C, E).
To more readily visualize the dynamics and elongation of individual microvilli during EPEC attachment in live cells, we took advantage of the fact that ΔEC1-PCDH24-GFP expression reduces microvillar packing density. Live cell imaging of attachment events revealed that microvillar elongation is an active and directed phenomenon, with the distal tips of microvilli extending toward bacteria (Fig. 2D-F). On infected cells, microvilli within ∼5 μm of a newly attached bacterium oriented their distal tips toward the pathogen. Rapid elongation immediately followed until the tip of the microvillus contacted the bacterium or surrounding membrane. In some instances microvilli retracted, returning to their original length almost instantaneously (Videos 2 and 7). Over time, additional protrusions made contact with a single bacterium, leading to the formation of a web of elongated microvilli around the microbe (Fig. 2E, 60′; Video 7). Directed elongation did not occur on cells incubated with 1.6 μm silica or polystyrene beads, demonstrating that this phenomenon is not a non-specific response to general mechanical perturbation (Videos 5 and 6). Our live cell studies also captured striking examples of microvilli elongating toward bacteria that were attached to neighboring cells (Fig. 2F; Video 7). This is noteworthy as it suggests that EPEC coordinates microvillar elongation, either by releasing a soluble cue that stimulates microvilli to grow toward the bacterium or by other mechanical pathways. Thus, in addition to long-range, directed flow of microvilli, directed elongation of microvilli may be a second mechanism EPEC uses to sequester pre-existing actin filaments around the site of pedestal formation.
Microvillar elongation is independent of T3SS-mediated injection of virulence factors
Microvillar elongation could be stimulated by injection of EPEC virulence factors through the T3SS, or through a mechanism that operates outside the host cell. To differentiate between these two possibilities, we performed live cell imaging of Caco-2BBE cells transfected with mCherry-UtrCH and ΔEC1-PCDH24-GFP, and incubated with a strain of EPEC lacking the T3SS ATPase, EscN (EPEC-ΔEscN).20 As expected, these bacteria could not form pedestals due to their inability to translocate Tir into host cells (Fig. 3A, B).21 We noted signal from ΔEC1-PCDH24-GFP, but not mCherry-UtrCH, in the characteristic oval shape surrounding bacterial cells, indicating accumulation of membrane at attachment sites without the actin enrichment typically observed during pedestal formation (Fig. 3A, B). Additionally, EPEC-ΔEscN induced microvillar elongation immediately following initial contact with the host cell (Video 8). We next investigated the detailed morphology of these brush borders using super-resolution microscopy. In un-transfected, differentiated Caco-2BBE cells fixed after 3 h incubation with EPEC-ΔEscN, we observed the formation of a dense network of elongated microvilli surrounding attached microbes (Fig. 3C). Arrays of elongated microvilli produced by EPEC-ΔEscN were much more elaborate than those produced by WT EPEC; microvilli were not only longer, but they also contained significantly higher levels of F-actin, as indicated by increased phalloidin staining relative to neighboring microvilli (Fig. 3C and D). Treating cells with cytochalasin B during infection with EPEC-ΔEscN diminished this response and resulted in shorter microvilli in contact with attached bacteria (Fig. S2A and B) relative to DMSO-treated cells (Fig. S2C), suggesting that elongation was a function of plus (barbed)-end incorporation of new G-actin subunits. These results are intriguing because they suggest that EPEC is able to manipulate actin dynamics and, specifically, polymerization at the plus ends of microvillar actin filaments, without direct access to the host cell cytosol through the T3SS.
Figure 3.
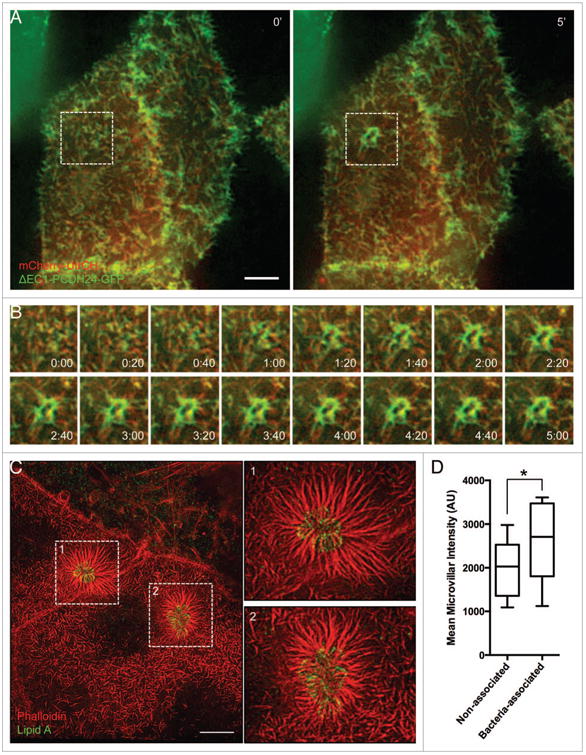
Loss of T3ss function eliminates pedestal formation, but not microvillar elongation. (A) Time-lapse images of ΔEscN-EPEC infected Caco-2BBE cells expressing ΔEC1-PCDH24-GFP (green) and mcherry-Utrch (red). (B) Individual frames of region outlined in (A). Microvillar elongation at sites of attachment occurs on the same time scale as cells infected with WT EPEC. Membrane accumulates around the bacteria, but actin pedestals do not form. (C) SIM image of dense network of microvilli accumulating and elongating around ΔEscN-EPEC. (D) Increased average actin intensity of microvilli that are infected with ΔEscN-EPEC compared with microvilli on the same cells that are not interacting with bacteria. Mean intensity of each microvillus was measured by line scan, and data represent the average of 6 cells, with 35–105 microvilli measured per cell. *P < 0.05. Scale bars = 5 μm.
