

Sir,
The human Ether-à -go-go-Related Gene (hERG) encodes potassium channels mediating the rapid delayed-rectifier K+ current, IKr, which is crucial for normal repolarization of the ventricles of the heart.[1] hERG is established to be a pharmacological target for Class Ia and Class III antiarrhythmic drugs and for numerous noncardiac drugs associated with acquired long QT syndrome and torsades de pointes arrhythmia.[1] Due to the pharmacological promiscuity of hERG channels, all novel pharmaceuticals must be tested for their propensity to inhibit hERG channel ionic current.[1]
The archetypal high-affinity hERG inhibitors come from the methanesulphonanilide drug family.[1,2] This family includes dofetilide (Tikosyn), a drug licensed in the USA for the treatment of supraventricular arrhythmias and an experimental Class III drug, E-4031 [Figure 1A]. Sotalol [Figure 1A], which is used to treat both supraventricular arrhythmias and serious ventricular arrhythmias in structurally normal hearts, differs from other methanesulphonanilides in that it binds with low affinity to hERG/IKr channels. For example, a study of displacement of tritiated dofetilide by E-4031, dofetilide, and D-sotalol from guinea-pig ventricular myocytes yielded respective Ki values of 38 nM, 47 nM, and 100 μM.[3] The structural basis for sotalol's low-affinity hERG/IKr block has not yet been established. Binding determinants of E-4031 and dofetilide, as well as another high-affinity methanesulphonanilide, MK499, have been mapped to the S6- and pore-helices that form the inner cavity of the hERG channel;[2,4] mutation of S6 aromatic residues (Y652 and F656) and of residues at the base of the pore helix (T623, S624, V625) markedly impaired the ability of these drugs to inhibit hERG current (IhERG).[2,4] Comparable information is not currently available for sotalol, perhaps in part due to difficulties in obtaining pure D-sotalol (which lacks the marked β-adrenoceptor-blocking properties of the racemic mixture) and in part because the low potency of the drug makes it difficult to study at concentrations required to produce profound IhERG inhibition.
Figure 1.
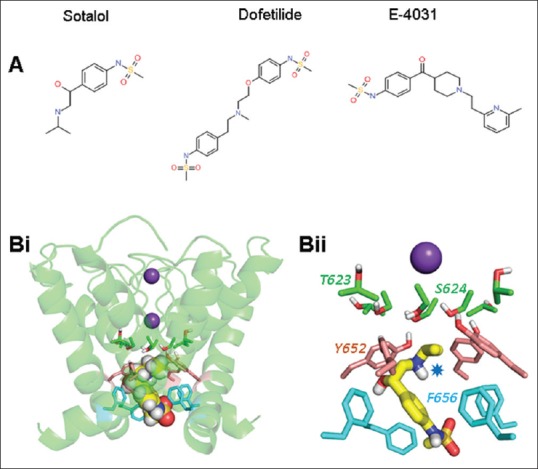
(A) Chemical structures of sotalol, dofetilide, and E-4031 generated using Symyx® Draw 3.3. (Bi) Low-energy score binding pose in the context of the whole channel pore. D-sotalol is shown as a space-filling structure colored according to atom type. The pore tetramer is represented by a green ribbon, and K+ ions occupy the S1 and S3 positions of the selectivity filter. Side chains of T623 and S624 (green), Y652 (pink), and F656 (blue) are displayed as sticks. (Bii) The same binding mode focusing on the side chains of amino acids lining the pore interior known to be determinants of high-potency methanesulphonanilide block. The charged secondary aliphatic amino group and aromatic moiety of D-sotalol can make cation-π and π-stacking interactions, respectively, with the side chains of F656 (blue) or Y652 (pink). In these poses, D-sotalol does not make simultaneous interactions with pore-helical residues T623 or S624
Comparison of the structures of E-4031 and dofetilide with sotalol [Figure 1A] shows that sotalol is a smaller molecule than the other drugs. We have previously compared structurally similar hERG-blocking drugs of different sizes (ranolazine and lidocaine) and found that, despite structural similarities, the smaller drug (lidocaine) was less well able to interact with hERG pore side chains than the larger molecule.[5] We hypothesized that a similar explanation accounts for relatively low-affinity IhERG block by sotalol. Accordingly, using a previously validated, MthK-based hERG pore model,[5,6] we have docked D-sotalol in the hERG channel inner cavity [Figure 1Bi and Bii]. Low-energy-score binding poses show an ability for sotalol to interact with the aromatic canonical drug-binding residues Y652 and F656. The charged secondary aliphatic amino group and aromatic moiety of D-sotalol can make cation-π and π-stacking interactions, respectively, with the side chains of F656 or Y652. In these poses, D-sotalol does not make simultaneous interactions with pore-helical residues (T623, S624). This contrasts markedly with the situation for high-affinity methanesulphonanilides.[2,4] To test this notion experimentally, we have performed experiments in which a D-sotalol concentration producing 50%–60% inhibition of wild-type (WT) channels was tested against alanine mutants of the S6 aromatic residue Y652 and pore-helical residue S624, expressed in HEK 293 cells. Recordings were made at physiological temperature using previously described conditions and protocols (voltage protocol shown in Figure 2B).[5] D-sotalol was obtained from Sequoia Research Products. Electrophysiological properties of the S624A and Y652A mutants allow them to be studied under identical conditions to the WT channel. 100 μM D-sotalol blocked WT IhERG by ~53% [Figure 2Ai and 2C], and this was not significantly reduced for S624A IhERG [Figure 2Aii and 2C]. In contrast, the Y652A mutation produced a moderate, statistically significant attenuation of IhERG block by D-sotalol [Figure 2Aiii and 2C].
