

Abstract
Aerosol-cloud interaction contributes to the largest uncertainties in the estimation and interpretation of the Earth’s changing energy budget. The present study explores experimentally the impacts of water condensation-evaporation events, mimicking processes occurring in atmospheric clouds, on the molecular composition of secondary organic aerosol (SOA) from the photooxidation of methacrolein. A range of on- and off-line mass spectrometry techniques were used to obtain a detailed chemical characterization of SOA formed in control experiments in dry conditions, in triphasic experiments simulating gas-particle-cloud droplet interactions (starting from dry conditions and from 60% relative humidity (RH)), and in bulk aqueous-phase experiments. We observed that cloud events trigger fast SOA formation accompanied by evaporative losses. These evaporative losses decreased SOA concentration in the simulation chamber by 25–32% upon RH increase, while aqueous SOA was found to be metastable and slowly evaporated after cloud dissipation. In the simulation chamber, SOA composition measured with a high-resolution time-of-flight aerosol mass spectrometer, did not change during cloud events compared with high RH conditions (RH > 80%). In all experiments, off-line mass spectrometry techniques emphasize the critical role of 2-methylglyceric acid as a major product of isoprene chemistry, as an important contributor to the total SOA mass (15–20%) and as a key building block of oligomers found in the particulate phase. Interestingly, the comparison between the series of oligomers obtained from experiments performed under different conditions show a markedly different reactivity. In particular, long reaction times at high RH seem to create the conditions for aqueous-phase processing to occur in a more efficient manner than during two relatively short cloud events.
1. Introduction
Many secondary organic aerosol (SOA) components and precursors are water-soluble, and some of them have been observed in cloudwater droplets in large field campaigns such as HCCT-2010,1 SOAS,2 and in the Po Valley.3−5 The atmospheric reactivity is markedly different in the water phase with respect to the gas phase. For example, clouds can promote oxidation of sulfur dioxide by ozone and/or by hydrogen peroxide,6,7 can promote oxidation of water-soluble organic compounds (WSOCs) with subsequent formation of organic acids and oligomers,8−11 and can create the conditions for photo-Fenton reactions to occur in the presence of metals.12
Photooxidation processes occurring in cloud droplets can be responsible for substantial modifications of the physical and chemical properties of both volatile organic compounds (VOCs) and particles, which initially served as cloud condensation nuclei (CCN). Such modifications are still largely unknown, especially for organic species, but they may alter the surface of the particles and hence their ability to serve as new CCN11 and thus trigger a feedback cycle on clouds.
Isoprene is the most abundant nonmethane hydrocarbon emitted in the atmosphere, and its atmospheric photooxidation is one of the largest sources of SOA at a global scale.13,14 Many studies have been conducted on isoprene oxidation in smog chambers,15−19 during field measurements,2,20−26 and through modeling27−30 to better estimate global SOA formation and characterize SOA composition at the molecular level. For example, isoprene was thought to be a weak aerosol producer, but in the presence of wet acidic sulfate aerosol high quantities of SOA have been observed in the southeastern U.S. formed through the isoprene epoxydiol (IEPOX) pathway.31−35 Because most of the photooxidation products of isoprene are water-soluble, their aqueous-phase processing has an important influence on SOA formation and aging.36 We investigated the impact of cloud events on an isoprene/NOx system in the presence of light and at different oxidation stages experimentally in a recent study.37 It was observed that a single and relatively short cloud event under irradiation led to a significant aqueous-SOA (aqSOA) production, with mass yields 2 to 4 times higher than those observed for isoprene photooxidation experiments performed under dry conditions. However, this aqSOA seemed to be metastable in the simulation chamber environment, as it evaporates slowly after cloud dissipation.37
Characterization of aqueous isoprene SOA was done in previous studies by collecting on filters SOA produced under high-NOx conditions in smog chambers, followed by photooxidation in aqueous-phase photoreactors.36,38 Liu et al.36 observed that the OHaq-radical aging of isoprene SOA induces a significant change in aerosol composition, characterized by higher hygroscopicity, formation of small organic acids (including formic, glyoxylic, glycolic, butyric, oxalic, and 2-methylglyceric acids), and enrichment in oligomers from reactions promoted by the OH radical (·OH). In similar experiments, Nguyen et al.38 observed photochemical production of furoxan-type and N-heterocyclic compounds but also degradation of high molecular-weight compounds.
High molecular-weight compounds, especially oligomers, have been the center of discussions for years concerning their importance in the ambient atmosphere. Kalberer et al.39 discovered that aged SOA from oxidation of aromatic compounds in a smog chamber contains a large fraction of oligomers; however, field studies in the ambient atmosphere reported only limited oligomer formation from biogenic precursors, that is, isoprene and monoterpenes. For example, Surratt et al.18 reported dimer formation from isoprene photooxidation, while Yasmeen et al.40 provided evidence for dimer formation from α-pinene photooxidation. Recently, Kourtchev et al.41 established that precursor concentrations and SOA mass are governing factors in triggering oligomerization of biogenic compounds, thus explaining the different findings between laboratory and field studies but highlighting that, in a future warmer climate, oligomers will become increasingly important SOA components.
Oligomerization can occur through different pathways, such as peroxyhemiacetal and acyl hydroperoxide formation, aldol and gem-diol reactions, and esterification.7,42 Evidence was also presented for oligomerization through secondary ozonide formation from bimolecular reactions involving Criegee intermediates, Criegee self-reactions, and heterogeneous reactions involving multifunctional hydroperoxides from stabilized Criegee intermediates.42−44 Concerning isoprene SOA, Zhang et al.45 and Nguyen et al.15 established that a high relative humidity (RH) suppresses oligomer formation not only in the case of condensation reactions (accompanied by loss of a water molecule) but also for hemiacetal formation. Conversely, Rodigast et al.46 highlighted a contradicting dependence of oligomer formation from methylglyoxal (an isoprene oxidation product) under varying RH and pH. Oligomerization from isoprene oxidation can occur also from acid-catalyzed ring-opening reactions of multifunctional epoxides.35,47 Studies on aqueous-phase photooxidation of methacrolein and methyl vinyl ketone, the two main first-generation gas-phase oxidation products of isoprene, have shown that they act as precursors for long homologous series of oligomers.48−50 In the case of methyl vinyl ketone, it has been suggested that oligomers are formed via radical reactions.38 In more complex mixtures of unsaturated conjugated precursors, co-oligomerization has been observed, forming complex oligomers bearing monomers of different structures.51 These detailed studies on aqueous-phase processing have been performed in bulk solutions and at high initial precursor concentrations; therefore, they need to be complemented with experiments that can simulate more realistic conditions in terms of gas-droplet-aerosol interactions.