Microvillar elongation is mediated by BFP retraction
We next sought to examine the mechanism of microvillar elongation. One candidate for stimulating elongation is the BFP, a critical component of the A/E process that is entirely separate from T3SS function. Mutant strains lacking the BFP can “burrow” between microvilli but inefficiently colonize the host cell surface, although they ultimately can form pedestals.13 Retraction of the BFP, which is mediated by the BfpF subunit, pulls the bacterium into close contact with the host cell and is important for efficient translocation of bacterial effector proteins by way of the T3SS.8 Remarkably, the Neisseria gonorrhoeae type IV pilus is capable of exerting transient retraction forces of 50–100 pN, while bundles of type IV pili produce persistent retraction forces of >200 pN.22,23 In A431 epidermoid carcinoma cells, retraction of the N. gonnorrhoeae type IV pilus induces protrusion formation and elongation.24 To determine if BFP retraction plays a role in the elongation of microvilli observed on the surface of differentiated Caco-2BBE cells, we utilized EPEC-bfpF+BfpFara, a strain in which an arabinose-inducible copy of BfpF compensates for a mutated variant of BfpF that cannot retract pili.8 We scored attachment events and binned them into the following categories: (1) microvilli that did not respond to presence of bacteria (Fig. 4A, no response), (2) microvilli that were oriented toward bacteria without significant elongation (Fig. 4B, directionality), and (3) robust microvillar elongation toward bacteria (Fig. 4C, elongation). In cells incubated with WT EPEC, directionality was observed around 22% of bacteria, with elongation occurring in 25% of attachment events. In cells infected with EPEC-bfpF+BfpFara in the absence of arabinose, however, directionality was reduced by half, and elongation events were almost completely absent (Fig. 4D). Incubation of bfpF+BfpFara EPEC with arabinose (i.e., retraction-permissive conditions) increased both directionality and elongation to levels comparable with WT EPEC (Fig. 4D). In cells exposed to EPEC-bfpF+BfpFara in the absence of arabinose, we also observed numerous examples of BFP, ranging from ∼100 nm to ∼1 μm long, interacting with the tips of microvilli that displayed little or no reorientation toward the bacterium (Fig. 4E and F). In conjunction with previously published work,23,24 our results suggest that directed microvillar elongation observed following initial EPEC attachment is most likely driven by BFP retraction, a process that has the potential to exert significant forces on surrounding microvilli. This mechanism would also explain how microvilli on one cell are able to elongate toward EPEC attached to a neighboring cell (Fig. 2F and 3C inset 1).
Figure 4.
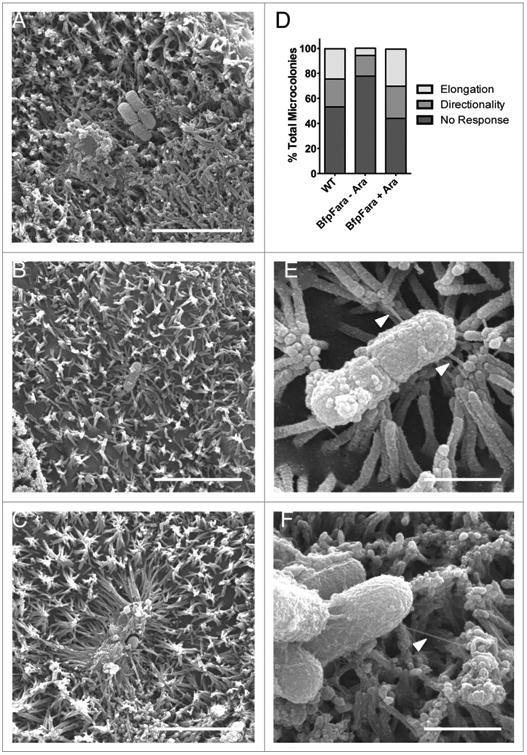
BFP retraction is necessary for directed microvillar elongation. (A-C) SEM images of WT EPEC inducing no response (A), directionality (B), and elongation (C) in surrounding microvilli. (D) Quantification of bacteria-induced microvillar responses in the presence of WT EPEC, retraction deficient bfpF+BfpFara EPEC, or bfpF+BfpFara EPEC + arabinose to induce retraction. (E, F) SEM images of BFP (white arrowheads) attaching to the tips of microvilli. Scale bars = 5 μm (A-C), 1 μm (E, F).