Figure 2.
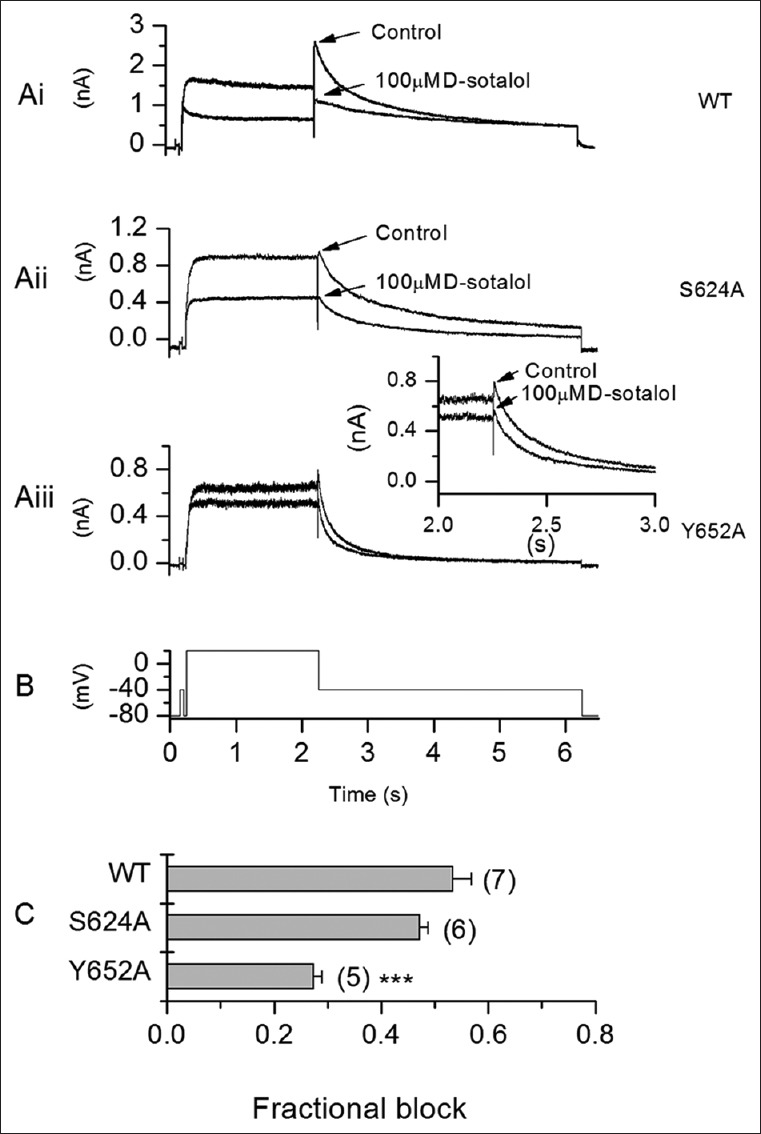
(Ai-iii) Representative traces of IhERG in control solution and following exposure to 100 μM D-sotalol (Ai) shows data for wild-type IhERG, (Aii) for S624A IhERG, (Aiii) for Y652A IhERG. The corresponding voltage protocol is shown in (B). (C) Bar charts showing the mean (± standard error of the mean) levels of inhibition (shown as fractional block) produced by 100 μM D-sotalol for wild-type, S624A, and Y652A hERG. Numbers of replicates (5–7) are given in brackets. There was no significant difference between wild-type (mean fractional block of 0.53) and S624A (mean fractional block of 0.47; P > 0.05), but Y652A (mean fractional block of 0.27) showed significantly attenuated inhibition (***P < 0.001); one-way analysis of variance with Bonferroni post-test. Recordings made at 37°C with K-based pipette solution and standard external Tyrode's solution.[5] IhERG: Ionic current carried by human Ether-à-go-go-Related Gene (hERG) channels
Our results support the notion that the smaller size of sotalol than of other methanesulphonanilides limits its ability to make simultaneous contacts with aromatic and pore-helix residues and that this, together with weaker contacts with key-binding determinants, underpins its low-affinity IhERG block. We suggest that a future detailed alanine scan of the inner cavity would be valuable to define more precisely the sotalol interaction site(s) on the hERG channel.
Financial support and sponsorship
The authors gratefully acknowledge the British Heart Foundation for funding (PG/10/017; PG/12/; PG/12/69/29784; PG/14/61/31015.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Hancox JC, McPate MJ, El Harchi A, Zhang YH. The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther. 2008;119:118–32. doi: 10.1016/j.pharmthera.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol. 2006;69:1709–16. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- 3.Lynch JJ, Jr, Baskin EP, Nutt EM, Guinosso PJ, Jr, Hamill T, Salata JJ, et al. Comparison of binding to rapidly activating delayed rectifier K + channel, IKr, and effects on myocardial refractoriness for class III antiarrhythmic agents. J Cardiovasc Pharmacol. 1995;25:336–40. doi: 10.1097/00005344-199502000-00021. [DOI] [PubMed] [Google Scholar]
- 4.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329–33. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du C, Zhang Y, El Harchi A, Dempsey CE, Hancox JC. Ranolazine inhibition of hERG potassium channels: Drug-pore interactions and reduced potency against inactivation mutants. J Mol Cell Cardiol. 2014;74:220–30. doi: 10.1016/j.yjmcc.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dempsey CE, Wright D, Colenso CK, Sessions RB, Hancox JC. Assessing hERG pore models as templates for drug docking using published experimental constraints: The inactivated state in the context of drug block. J Chem Inf Model. 2014;54:601–12. doi: 10.1021/ci400707h. [DOI] [PMC free article] [PubMed] [Google Scholar]