The aim of this work is to study the formation and characterize the composition of SOA derived from the photooxidation of methacrolein during cloud condensation-evaporation cycles to better simulate the interaction between VOCs, aerosol, and cloud droplets under atmospherically realistic conditions. A range of various on- and off-line mass spectrometry techniques were used to obtain a detailed characterization of SOA at the molecular level in dry conditions, during cloud events and after cloud evaporation, and as such to obtain insights into the reaction processes involved in SOA processing in an aqueous/multiphase environment.
2. Experimental Section
2.1. Experimental Protocols and Online Measurements
Experiments were performed in the CESAM chamber (French acronym for Experimental Multiphasic Atmospheric Simulation Chamber), a 4.2 m3 stainless steel smog chamber equipped with three high-pressure xenon arc lamps and Pyrex filters of 6.5 mm thickness that produce an irradiation spectrum very similar to the solar spectrum at ground level.52,53 Details of experimental protocols and measurements are reported in previous studies.37,53 Briefly, methacrolein was reacted with ·OH in the presence of NOx under irradiation in dry conditions (RH < 5%) and at constant temperature (Table 1) without seed particles. Cloud experiments were performed by generating clouds after the SOA mass reached its maximum concentration in dry conditions. In a first step, RH was increased to greater than 80%, shortly after the first cloud was generated using the protocol (triphasic protocol gas-particulate-cloud) already described elsewhere,37,52 and a second cloud was generated ∼1 h after the evaporation of the first cloud. Control experiments of isoprene and methacrolein photooxidation in dry conditions (without cloud formation or increased RH) and blank experiments (without VOC injection but with and without cloud formation) were also done. Additionally, a triphasic experiment starting at 60% RH rather than at dry conditions was also performed. A scheme of the different methacrolein photooxidation experiments is reported in Figure 1. Chamber blanks were checked at the beginning of each experiment with online instruments; VOCs and particle concentrations were always below detection limits.
Table 1. Type of Experiments, Experimental Conditions, SOA Yields, Generated Clouds, and Instrumental Analysis (Mass Spectrometry) of SOA from Methacrolein Photooxidation.
| [VOC]0a | [HONO]0 | [NO]0 | [NO2]0 | [O3]max | ΔM0b,c | T | RH | MS
measurementsd |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| experiments | ppb | ppb | ppb | ppb | ppb | μg/m3 | °C | % | N clouds | AMS | GC-MS | ESI-HRMS |
| blanks | ||||||||||||
| BL-120115 | 0 | 137 | 195 | 104 | 2 | <0.3 | 19.6 | 0 | 0 | N | Y | Y |
| BL-140115 | 0 | 232 | 162 | 108 | 3.2 | 22.5 | 20.5 | 71 | 1 | N | Y | Y |
| control experiments | ||||||||||||
| MC-020315 | 704 | 120 | 233 | 92 | 214 | 137 | 20.5 | 4–5 | 0 | N | Y | Y |
| triphasic experiments | ||||||||||||
| MT-180113 | 735 | 124 | 88 | 25 | 94 | 58.8 | 19.8 | 0 | 2 | Y | Y | Y |
| MT-210113 | 927 | 150 | 118 | 81 | 123 | 65.8 | 19.4 | 5–3 | 2 | Y | Y | Y |
| MT-230113 | 396 | 125 | 67 | 5 | 51 | 27.3 | 19.6 | 5–3 | 2 | Y | Y | N |
| MT-130614 | 874 | 110 | 146 | 191 | 137 | 37 | 21.9 | 67 | 2 | Y | N | Y |
Measurement uncertainty is 15 ppb.
SOA concentration with effective density of 1.4 g/cm3.
Measurement uncertainty is 0.1 μg/m3.
Y = yes, N = no.
Figure 1.

Scheme of methacrolein photooxidation experiments performed in the CESAM chamber and in the aqueous phase photoreactor. RHt0 refers to RH at the beginning of the experiment, and RHtf refers to RH at the end of the experiments, after the cloud events.
VOC measurements were done online with a Fourier transform infrared spectrometer (FTIR, Bruker, TENSOR 37) and a proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS 8000, Ionicon Analytik). HONO was measured with NitroMAC, an instrument developed in-house.54 Ozone was monitored with a Horiba APOA-370 analyzer, and NOx was measured using a Horiba APNA-370. SOA size distributions were measured using a Scanning Mobility Particle Sizer (SMPS) composed of a Differential Mobility Analyzer (DMA, TSI, model 3080) coupled to a Condensation Particle Counter (CPC, TSI, model 3010), and a high-resolution time-of-flight aerosol mass spectrometer (HR-TOF-AMS, Aerodyne). The SMPS and the HR-TOF-AMS instruments were connected to the chamber through the same sampling line, dried with a 60 cm Nafion tube (Permapure, model MD-110). The RH was continuously measured downstream of the Nafion tube and was always less than 22%, maintaining the RH in the sampling line lower than the efflorescence point of any expected aerosol species.37 Drying the sampling line has the advantage of preventing clogging of the critical orifice of the instruments and possible electrical discharges that may happen in the DMA at high RH; however, it may lead to partial losses of reversible aqSOA.55 More details about experimental protocols and online measurements can be found in previous studies.37,52,53
2.2. Aerosol Collection and Offline Analyses
2.2.1. Collection of Aerosol Samples on Filters
At the end of each experiment, the aerosol was collected on a prebaked (650 °C for 24 h) quartz fiber filter (diameter 47 mm, Tissuquartz 2500 QATUP, Pall Life Sciences), as used in previous studies,56 using a stainless-steel filter holder (Pall Life Sciences) fitted with an upstream charcoal denuder directly connected to the chamber. The denuder was purged using pure N2 for at least 8 h prior to each experiment. Sampling was done at a flow rate of ca. 18 L min–1 for at least 8 h (overnight sampling at the end of each experiment to collect all SOA present in the smog chamber). Filters were then wrapped in clean aluminum foil and kept at −20 °C until analysis. Filter blanks, chamber blanks, and experimental blanks were also collected routinely during the campaigns.
2.2.2. GC-MS Analysis
For gas chromatography–mass spectrometry (GC-MS) analysis, sections of the filters (1/4, 1/8, or 1/16, depending on the aerosol mass collected on the filters) were spiked with recovery standard (methyl O-l-xylanopyranoside; Sigma-Aldrich) and extracted three times with 10 mL of methanol for 5 min under ultrasonic agitation. The combined extracts were reduced with a rotary evaporator to ∼1 mL. Filtered concentrates (Teflon syringe filter, 0.45 μm) were completely dried under a gentle stream of N2. The trimethylsilylation reagent used was N,O-bis(trimethylsilyl)trifluoroacetamide containing 1% trimethylchlorosilane as a catalyst. A mixture of the reagent and pyridine (2/1; v/v) was added to the dried samples, and the mixtures were reacted at 70 °C for 1 h and left at room temperature overnight. An aliquot of 1 μL was analyzed in a system comprising a TRACE GC2000 gas chromatograph, which was coupled to a Polaris Q ion trap mass spectrometer equipped with an electron ionization (EI) source (Thermo Scientific, San Jose, CA). A Heliflex AT-5MS fused silica capillary column (5% phenyl, 95% methylpolysiloxane, 0.25 μm film thickness, 30 m × 0.25 mm internal diameter) preceded by a deactivated fused silica precolumn (2 m × 0.25 μm i.d.; Alltech, Deerfield, IL) was used to separate the derivatized extracts. Helium was used as carrier gas at a flow rate of 1.1 mL min–1. The temperature program was as follows: isothermal hold at 50 °C for 5 min, temperature ramp of 3 °C min–1 up to 200 °C, isothermal hold at 200 °C for 2 min, temperature ramp of 30 °C min–1 to 310 °C, and isothermal hold at 310 °C for 2 min. The analyses were performed in the full scan mode (mass range: m/z 50–650). The ion source was operated at electron energy of 70 eV and a temperature of 200 °C. The temperatures of the GC injector and the GC-MS transfer line were 250 and 280 °C, respectively.