Microvillar actin bundles are incorporated into EPEC pedestals
Our data indicate that EPEC stimulate a combination of long-and short-range dynamics to recruit actin filaments to sites of attachment and pedestal formation. By inducing elongation of nearby microvilli toward newly attached bacteria and by drawing microvilli at distal locations toward the site of attachment, EPEC is able to sequester large amounts of pre-existing actin filaments that could facilitate pedestal assembly. If microvillar F-actin bundles are used by EPEC to build pedestals, microvillar coalescence into these structures should be evident during live cell imaging. Additionally, microvillar F-actin associated proteins should localize to pedestals. To test these predictions, we performed live cell attachment assays with Caco-2BBE cells expressing mCherry-tagged espin. Espin is an actin bundling protein that targets preferentially to parallel microvillar actin bundles, where it stabilizes these structures; overexpression of espin allows microvilli to reach >5 μm in length.25 Live cell imaging of EPEC attachment and pedestal formation revealed a rapid accumulation of mCherry-espin signal in characteristic oval shaped pedestals immediately underneath attaching microbes (Fig. 5A, B; Video 9). In fixed samples stained with phalloidin, signals from actin (phalloidin) and mCherry-espin colocalized (Fig. 5C), and the relative ratios of espin to actin in pedestals were similar to those found in microvilli in the same cells (Fig. 5D). Endogenous espin was also localized to pedestals using immunofluorescence (Fig. S3A). Other microvillar-specific proteins, particularly those involved in stabilizing the core actin bundle, might also be expected to localize to pedestals as microvilli coalesce. Indeed, consistent with a previous report,26 we observed striking recruitment of the bundling protein villin to mature pedestals following 3 h infection by EPEC (Fig. S3B). We also found the bi-functional capping/bundling protein epidermal growth factor receptor substrate 8 (Eps8), which primarily localizes to microvillar tips, in discreet puncta surrounding pedestals (Fig. S3C).27,28 In combination, these data suggest that microvillar core bundles and associated components are found in assembling pedestals.
Figure 5.
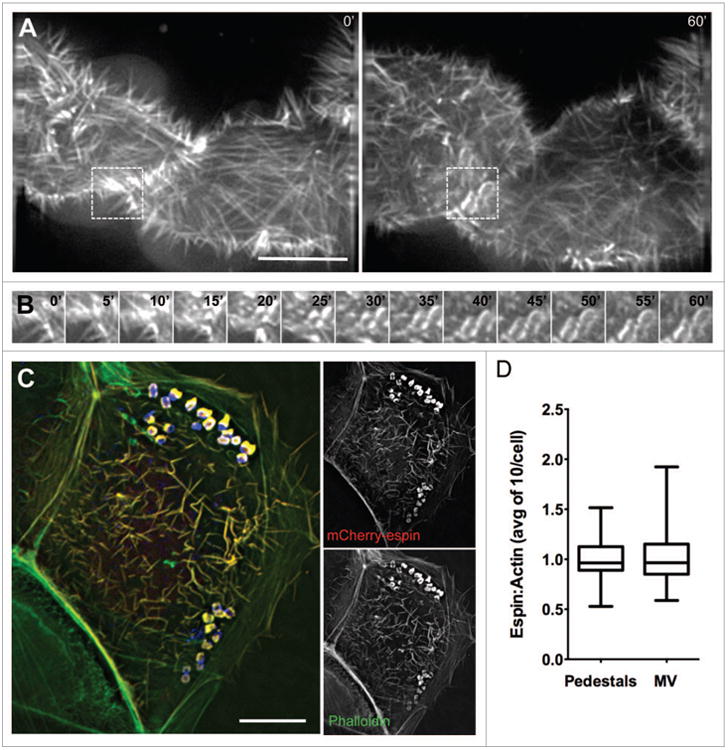
The parallel actin-bundling protein espin localizes to nascent pedestals in developing A/e lesions. (A) Deconvolution images of mcherry-espin-expressing Caco-2BBE cell at 0 min (Left) and 60 min (Right) after initial EPEC attachment. (B) Frames at 5 min intervals from boxed region in (A), showing espin accumulation in the developing pedestal. (C) Deconvolution image of fixed, EPEC infected Caco-2BBE cell expressing mcherry-espin (red) and stained with phalloidin (green) and α-LipidA (blue). (D) Quantification of the ratio of mcherry-espin to actin signal in pedestals and microvilli of fixed, EPEC-infected Caco-2BBe cells. Scale bars in (A, C) = 10 μm.
Although EPEC pedestals formed on the surface of Caco-2BBE cells exhibited the characteristic “hot dog bun” morphology in both light micrographs and SEM images, we performed additional ultrastructural analysis to determine if newly forming pedestals demonstrated features indicative of microvillar incorporation (Fig. 6A). Interestingly, we found numerous examples of protrusions of varying length, with diameters similar to those of microvilli, extending from the apical edge of nascent pedestals (Fig. 6A-C). TEM revealed similar structures with short actin bundles extending from the surface of pedestals (Fig. 6B′ and C′). These observations strongly suggest that microvilli immediately surrounding attached bacteria are incorporated into the pedestal wall, a conclusion consistent with our light microscopy results (Fig. 5A and B; Video 9).
Figure 6.

For figure legend see page 513. Microvilli are incorporated into nascent pedestals, forming a “hand-of-doom” morphology. (A) SEM images of micro-colonies 3 h after infection. Boxed regions magnified in panels 1–3, showing microvilli protruding from pedestals. (B) SEM (B) and TEM (B′) of microvillar tips protruding from the top of a pedestal (arrowheads). (C) SEM (C) and TEM (C′) of microvillar tips protruding from the side of a pedestal overlying a bacterium (arrowheads). Scale bars = 2 μm (A), 1 μm (B, C), 500 nm (B′, C′).