The signals (peak areas) of the total ion chromatograms served as input data for the quantitative determinations. The calibration was done using glyceric acid as surrogate standard for 2-methylglyceric acid (2-MG); the amounts given for the 2-MG-related compounds are thus in 2-MG equivalents. For the quantification of the 2-methyltetrols, meso-erythritol was used as surrogate standard.
Additional samples were analyzed with GC-MS from isoprene photooxidation experiments already described elsewhere37,53 and whose experimental details are reported in Table S1 of the Supporting Information. Briefly, experiments were performed with both the triphasic (gas-particulate-cloud) protocol (described in Section 2.1) and a diphasic protocol (gas-cloud) in which the RH was increased and clouds were formed during the first stages of isoprene photooxidation (after 2 h of irradiation) prior to any SOA formation. Control experiments in dry conditions and blank experiments were also done. Samples were collected following the same procedure described in Section 2.2.1.
2.2.3. NanoESI-HRMS Analysis
2.2.3.1. Sample Preparation and Analysis
Filters were extracted in methanol (Optima LC/MS grade, Fisher Scientific) in slurry ice in an ultrasonic bath and then filtered according to the procedure described elsewhere.56 The volume of each extract was then adjusted to a concentration of ∼0.2 μg/μL of SOA by evaporating the solvent to ∼30–1300 μL (depending on the total mass of SOA collected) under a gentle flow of N2.
A high-resolution mass spectrometer (LTQ Velos Orbitrap, Thermo Scientific, Bremen, Germany) with a resolution of 100 000 at m/z 400 and a typical mass accuracy within ±2 ppm equipped with a chip-based nanoESI source (Triversa NanoMate Advion, Ithaca, NY) was used to analyze the extracts. Samples were sprayed at a gas (N2) pressure of 0.30 psi at 1.8 kV in positive ionization mode and 0.80 psi at −1.4 kV in negative ionization mode with a transfer capillary temperature of 210 °C. Data were acquired in the full scan mode in the m/z ranges of 50–600, 150–900, and 200–2000 (1 min acquisition each, corresponding to 36–38 scans). The instrument was calibrated routinely within an accuracy of ±2 ppm, using a Pierce LTQ Velos electrospray ionization (ESI) Positive Ion Calibration Solution and a Pierce ESI Negative Ion Calibration Solution (Thermo Scientific). Lock masses of the background ions C17H25O3S– (m/z 311.16864) in negative ionization and C12H26O7Na+ (m/z 305.15707), C14H30O8Na+ (m/z 349.18329), and C24H30O6Na+ (m/z 437.19346) in positive ionization were used for internal mass calibration.
2.2.3.2. Data Treatment
Molecular formulas were assigned using Xcalibur 2.2 (Thermo Scientific, Bremen, Germany) within a ±4 ppm error and under the following restrictions: number of 12C = 1–100 and 13C = 0–1, H = 1–200, O = 0–50, N = 0–5, 32S = 0–1 and 34S = 0–1 (only in negative mode), and Na = 0–1 (only in positive mode). For each peak, the 40 assignments with the lowest mass error were retained at this stage. Data were then filtered using a Mathematica 10 (Wolfram Research Inc., UK) code developed in-house and described in detail elsewhere.57 A mass shift tolerance was defined by comparing theoretical and experimental masses of 10–20 compounds, which are either known background contaminations in the mass spectra or known oxidation products from isoprene and methacrolein. Formula assignments were selected based on widely accepted rules for filtering molecular formulas from accurate mass measurements.58 Only peaks with intensities 10 times higher than in the blanks (both procedural and experimental blanks), O/C ≤ 2, 0.3 ≤ H/C ≤ 2.5, N/C ≤ 0.5, and S/C ≤ 0.2 were further considered in the data analysis. When several formulas satisfied all restrictions within a 2 ppm accuracy, the formula with the lowest mass error (corrected for the average mass shift calculated from the 10–20 known compounds) was selected. Multiple molecular assignments are often found for peaks above 500 Da within the instrumental accuracy causing the resulting molecular assignment selecting the formula with the lowest mass error to be misleading. For this reason, peaks above 500 Da were still included in the dataset but discussed only to examine the length of homologous oligomer series identified at lower masses, using the method described in Stenson et al.,59 and they are not discussed in detail otherwise.
2.2.3.3. MonomerHunter
Repeating monomeric units within oligomers were investigated using the program “MonomerHunter”60 available online (fgcz-m0n0merhunter.uzh.ch/cgi/main.pl). MonomerHunter takes a mass spectrum peak list as input and computes all mass differences ordered according to their frequency of occurrence. MonomerHunter was run on theoretical masses of the final peak lists of each high-resolution mass spectrometry (HRMS) sample on the entire mass range for peaks larger than 10% of the mean intensity, with a mass accuracy of 0.001 Da and for mass differences in the range of 1–320 Da. A molecular formula was assigned to each exact mass difference using the package “Rcdk” for R 3.2.5.
3. Results and Discussion
3.1. SOA Formation from Methacrolein Photooxidation in the Presence of Clouds
Brégonzio-Rozier et al.53 reports on all the results obtained under dry conditions in which SOA mass yields were around the lowest values reported in literature and comparable with experiments performed with natural light or light that simulates solar irradiation over UV–visible wavelengths. In contrast to what has been observed in isoprene experiments,37 in methacrolein experiments when RH was increased from 2−5% to more than 80% in less than a minute (without cloud formation) the SOA mass concentration sharply decreased (Figure 2a and Table S2). The decrease in mass concentration was accompanied by a shift of the size distribution toward smaller diameters (from ∼200 nm to ∼150 nm in mobility diameter), while the number concentration remains constant. At the same time, the concentration of VOCs increased sharply upon RH increase (Figure 3).
Figure 2.
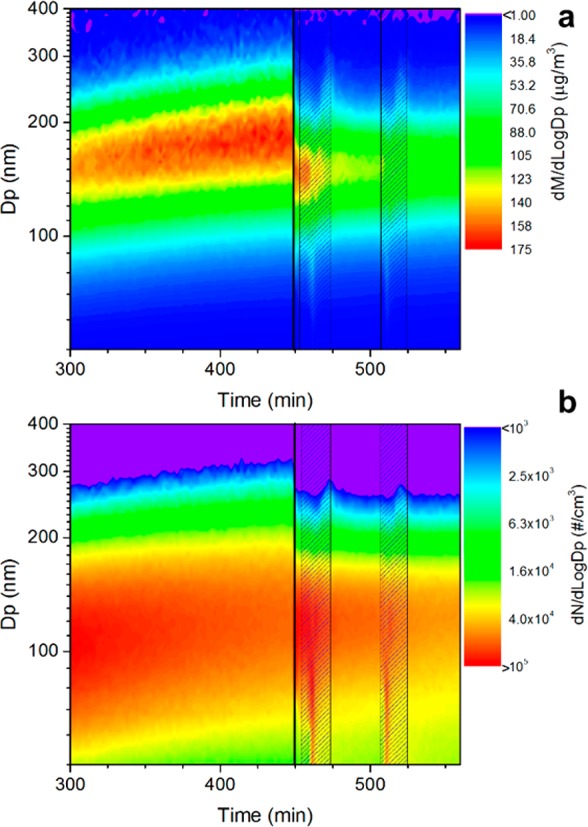
Particle mass (a) and number (b) size distributions for the experiment MT-18012013. Black line indicates time at which the RH was increased from 3 to 5% to >80%; hatched areas indicate cloud events.
Figure 3.
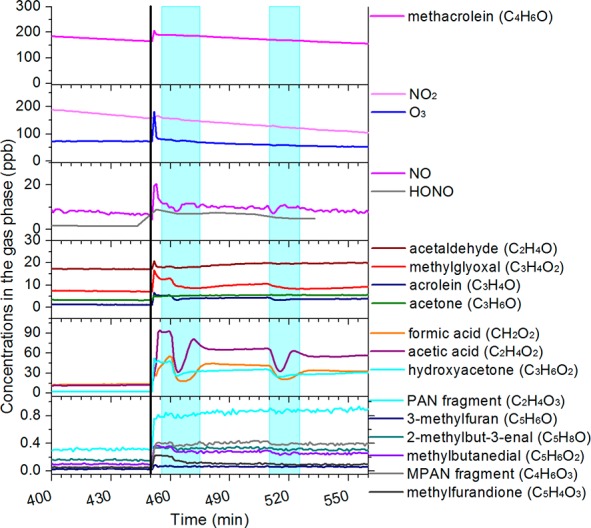
Time series of gaseous compounds measured with a PTR-TOF-MS, FTIR, and NOx, O3, and HONO analyzers for the experiment MT-18012013. Black line indicates time at which RH was increased from 2−5% to >80%; light blue areas indicate cloud events. Measurement uncertainty for HONO is 10%.
It was observed in a previous study that evaporative losses for fresh α-pinene and limonene SOA are larger and faster at high relative humidity due to the lower particle viscosity at high RH.61 Isoprene-derived SOA has a lower viscosity than α-pinene-derived SOA. In fact, isoprene SOA is semisolid at RH < 30%, while it is liquid at RH > 60%62 supporting the hypothesis that evaporative losses may occur on the particles when RH sharply increases from 25% to more than 80% as suggested also by the increase of VOC concentrations (Figure 3). In our experimental conditions, SOA is likely to be formed predominantly through oxidation of methacryloyl peroxynitrate (MPAN) forming 2-methylglyceric acid (2-MG) and its oligomers as observed in previous studies33,35,63,64 and confirmed by GC-MS measurements (see Section 3.3.1). Zhang et al.45 and Nguyen et al.15 observed that high RH suppresses 2-MG formation and oligomerization in the isoprene-NOx system. Nguyen et al.64 also observed that in an aerosol-droplet system acidity promotes re-evaporation of 2-MG to the gas phase. This also supports the hypothesis that evaporative losses may occur on the particles, and it may partly explain the low methacrolein-derived SOA measured in southeastern U.S. during the SOAS campaign.25,65
On the one hand, the increase of VOCs is even higher than the measured decrease in particle mass (Table S2), so gas-wall repartitioning of VOCs66 and higher wall losses of SOA at high RH may also play a role. On the other hand, the characteristic very fast water injection may also be responsible for the observed losses. During water injection, a transient cloud may form, which can activate particles into very large droplets. Those droplets exhibit a very short lifetime in the chamber (lifetime of particles in the 200 nm range is on the order of several days, but in the 40 μm range it is only a few minutes). These ongoing losses of SOA mass also affected observations throughout the experiment, including during cloud events.
During cloud events, a slower decrease in dried SOA mass and an increase in number of particles were observed, suggesting that fresh SOA is produced although partly hindered by the ongoing evaporative losses. Concurrently, the water-soluble VOC concentration decreased (blue zones in Figures 3 and 4) as observed for isoprene SOA, suggesting a repartitioning of water-soluble VOCs into the cloudwater droplets. This can also be observed in Figure S1, showing that the decrease in gaseous concentrations of VOCs is proportional to the Henry’s law constant, indicating partitioning into the cloud droplets. As previously observed for isoprene SOA,37 during cloud events a second mode at larger diameters (centered at ∼350 nm in vacuum aerodynamic diameter) forms (Figure 5b). This second mode has been associated with the “droplet” mode observed in the atmosphere.67−69 In our experiments, this second mode is overlapping with that formed in dry conditions (centered at ∼220 nm in vacuum aerodynamic diameter) and is composed of ammonium and nitrate as well as additional organic compounds (Figure 5b). SOA formed during cloud events is metastable in the smog chamber and gradually decreases in 20–30 min after cloud dissipation (Org-PC1 trace in Figure 4 and Figure 5) probably as a result of gas-particles-wall repartitioning of the SVOCs formed during the cloud event.37
Figure 4.