Stabilizing microvillar bundles inhibits pedestal formation
Further imaging of mCherry-espin expressing Caco-2BBE cells revealed that not all cells with EPEC contacting the brush border formed robust pedestals. In several instances, bacteria settled onto microvilli of espin-expressing cells and went through normal division over the course of a 1 h assay. Minimal flow and elongation was observed, and the reorientation of microvillar tips toward bacteria was reduced (Fig. 7A; Video 10). After 1 h incubation with EPEC, we binned attachment sites into three stages based on intensity of pedestal labeling and continuity of structure (Fig. 7B; Fig. S4). In control cells, almost all individual bacteria contacting the host cell surface produced a pedestal, compared with 45% in espin-expressing cells (stages 1 and 2 combined). Only 22% of the pedestals that did form reached maturity (stage 2) compared with 70% in control cells, as indicated by a continuous ring of F-actin encompassing the bacterium at an intensity greater than that of surrounding microvilli (Fig. 7B; Fig. S4). Additionally, the rate at which pedestals appeared in espin-expressing cells varied widely, from ∼1–5 min (comparable to non-espin-expressing cells) to >15 min, while the mean time to the first visible sign of a pedestal increased from <3.8 min to ∼6.9 min (Fig. 7C). In the remaining espin-expressing cells (stage 0), microvilli appeared unperturbed with no subsequent remodeling, and pedestal formation did not occur (Fig. 7A, B; Fig. S4). We also co-transfected cells with mCherry-espin and EGFP-β-actin to confirm that this result was not simply a matter of espin being excluded from assembling pedestals. Although pedestal formation was reduced in cells transfected with mCherry-espin, pedestals that did form were always visible in both channels (i.e., there were no pedestals that contained only EGFP-β-actin) and, as discussed above, endogenous espin localized to pedestals in fixed samples (Fig. 5C; Fig. S3A). Together, these data indicate that incorporation of F-actin bundles from microvilli into pedestals becomes rate limiting when the bundle-stabilizing protein, espin is overexpressed.
Figure 7.

Stabilization of microvillar actin bundles slows pedestal formation. (A) Deconvolution (Left) images of an mcherry-espin-expressing Caco-2BBE cell and attaching EPEC (DIC, Right) 0 min and 60 min after initial bacterial contact. Scale bar = 5 μm. (B) Percentage of microcolonies attached to mcherry-espin or mcherry-Utrch expressing cells that develop no pedestal (0), an intermediate pedestal (1), or a fully mature pedestal (2) after 60 min. (C) Time at which actin first begins to accumulate in each pedestal under microcolonies categorized as 1 or 2 in (B), *P < 0.05
Discussion
Many of the early studies investigating EPEC pedestal formation focused on elucidating pathway biochemistry and relied upon HeLa or other non-polarized cell lines including MEFs, COS1, and HEp-2 (for example, refs. 29–33). Although these cell lines are attractive due to their experimental flexibility, they lack a brush border, building instead a sparse network of filopodia on their dorsal surface.34 For this reason, we used the Caco-2BBE intestinal epithelial cell culture model to investigate apical cyto-skeletal dynamics and morphological changes induced by EPEC infection. As part of the differentiation process, Caco-2BBE cells develop hallmarks of epithelial polarity including a well-formed brush border, junctional complexes, and robust apical and/or basolateral trafficking.35-38 Employing this cell culture model for our high-resolution live imaging studies of EPEC infection enabled us to observe previously unreported features of the A/E process, which likely hold direct relevance for understanding how EPEC infects enterocytes in vivo.
A number of earlier studies suggested that EPEC pedestals arise entirely from de novo assembly of a branched actin network nucleated by Arp2/3. However, the relationship between newly forming pedestals and other pre-existing actin-based structures on the apical surface has remained unclear, despite extensive biochemical and ultrastructural analyses on the pathogenic mechanism of this bacterium.15,16,39,40 For the work described here, we focused on investigating microvillar dynamics and real-time morphological changes during EPEC attachment to complement existing studies. We found that microvilli flow across the epithelial cell surface and elongate toward sites of newly attached microbes. Long-range flow requires intermicrovillar adhesion, while elongation is driven by BFP retraction. Additionally, we found that microvillar actin-associated proteins, in particular the bundling protein espin, localize to pedestals. Espin overexpression also stabilizes microvillar F-actin bundles and delays pedestal formation.
We propose that microvillar flow and elongation are two mechanisms that allow EPEC to repurpose the large reservoir of microvillar actin filaments into bacterial lesions on the host cell apical surface. Microvillar flow could function to move existing F-actin to sites of attachment, whereas directed microvillar elongation might allow EPEC to tether individual protrusions and pull them into place for repurposing or remodeling of the underlying filaments. In turn, microvillar F-actin could be directly incorporated into pedestals, or perhaps remodeled or recycled, which would require severing by other actin-binding proteins. Consistent with this latter idea, proteomic analysis of parental Caco-2 cells revealed that expression of the severing proteins gelsolin and cofilin is upregulated by >3.5- and 2.5-fold, respectively, during EPEC infection.41 The energy cost associated with either direct incorporation or remodeling would likely be significantly lower than that required to polymerize a branched F-actin network de novo using Arp2/3.
Our studies also revealed that the intermicrovillar adhesion protein, PCDH24, is required for EPEC-stimulated microvillar flow. PCDH24 forms an adhesion complex by interacting in trans with MLPCDH, in turn providing a physical link between adjacent microvilli.19 EPEC might exploit the interconnected nature of brush border microvilli to “reel in” microvilli from distal sites. Although the driving force behind microvillar flow remains unknown, one candidate is the motor protein, myosin-2. This motor is activated during EPEC infection and disrupts tight junctions by exerting force on junction-associated actin.42 Because myosin-2 is also found in the terminal web at the base of the brush border,43 it could interact with the minus (pointed) ends of microvillar actin filaments and generate force to drive flow over the apical surface. Interestingly, cells infected with EPEC-ΔEscN did not exhibit robust long-range flow of microvilli across their surface (data not shown), suggesting that injection of Tir or other virulence factors may be required. Additional work will be needed to determine the involvement of myosin-2 and other host cell machinery in the dynamics reported here.