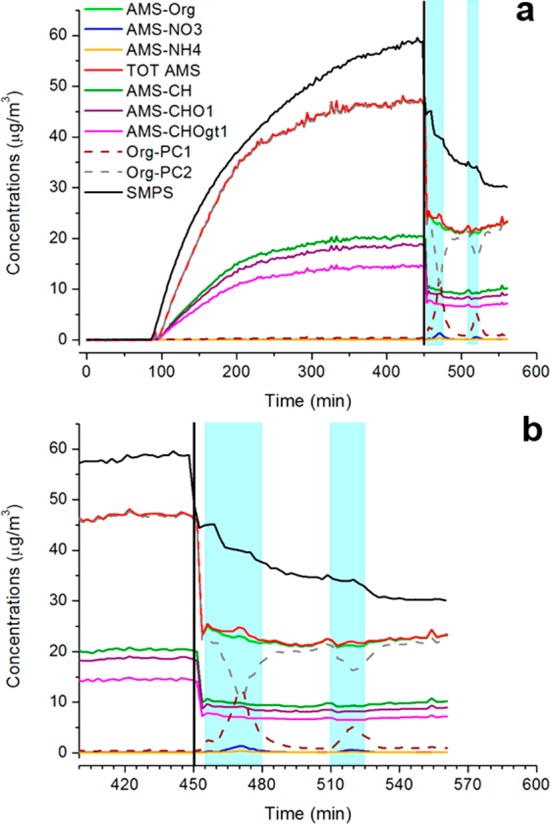
Time series of aerosol mass concentration measured with the SMPS and the AMS, aerosol components measured with the AMS and organic mass corrected for evaporative losses at high RH using PCA (a) and zoomed-in figure during cloud events (b). Example from experiment MT-18012013. AMS-Org stands for organics, AMS-CH stands for CH fragments, AMS-CHO1 stands for CHO fragments with only one oxygen atom, AMS-CHOgt1 stands for CHO fragments with more than one oxygen atom. Light blue areas indicate cloud events.
Figure 5.

SOA compositions and size distributions for the experiment MT-18012013 measured with an HR-TOF-AMS instrument at high RH before any cloud event (a), during the first cloud event (b), and after the first cloud event (c). (left) Normalized mass spectra of the organic components of dried aerosol. (right) Dried aerosol size distributions. Cx stands for C-containing fragments, CH stands for CH fragments, CHO1 stands for CHO fragments with only one oxygen atom, CHOgt1 stands for CHO fragments with more than one oxygen atom, CHN stands for CHN fragments, CHO1N stands for CHON fragments with only one oxygen atom, CHOgt1N stands for CHON fragments with more than one oxygen atom, HO stands for OH+, H2O+, H3O+ fragments and their isotopes.
Two approaches were used to calculate the production of transient SOA mass during cloud events (i.e., the mass of aqSOA formed). First, a bimodal log-normal fitting was performed to evaluate the contribution of the second mode formed during cloud events to the total aerosol mass measured. Results are compared in Table S3 and show a SOA production ranging between 2.2 and 33 μg m–3 with an associated yield ranging between 1.3 and 16 × 10–3.
Second, principal component analysis (PCA) was used to decompose the time series of organic compounds (HR-TOF-AMS measurements) into the contribution of two latent variables: PC 1, explaining the variance due to production of SOA during cloud events and PC 2, explaining the variance due to production of SOA in dry conditions and the evaporative losses at high RH (Figure 4). With this method, it is possible to extract the production of organic aerosol partly masked by the evaporative losses that range between 6 and 45 μg m–3 with an associated yield ranging between 2.9 and 26 × 10–3 (Table S3, Figure 4). The decrease of water-soluble VOCs from the gas phase is higher than both estimates, thus explaining the production of additional SOA mass (Table S3). However, both calculations are affected by large uncertainties and should be taken as indicative estimates rather than quantitative estimates.
3.2. Bulk Chemical Composition of SOA
The droplet mode contains ammonium and nitrate together with organic compounds as shown by HR-TOF-AMS measurements (Figure 5b). To determine the possible contribution of nitrogenous organic compounds to the measured concentrations of ammonium and nitrate, molar ratios of NH4+/NO3– and mass ratios of NO+/NO2+ fragments were measured and compared with those from different nitrate and nitrite salts and nitrogenous organic compounds. Results are shown in Table S4 of the Supporting Information.
From the molar NH4+/NO3– close to 1 and NO+/NO2+ fragments close to that expected for ammonium nitrate (Table S4) we can confirm that there is production of ammonium nitrate during cloud events. While formation of nitrate in a NOx-rich environment is expected, ammonium formation is much more surprising. The quantity formed is quite small and may be due to background contaminations of ammonia, as suggested previously for the smog chamber used in these experiments37 and measured in another smog chamber of comparable characteristics.70
Concerning organic aerosol, HR-TOF-AMS results show that its composition does not change significantly during cloud formation (Figures 5 and 6). Organic material is composed mainly of CHO compounds, while only one nitrogenous organic compound, the fragment C4H7N2O3+, was detected (Figure 5). This fragment has been previously reported for isoprene/NOx SOA,71 but it was not detected in GC-MS and HRMS measurements (Section 3.3), indicating that it may be a fragment of higher molecular-weight compounds.
Figure 6.
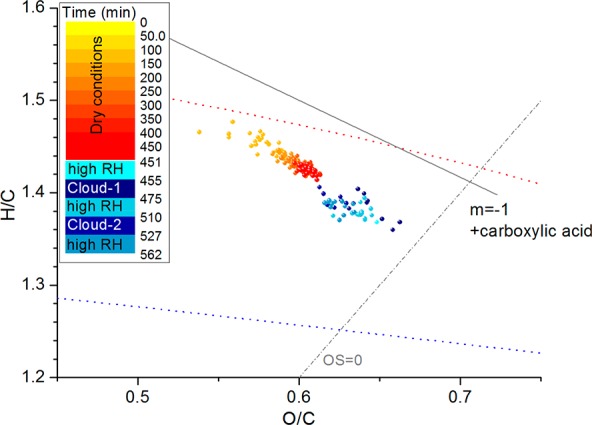
Van Krevelen diagram from HR-TOF-AMS measurements of SOA from the photooxidation of methacrolein. Example from the experiment MT-18012013. Red and blue dotted lines define the space in which ambient oxidized organic aerosol usually falls.72,73
The oxidation level of the organic compounds changes from dry conditions to humid conditions (high RH and cloud events). Figure 6 shows that, during SOA formation and aging in dry conditions, the oxidation level increases following the slope of formation of organic acids in the van Krevelen diagram. Increasing RH seems to accelerate the oxidation, as the time scale of SOA processing in high RH conditions is significantly smaller than in dry conditions (i.e., 10–15 min cloud event vs 450 min in dry conditions), but the change in O/C is comparable. However, no significant differences can be observed between high RH conditions and cloud events. Conversely, Figure 4 shows that concentrations of CHO fragments with more than one oxygen atom (i.e., AMS-CHOgt1 trace in Figure 4) decrease less than the other compound classes when the RH is raised, partially explaining the higher O/C in high humidity conditions (Figure 6) and a change in density (of dried aerosol) from 1.39 ± 0.01 g cm–3 in dry conditions to 1.42 ± 0.01 g cm–3 in humid conditions. Particle organic composition does not change during and after cloud events (RH > 90%; Figure 5b,c), which can be explained by the fact that (i) SOA formed during clouds has a similar elemental composition to SOA formed in humid conditions and (ii) SOA produced during clouds is characterized by semivolatile compounds, which re-evaporate gradually after cloud dissipation resulting in SOA with a similar composition and size distribution as before the cloud events, as previously observed by Brégonzio-Rozier et al.37 for isoprene photooxidation experiments.
3.3. SOA Composition at Molecular Level
3.3.1. Molecular Composition of SOA and Behavior of Isoprene SOA Markers
GC-MS measurements were done for SOA formed from the photooxidation of both methacrolein and isoprene37,53 in dry, control experiments (methacrolein, isoprene), triphasic gas-particles-cloud experiments (methacrolein, isoprene) and diphasic gas-cloud experiments (isoprene only).
Results show that, in all experiments, SOA mass is dominated by a relatively small number of compounds that make up to ∼50% of the total aerosol mass (Figure 7). The most abundant compound is 2-MG, followed by its dimer (2-MG-dimer). A linear and a branched trimer of 2-MG were also detected in all experiments. In the control and triphasic experiments with isoprene as the precursor, small amounts of 2-methyltetrols were also detected. These structural elucidations were based on a previous MS characterization study by Szmigielski et al.74 Three other compounds previously unreported were also detected: 2-methylglyceraldehyde (2-MG-aldehyde), a dimer of both 2-MG and 2-MG-aldehyde, and a compound formed from the esterification of 2-MG with ethylene glycol (2-MG-EGE), although the latter was not detected in all experimental repeats. For all these compounds, authentic standards were not available. The concentrations reported for 2-MG oligomers are likely to represent a lower limit, which does not take into account possible thermal degradation in the GC inlet and hydrolysis of 2-MG oligomers during trimethylsilylation prior to GC-MS analysis.
Figure 7.
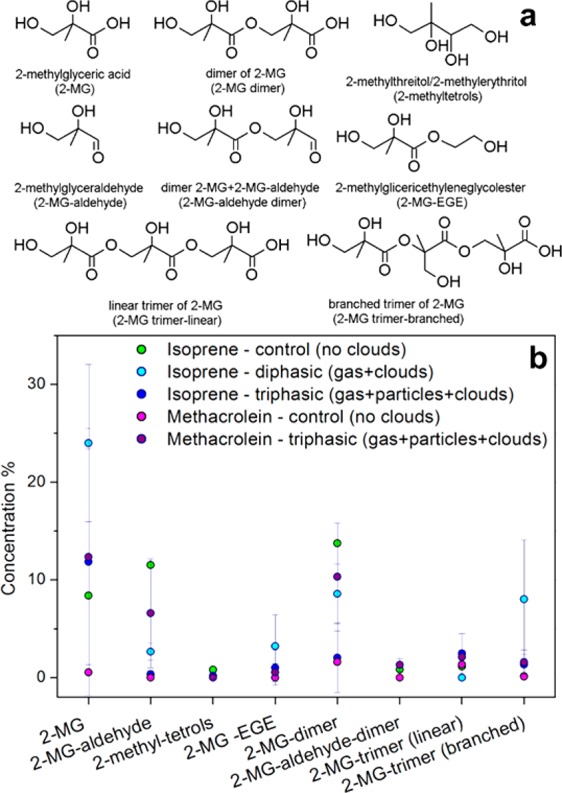
Structures of the compounds detected in GC-MS (a) and their relative abundance in mass (b) in SOA formed from the photooxidation of isoprene and methacrolein in different types of experiments (i.e., control, diphasic, and triphasic experiments). Error bars show standard deviation of experimental repetitions (2–4 repeats) in the same conditions.
Nevertheless, in general GC-MS results show no significant quantitative differences in composition between isoprene control, isoprene diphasic, isoprene triphasic, methacrolein control, and methacrolein triphasic experiments, taking into account the variability of the experimental repeats. This result supports the hypothesis that during cloud events, metastable aqSOA is formed from dissolution of water-soluble VOCs onto cloud droplets via reversible reactions, with no significant change in aerosol composition after cloud evaporation. However, in the ambient atmosphere aerosol composition is more complex, and the presence of inorganic components can drastically influence the solubility and the reactivity of the organic compounds in the aqueous phase. For example, it was observed that methacrolein-derived SOA was suppressed in the presence of acidic sulfate seeds in southeastern U.S.25,65
Our results also highlight the importance of 2-methylglyceric acid and related compounds in isoprene-derived SOA. In field measurements, 2-methylglyceric acid, together with 2-methyltetrols, is often used as a tracer for isoprene SOA.24,75,76 For example, Kleindienst et al.76 used laboratory-based mass fractions of tracers to estimate the contribution of different precursors to SOA in a U.S. location. However, Hu et al.75 found that the NOx level and RH were not sufficient to explain changes in the ratio of 2-methylglyceric acid to 2-methyltetrols in aerosol of land, ocean, and Antarctic origin, suggesting that temperature may play a pivotal role. Our study shows that oligomerization of 2-methylglyceric acid is very important, with high concentrations of dimers and trimers, but also co-oligomers, reaching up to 15–20% of SOA mass under NOx conditions, so that this process should be taken into account in the ambient atmosphere. Such a mechanism is likely to become increasingly important in a warmer atmosphere, which enhances emissions of biogenic VOCs and oligomer formation, as observed recently by Kourtchev et al.41 Complementary analyses revealed the presence of longer oligomers and provided more detailed information on the differences among different experimental conditions as discussed below.
3.3.2. Oligomerization in Multiphase and in Aqueous-Phase Experiments
Samples from the photooxidation of methacrolein in control and triphasic experiments were also analyzed with nanoESI-HRMS, and results are compared here with a triphasic experiment performed at ∼60% RH from the start of the reaction and also with aqueous-phase photooxidation experiments of methacrolein(aq) by ·OH(aq) (without NOx). Details of the experiments done in the aqueous photoreactor are discussed in the Supporting Information.
In control experiments of methacrolein photooxidation in dry conditions, 1016 CHO compounds were detected, while in triphasic experiments, only 759 CHO compounds were detected. On the one hand, the detection of a smaller number of compounds in triphasic experiments is likely caused by evaporative losses at high RH. On the other hand, in triphasic experiments performed at ∼60% RH from the start of the reaction, 1573 CHO compounds were detected, more than in the control and triphasic experiments started in dry conditions, suggesting that long-term oxidation in humid conditions favors the formation of a larger variety of organic compounds and affects SOA composition more than two relatively short cloud events. The number of organic compounds produced in aqueous-phase experiments (in NOx-free conditions) was 483 after a reaction time of 15 min and increased to 831 after a reaction time of ∼3 h, illustrating how the chemical system becomes increasingly complex with time. Unexpectedly in triphasic experiments, SOA seems to be less oxidized than in control experiments (Figure 8), while HR-TOF-AMS results suggest the opposite (Figure 6). This can be explained by the fact that the HR-TOF-AMS instrument measures the bulk composition of the aerosol quantitatively, while direct infusion in nanoESI-HRMS measures the composition of the aerosol qualitatively at the molecular level. Conversely, we can exclude that this difference is due to sampling/extraction/collection artifacts. It is noted that the HR-TOF-AMS technique has a lower collection efficiency for oxidized compounds compared to non-oxidized compounds,77 while methanol used for filter extraction provides a better extraction efficiency for oxidized compounds so that instrumental artifacts would lead to the opposite result. In addition, GC-MS results reveal that the SOA mass is dominated by a rather small number of compounds, which are likely to give a strong contribution to the bulk oxidation level measured by the HR-TOF-AMS technique.