Our imaging studies also revealed that EPEC stimulates dramatic elongation of individual microvillar protrusions through a mechanism that requires BFP retraction. Several studies have previously demonstrated that the type IV pilus is capable of exerting forces high enough to draw out protrusions from the host cell surface during retraction.23,24 Thus, our observations could be explained by BFP attaching at or near the tips of microvilli, putting the protrusions under sufficient strain to induce their reorientation toward the bacteria and leading to the apparent “blast radius” observed in our electron micrographs (Fig. 2A). Subsequent BFP retraction could then pull microvillar tips in toward the bacterium, inducing elongation. In this way, BFP retraction could serve to not only pull EPEC into close contact with the host cell surface,8 but also to organize microvillar F-actin for remodeling into pedestals. Future work to investigate the molecular links between bacterial effector molecules, microvillar cytoskeletal proteins, and structural dynamics of the enterocyte brush border will be important for fully understanding the mechanisms that drive EPEC infection.
Materials and Methods
Cell culture and bacterial strains
Caco-2BBE cells were grown in DMEM (Gibco, 11995–065) supplemented with 18% defined fetal bovine serum (Atlanta Biologicals, S11550) and 1 mM L-glutamine (Gibco, 25030164) at 37 °C, 5% CO2. All cells were maintained in T75 flasks (Santa Cruz, sc-200263). EPEC were grown overnight in Luria-Bertani broth with kanamycin (WT), ampicillin (EPEC-bfpF+BfpFara), or chloramphenicol (ΔEscN) shaking at 225 rpm, 37 °C. Prior to use in attachment assays, overnight EPEC cultures were diluted and grown for 2 h in recovery media as described.20 For induction of BFP retraction, attachment assays were performed in the presence of 0.2% arabinose as previously described.8
Drug treatment assays
Caco-2BBE cells were grown on glass coverslips for 14 d in order to reach late differentiation. For EPEC assays, prior to the experiment cells were treated for 2 h with either DMSO (1:1000; Sigma, D8418) or 30 μM cytochalasin B (Sigma) reconstituted in DMSO. Following pretreatment, cell culture media was replaced by serum-free DMEM containing recovered EPEC (2 × 105 CFU/ml) and fresh DMSO or cytochalasin B. Samples were incubated for 3 h, fixed, processed for immunofluorescence, and imaged as described below. For quantification, z-stacks were first converted to max intensity projections, then the number of bacterial clusters with and without associated pedestal actin was counted.
Sample processing for SEM and TEM imaging
Caco-2BBE cells were grown on collagen-coated Transwell filters (Corning, 3493) for 14 d. EPEC at 2 × 105 CFU/ml were added to monolayers and incubated for 1 h. Sample preparation for SEM was performed as described previously,44 and samples were imaged using an FEI Quanta 250 ESEM managed by the Vanderbilt University Medical Center Cell Imaging Shared Resource Core.
Specimens were processed for TEM and imaged in the Vanderbilt Cell Imaging Shared Resource Electron Microscope facility. Cells were fixed in 2.5% gluteraldehyde in 0.1 M cacodylate buffer, pH 7.4 at room temperature (RT) for 1 h and then transferred to 4 °C and incubated overnight. The samples were washed 3× in 0.1 M cacodylate buffer, then incubated for 1 h in 1% osmium tetraoxide at RT, then washed 3× with 0.1 M cacodylate buffer. During the second wash, the Transwell was removed from the dish and Swiss-rolled into a tube, secured with a strand of hair, transferred to a clean 1.5 ml Eppendorf tube and washed again with cacodylate buffer. The samples were then dehydrated through a graded ethanol series: 30%, 50%, 70%, 80%, 95%, and then 3 exchanges of 100% ethanol. Next, the samples were incubated in a mixture of 100% ethanol and propylene oxide (PO) followed by 2 exchanges of pure PO. Samples were then infiltrated with 25% Epon 812 resin and 75% PO at RT. Next, they were infiltrated with 50% Epon 812 resin and 50% PO for 1 h at RT, then exchanged with new 50% Epon 812 resin and 50% PO, and incubated overnight at RT. Subsequently, the samples were put through a 75%:25% (resin:PO) exchange, then exchanged into pure epoxy resin for 4 h and incubated with pure epoxy resin overnight. Next, the resin was exchanged with fresh, pure epoxy resin and incubated for 3 h, then embedded in epoxy resin and polymerized at 60 °C for 48 h. Thick sections of 500–1000 nm were cut and imaged on a Nikon AZ100M. Regions of interests were selected and 70–80 nm ultra-thin sections were then cut from the block and collected on 300-mesh copper grids. The copper grids were post-section stained at RT with 2% uranyl acetate and lead citrate. Samples were subsequently imaged on the Philips/FEI Tecnai T12 electron microscope.