Figure 8.
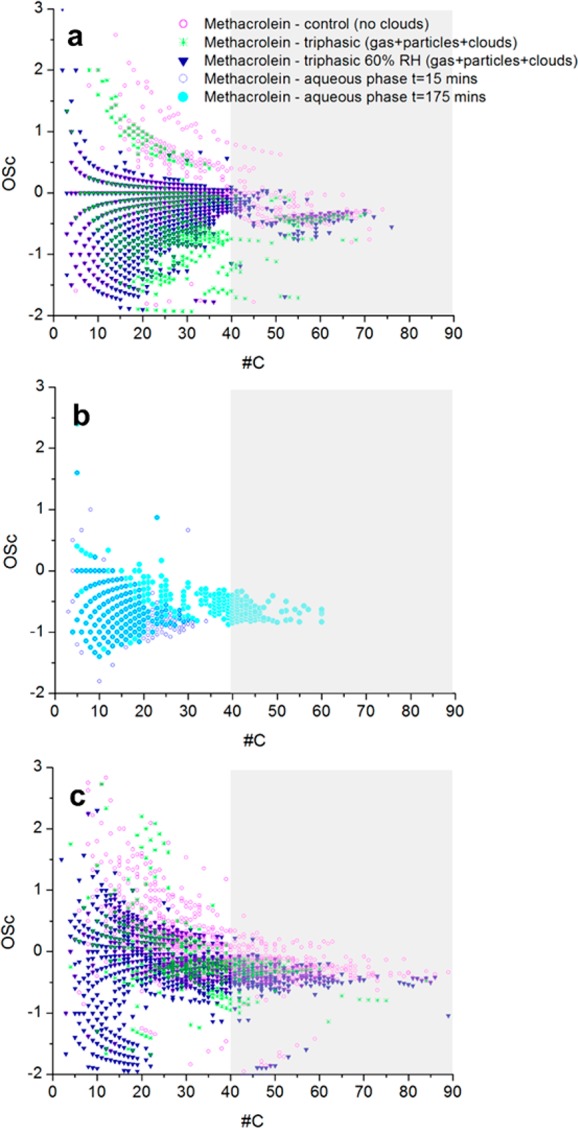
Carbon oxidation state plot of CHO compounds in smog-chamber experiments (a), aqueous-phase experiments (b), and CHNO compounds in smog-chamber experiments (c). Smog-chamber experiments were from the photooxidation of methacrolein in the presence of NOx, and aqueous-phase experiments were from the photooxidation of methacrolein in NOx-free conditions. The gray area indicates the part of the plot showing data more affected by uncertainty in formula assignments, where a unique formula assignment with a resolving power of 100 000 is not possible.
Figure 8 also illustrates that aqueous-phase experiments (Figure 8b) show a markedly different qualitative composition compared to smog chamber experiments (Figure 8a), albeit more similar to the triphasic experiments. These differences were investigated in more depth in terms of composition of the oligomers produced in the different experimental conditions.
HRMS data of SOA in control experiments, triphasic experiments, triphasic experiments at high RH, and aqueous photooxidation experiments were analyzed with MonomerHunter to detect the most common recurrent mass differences (i.e., monomers) between two peaks in the mass spectra. The MonomerHunter results are simply calculated mathematical monomers in terms of exact mass differences between two peaks. It is therefore an upper-limit estimate of the total number of monomers, because some calculated “monomers” are not actual monomers in a chemical sense. Results are shown in Figures 9 and 10 and Tables S5 and S6. Very long homologous series of peaks, with mass ranging up to ∼1500 Da with three or more members and separated by the same repeating unit (“monomer”) were detected in all types of experiments, as observed in previous studies of isoprene photooxidation,15,16 isoprene ozonolysis in a smog chamber,78 and isoprene, methacrolein, and methyl vinyl ketone bulk aqueous photooxidation.48−50,79,80 Formation of such long oligomers can be explained by high concentrations of VOC precursor used in these experiments. As shown by Kourtchev et al.,41 the formation of oligomers is correlated with the initial precursor concentration (excluding a strong influence from instrumental artifacts and) confirmed by liquid chromatography mass spectrometry (LC-MS) measurements of monoterpene SOA.56
Figure 9.
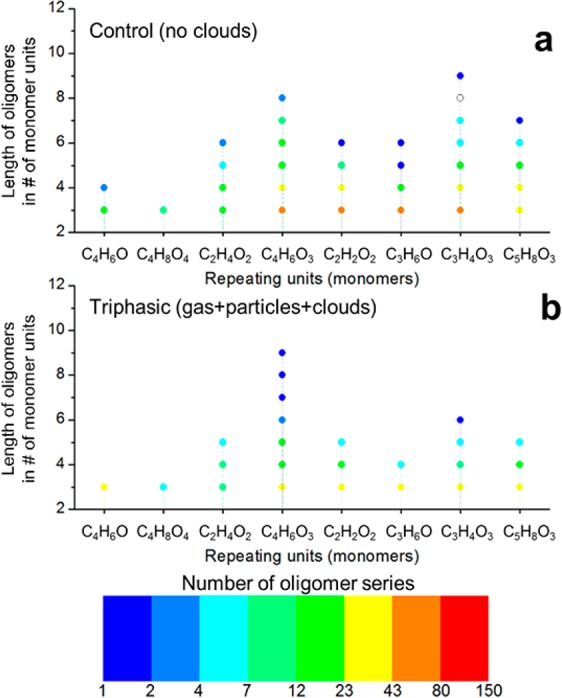
Trimers and longer oligomers (CHO compounds only) detected in methacrolein photooxidation experiments in the smog chamber in control conditions without clouds (a) and in triphasic conditions with two cloud events (b). Molecular formulas of the repeating units can be associated with (left to right) methacrolein (C4H6O), 2-methylglyceric acid (C4H8O4), glycolaldehyde (C2H4O2), methylglyceric acid (C4H6O3 because of H2O loss), hydroxyacetic acid (C2H2O2), acetone (C3H6O), pyruvic acid/hydrolyzed methylglyoxal (C3H4O3), and 2-hydroxy-2-methylbutanedial (C5H8O3).
Figure 10.