Sample processing for confocal imaging
Coverslips were fixed with 4% paraformaldehyde/PBS for 15 min, washed, and blocked for 1 h in 5% BSA/PBS. For EPEC attachment assays, goat polyclonal anti-LipidA (Abcam, ab20001) was diluted 1:500 and incubated on sample cover-slips for 2 h. Coverslips were then washed (3× PBS) and treated for 30 min with donkey-anti-goat secondary antibody (1:200; Molecular Probes, A11055) and Alexa-fluor 568-conjugated phalloidin (1:200; Molecular Probes, A12380) to delineate pedestals and cell morphology. For EPEC assays, images were acquired using LSCM (Leica TCS SP5) and SIM (Applied Precision OMX), and processed using ImageJ version 1.47h. Coverslips were imaged using deconvolution (Nikon) and SIM, and processed using ImageJ version 1.47h. Immunofluorescence for endogenous actin bundling proteins was performed as above, using rabbit polyclonal anti-espin (1:200; Sigma, HPA028674), mouse monoclonal anti-villin (Pierce, MA1–80757), or rabbit polyclonal anti-EPS8 (1:200; Sigma, HPA 003897), and imaged on a Nikon TiE widefield microscope with 2D deconvolution.
Live cell imaging
Caco-2BBE cells were seeded on glass-bottom dishes (MatTek, D35–20–1.5-N) and simultaneously transfected (Effectene; Qiagen, 301425) with mCherry-UtrCH18 or mCherry-espin25 and/or PCDH24-GFP, AEC1-PCDH24-GFP, or GFP-β-actin, then grown for 2 d. EPEC were grown and activated as described above. Before imaging, DMEM cell culture media was replaced with CO2-independent media (Gibco, 18045088). In order increase the chances of imaging productive binding events, high numbers of bacteria (∼1 × 108 CFU) were added to the dishes. Silica beads of approximately the same diameter as bacteria (1.57 μm; Bangs Laboratories, SS04N) were used for control experiments. Cells were imaged using an Applied Precision DeltaVision deconvolution microscope with environmental chamber heated to 37 °C, with z-stacks through the apical domain and time intervals of 20 s or 1 min. A DIC reference image was taken at the middle z-position for each time point. Image files were deconvolved then exported, and processed using FIJI version 1.48b. Microvilli were tracked using the MTrackJ plugin, following the distal tips of microvilli across frames. For display purposes, all movie files were cropped to show only attachment events.
Supplementary Material
Acknowledgments
The authors thank all members of the Tyska laboratory and the Vanderbilt Epithelial Biology Center for advice and support. Gail Hecht and Michael Donnenberg graciously provided bacterial strains, and Nicholas Biais contributed valuable discussion. Live cell imaging was performed with the support of Joseph Roland and the Vanderbilt Digital Histology Shared Resource. Electron and light microscopy were performed in part through the use of the VUMC Cell Imaging Shared Resource, with the assistance of Janice Williams and Matthew Stephenson. This work was supported by American Heart Association (AHA) pre-and post-doctoral fellowships (D.A.S., S.W.C.), and National Institutes of Health grants DK075555 and DK095811 (M.J.T.).
Abbreviations
- A/E
Attaching/effacing
- Arp
Actin-related protein
- BFP
Bundle forming pilus
- EC
extracellular cadherin
- EPEC
Enteropathogenic E. coli
- Eps8
epidermal growth factor receptor substrate 8
- GFP
green fluorescent protein
- MLP
Microvillar-like process
- MLPCDH
Mucin-like protocadherin
- PCDH24
Protocadherin-24
- RT
Room temperature
- SEM
Scanning electron microscopy
- SIM
Structured illumination microscopy
- T3SS
Type III secretion system
- TEM
Transmission electron microscopy
- UtrCH
Utrophin calponin homology domain
- WT
Wild type
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflict of interest was disclosed.
References
- 1.Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–33. doi: 10.1146/annurev.mi.31.100177.000543. http://dx.doi.org/10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 2.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut micro-biota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–23. doi: 10.1073/pnas.0407076101. http://dx.doi.org/10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–20. doi: 10.1126/science.1104816. http://dx.doi.org/10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 4.Evans DJ, Evans DG. Escherichia Coli in Diarrheal Disease. In: Baron S, editor. Medical Microbiology. Galveston (TX): 1996. [PubMed] [Google Scholar]
- 5.Shifrin DA, Jr, McConnell RE, Nambiar R, Higginbotham JN, Coffey RJ, Tyska MJ. Enterocyte microvillus-derived vesicles detoxify bacterial products and regulate epithelial-microbial interactions. Curr Biol. 2012;22:627–31. doi: 10.1016/j.cub.2012.02.022. http://dx.doi.org/10.1016/j.cub.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hicks S, Frankel G, Kaper JB, Dougan G, Phillips AD. Role of intimin and bundle-forming pili in enteropathogenic Escherichia coli adhesion to pediatric intestinal tissue in vitro. Infect Immun. 1998;66:1570–8. doi: 10.1128/iai.66.4.1570-1578.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cleary J, Lai LC, Shaw RK, Straatman-Iwanowska A, Donnenberg MS, Frankel G, Knutton S. Enteropathogenic Escherichia coli (EPEC) adhesion to intestinal epithelial cells: role of bundle-forming pili (BFP), EspA filaments and intimin. Microbiology. 2004;150:527–38. doi: 10.1099/mic.0.26740-0. http://dx.doi.org/10.1099/mic.0.26740-0. [DOI] [PubMed] [Google Scholar]
- 8.Zahavi EE, Lieberman JA, Donnenberg MS, Nitzan M, Baruch K, Rosenshine I, Turner JR, Melamed-Book N, Feinstein N, Zlotkin-Rivkin E, et al. Bundle-forming pilus retraction enhances enteropathogenic Escherichia coli infectivity. Mol Biol Cell. 2011;22:2436–47. doi: 10.1091/mbc.E11-01-0001. http://dx.doi.org/10.1091/mbc.E11-01-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevens JM, Galyov EE, Stevens MP. Actin-dependent movement of bacterial pathogens. Nat Rev Microbiol. 2006;4:91–101. doi: 10.1038/nrmicro1320. http://dx.doi.org/10.1038/nrmicro1320. [DOI] [PubMed] [Google Scholar]
- 10.Dean P, Maresca M, Schüller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc Natl Acad Sci U S A. 2006;103:1876–81. doi: 10.1073/pnas.0509451103. http://dx.doi.org/10.1073/pnas.0509451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernet-Camard MF, Coconnier MH, Hudault S, Servin AL. Pathogenicity of the diffusely adhering strain Escherichia coli C1845: F1845 adhesin-decay accelerating factor interaction, brush border microvillus injury, and actin disassembly in cultured human intestinal epithelial cells. Infect Immun. 1996;64:1918–28. doi: 10.1128/iai.64.6.1918-1928.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phillips AD, Giròn J, Hicks S, Dougan G, Frankel G. Intimin from enteropathogenic Escherichia coli mediates remodelling of the eukaryotic cell surface. Microbiology. 2000;146:1333–44. doi: 10.1099/00221287-146-6-1333. [DOI] [PubMed] [Google Scholar]
- 13.Shaw RK, Cleary J, Murphy MS, Frankel G, Knutton S. Interaction of enteropathogenic Escherichia coli with human intestinal mucosa: role of effector proteins in brush border remodeling and formation of attaching and effacing lesions. Infect Immun. 2005;73:1243–51. doi: 10.1128/IAI.73.2.1243-1251.2005. http://dx.doi.org/10.1128/IAI.73.2.1243-1251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dean P, Young L, Quitard S, Kenny B. Insights into the pathogenesis of enteropathogenic E. coli using an improved intestinal enterocyte model PLoS One. 2013;8:e55284. doi: 10.1371/journal.pone.0055284. http://dx.doi.org/10.1371/journal.pone.0055284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campellone KG. Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia coli: Tir, EspFU and actin pedestal assembly. FEBS J. 2010;277:2390–402. doi: 10.1111/j.1742-4658.2010.07653.x. http://dx.doi.org/10.1111/j.1742-4658.2010.07653.x. [DOI] [PubMed] [Google Scholar]
- 16.Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol. 2011;80:1420–38. doi: 10.1111/j.1365-2958.2011.07661.x. http://dx.doi.org/10.1111/j.1365-2958.2011.07661.x. [DOI] [PubMed] [Google Scholar]
- 17.Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–76. doi: 10.1146/annurev.biophys.29.1.545. http://dx.doi.org/10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 18.Burkel BM, von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton. 2007;64:822–32. doi: 10.1002/cm.20226. http://dx.doi.org/10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawley SW, Shifrin DA, Jr, Grega-Larson NE, McConnell RE, Benesh AE, Mao S, Zheng Y, Zheng QY, Nam KT, Millis BA, et al. Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell. 2014;157:433–46. doi: 10.1016/j.cell.2014.01.067. http://dx.doi.org/10.1016/j.cell.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma R, Tesfay S, Tomson FL, Kanteti RP, Viswanathan VK, Hecht G. Balance of bacterial pro-and anti-inflammatory mediators dictates net effect of enteropathogenic Escherichia coli on intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:G685–94. doi: 10.1152/ajpgi.00404.2005. http://dx.doi.org/10.1152/ajpgi.00404.2005. [DOI] [PubMed] [Google Scholar]
- 21.Gauthier A, Puente JL, Finlay BB. Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization. Infect Immun. 2003;71:3310–9. doi: 10.1128/IAI.71.6.3310-3319.2003. http://dx.doi.org/10.1128/IAI.71.6.3310-3319.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maier B, Potter L, So M, Long CD, Seifert HS, Sheetz MP. Single pilus motor forces exceed 100 pN. Proc Natl Acad Sci U S A. 2002;99:16012–7. doi: 10.1073/pnas.242523299. http://dx.doi.org/10.1073/pnas.242523299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biais N, Ladoux B, Higashi D, So M, Sheetz M. Cooperative retraction of bundled type IV pili enables nanonewton force generation. PLoS Biol. 2008;6:e87. doi: 10.1371/journal.pbio.0060087. http://dx.doi.org/10.1371/journal.pbio.0060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higashi DL, Zhang GH, Biais N, Myers LR, Weyand NJ, Elliott DA, So M. Influence of type IV pilus retraction on the architecture of the Neisseria gonorrhoeae-infected cell cortex. Microbiology. 2009;155:4084–92. doi: 10.1099/mic.0.032656-0. http://dx.doi.org/10.1099/mic.0.032656-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loomis PA, Zheng L, Sekerková G, Changyaleket B, Mugnaini E, Bartles JR. Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J Cell Biol. 2003;163:1045–55. doi: 10.1083/jcb.200309093. http://dx.doi.org/10.1083/jcb.200309093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanger JM, Chang R, Ashton F, Kaper JB, Sanger JW. Novel form of actin-based motility transports bacteria on the surfaces of infected cells. Cell Motil Cytoskeleton. 1996;34:279–87. doi: 10.1002/(SICI)1097-0169(1996)34:4<279::AID-CM3>3.0.CO;2-3. http://dx.doi.org/10.1002/(SICI)1097-0169(1996)34:4<279∷AID-CM3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Disanza A, Carlier MF, Stradal TE, Didry D, Frittoli E, Confalonieri S, Croce A, Wehland J, Di Fiore PP, Scita G. Eps8 controls actin-based motility by capping the barbed ends of actin filaments. Nat Cell Biol. 2004;6:1180–8. doi: 10.1038/ncb1199. http://dx.doi.org/10.1038/ncb1199. [DOI] [PubMed] [Google Scholar]
- 28.Disanza A, Mantoani S, Hertzog M, Gerboth S, Frittoli E, Steffen A, Berhoerster K, Kreienkamp HJ, Milanesi F, Di Fiore PP, et al. Regulation of cell shape by Cdc42 is mediated by the synergic actin-bundling activity of the Eps8-IRSp53 complex. Nat Cell Biol. 2006;8:1337–47. doi: 10.1038/ncb1502. http://dx.doi.org/10.1038/ncb1502. [DOI] [PubMed] [Google Scholar]
- 29.Campellone KG, Leong JM. Nck-independent actin assembly is mediated by two phosphorylated tyrosines within enteropathogenic Escherichia coli Tir. Mol Microbiol. 2005;56:416–32. doi: 10.1111/j.1365-2958.2005.04558.x. http://dx.doi.org/10.1111/j.1365-2958.2005.04558.x. [DOI] [PubMed] [Google Scholar]
- 30.Campellone KG, Giese A, Tipper DJ, Leong JM. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol Microbiol. 2002;43:1227–41. doi: 10.1046/j.1365-2958.2002.02817.x. http://dx.doi.org/10.1046/j.1365-2958.2002.02817.x. [DOI] [PubMed] [Google Scholar]
- 31.Brown MD, Bry L, Li Z, Sacks DB. Actin pedestal formation by enteropathogenic Escherichia coli is regulated by IQGAP1, calcium, and calmodulin. J Biol Chem. 2008;283:35212–22. doi: 10.1074/jbc.M803477200. http://dx.doi.org/10.1074/jbc.M803477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finlay BB, Rosenshine I, Donnenberg MS, Kaper JB. Cytoskeletal composition of attaching and effacing lesions associated with enteropathogenic Escherichia coli adherence to HeLa cells. Infect Immun. 1992;60:2541–3. doi: 10.1128/iai.60.6.2541-2543.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campellone KG, Rankin S, Pawson T, Kirschner MW, Tipper DJ, Leong JM. Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J Cell Biol. 2004;164:407–16. doi: 10.1083/jcb.200306032. http://dx.doi.org/10.1083/jcb.200306032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohil AB, Robertson BW, Cheney RE. Myosin-X is a molecular motor that functions in filopodia formation. Proc Natl Acad Sci U S A. 2006;103:12411–6. doi: 10.1073/pnas.0602443103. http://dx.doi.org/10.1073/pnas.0602443103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao X, Surma MA, Simons K. Polarized sorting and trafficking in epithelial cells. Cell Res. 2012;22:793–805. doi: 10.1038/cr.2012.64. http://dx.doi.org/10.1038/cr.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson MD, Bement WM, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. II. Changes in expression and localization of brush border proteins during cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:461–72. doi: 10.1242/jcs.105.2.461. [DOI] [PubMed] [Google Scholar]
- 37.Peterson MD, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. I. Ultrastructural analysis of cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:445–60. doi: 10.1242/jcs.105.2.445. [DOI] [PubMed] [Google Scholar]
- 38.Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- 39.Dean P, Kenny B. The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr Opin Microbiol. 2009;12:101–9. doi: 10.1016/j.mib.2008.11.006. http://dx.doi.org/10.1016/j.mib.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frankel G, Phillips AD, Rosenshine I, Dougan G, Kaper JB, Knutton S. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol Microbiol. 1998;30:911–21. doi: 10.1046/j.1365-2958.1998.01144.x. http://dx.doi.org/10.1046/j.1365-2958.1998.01144.x. [DOI] [PubMed] [Google Scholar]
- 41.Hardwidge PR, Rodriguez-Escudero I, Goode D, Donohoe S, Eng J, Goodlett DR, Aebersold R, Finlay BB. Proteomic analysis of the intestinal epithelial cell response to enteropathogenic Escherichia coli. J Biol Chem. 2004;279:20127–36. doi: 10.1074/jbc.M401228200. http://dx.doi.org/10.1074/jbc.M401228200. [DOI] [PubMed] [Google Scholar]
- 42.Yuhan R, Koutsouris A, Savkovic SD, Hecht G. Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology. 1997;113:1873–82. doi: 10.1016/s0016-5085(97)70006-4. http://dx.doi.org/10.1016/S0016-5085(97)70006-4. [DOI] [PubMed] [Google Scholar]
- 43.Bement WM, Mooseker MS. The cytoskeleton of the intestinal epithelium: Components, assembly and dynamic rearrangements. In: Hesketh JE, P IF, editors. The cytoskeleton. Greenwich, CT: Jai Press; 1996. pp. 359–404. [Google Scholar]
- 44.McConnell RE, Higginbotham JN, Shifrin DA, Jr, Tabb DL, Coffey RJ, Tyska MJ. The enterocyte microvillus is a vesicle-generating organelle. J Cell Biol. 2009;185:1285–98. doi: 10.1083/jcb.200902147. http://dx.doi.org/10.1083/jcb.200902147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.