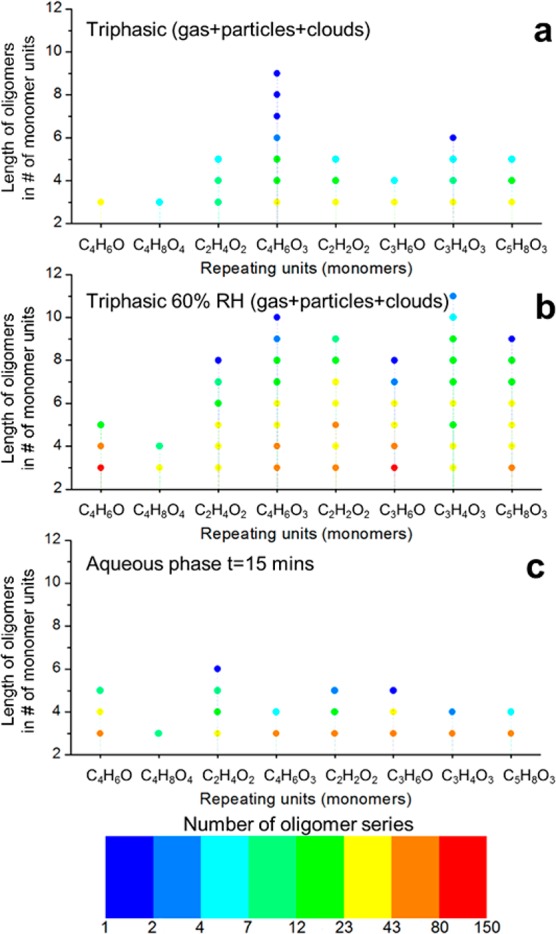
Trimers and longer oligomers (CHO compounds only) detected in methacrolein photooxidation experiments in the smog chamber in triphasic conditions with two cloud events (a), in triphasic conditions starting at 60% RH with two cloud events (b), and methacrolein photooxidation in NOx-free conditions in the aqueous phase with 15 min of reaction time (c). Molecular formulas of the repeating units can be associated with (left to right) methacrolein (C4H6O), 2-methylglyceric acid (C4H8O4), glycolaldehyde (C2H4O2), methylglyceric acid (C4H6O3 because of H2O loss), hydroxyacetic acid (C2H2O2), acetone (C3H6O), pyruvic acid/hydrolyzed methylglyoxal (C3H4O3), and 2-hydroxy-2-methylbutanedial (C5H8O3).
The most important repeating units correspond to 2-methylglyceric acid (C4H8O4 and C4H6O3), acetaldehyde (C2H4O), glycolaldehyde (C2H4O2 and C2H2O), hydroxyacetone (C3H4O), methyglyoxal/lactic acid/acrylic acid (C3H4O2), and pyruvic acid (C3H4O3). Other common but small repeating units are methyl, oxygen, water, and formaldehyde. The extremely large number of pairs that they connect, up to ∼700 pairs (Tables S5 and S6), point out the complexity of aerosol composition characterized not only by homologous series but by a large co-oligomerized system containing monomers of different structures, also observed by Renard et al.81 in the aqueous-phase oligomerization of mixtures of α,β-unsaturated carbonyls.
A lower number of oligomer series and shorter series (with a smaller number of repeating units) were observed in triphasic experiments starting from dry conditions compared with control experiments probably due to hydrolysis during cloud events (Figure 9). However, triphasic experiments starting at 60% RH were characterized by many more and longer oligomers, up to 120 oligomers with more than 4 monomer units and up to 11 monomer units (Figure 10).
A markedly different reactivity was observed for oligomers produced in dry conditions, multiphase conditions starting at 60% RH, and in the aqueous phase. As shown in Figure 10, 2-methylglyceric acid is the most recurrent monomer in SOA formed in experiments starting in dry conditions, and its oligomers are likely formed predominantly by esterification (repeating unit C4H6O3 from loss of a water molecule). Conversely, methacrolein (C4H6O) and acetone (C2H6O) are important monomers in aqueous-phase oxidation, while they do not lead to a long homologous series in the control and triphasic smog chamber experiments. This could be due to their higher volatility and low solubility and therefore scarce repartitioning into water droplets, as shown in Figure 3. The mechanism of oligomerization for methyl vinyl ketone in the aqueous phase has been shown to proceed via radical initiation and propagation;48,50,82 hence, O2 dissolved in the water droplets could also have an effect in smog chamber conditions. In this respect, it has been shown in a previous study that dissolved O2 inhibits radical oligomerization,48 and water droplets in atmospheric conditions are likely to be saturated with O2 assuming partitioning equilibrium.49 In addition, oxidation in the condensed phase is limited by the uptake of ·OH from the gas phase.83
In smog-chamber experiments, 2-hydroxy-2-methylbutanedial (C5H8O3)16 is another important monomer making up to 96 (>4 units) homologous series with up to 9 monomer units, producing hemiacetals through addition chemistry,16 while it is not as important in aqueous-phase oxidation likely due to hydrolysis of hemiacetals. However, glycolaldehyde, which is expected to react via a similar mechanism,16 is an important monomer in all tested conditions.
Results for the triphasic experiments at ∼60% RH show that SOA presents characteristic homologous series of all three control, triphasic, and aqueous-phase experiments. This suggests that a long-term oxidation at high RH (7–8 h) affects SOA composition to a higher degree than a couple of relatively short cloud events (∼15 min). In fact, at RH > 60% isoprene SOA has a viscosity of a liquid, therefore creating the conditions for reactions in the condensed phase (wet aerosol) to occur.62,84 This is supported by aqueous-phase experiments performed for a longer reaction time, where we have observed a wider variety of different and longer oligomers compared to experiments performed with a shorter reaction time (Figure S3). In general, on the one hand, SOA produced at high RH is characterized by many different and longer oligomers compared to experiments started in dry conditions in contrast to what was observed in previous experiments by Nguyen et al.15 On the other hand, Rodigast et al.46 observed a dependence of oligomer formation from methylglyoxal not only on RH but also on pH of seed particles, which may partly explain our results, as methylglyoxal is one of the reaction products.
In addition, the aerosol liquid water content (ALW) at 60% RH in our experiments (before RH increase to 80%) was ∼4–5 μg m–3 as estimated using f44 (from HR-TOF-AMS data)85 and the calculation described in Guo et al.86 This value is in line with an average ALW of 3.2 μg m–3 in rural sites, reported by Nguyen et al.87 With such an ALW, it is sufficient to dissolve 1/1000 of the organic aerosol matter to obtain concentrations in the water layer of ∼1.5–3 × 103 mgC L–1 that are comparable with the methacrolein concentrations used in the aqueous-phase experiments (see Section S1.3). Such a partitioning between organic and aqueous phase in the aerosol seems quite easy to get to. Our results suggest that such ALW content is sufficient to promote aqueous-phase oligomerization in addition to the gas-phase mechanism, if the reaction time is comparable (long-term reaction at high RH rather than two relatively short cloud events).
Concerning organonitrogen compounds, the photooxidation of methacrolein produced 2527, 1147, and 1806 CHNO compounds in dry control experiments, triphasic experiments, and triphasic experiments performed at ∼60% RH from the start, which is consistent with the results discussed above. The majority of monomers extracted with MonomerHunter do not contain nitrogen. Only two mass differences corresponding to nitrogenated compounds were found in the top 50 monomers for CHNO compounds: CHNO2 (likely to be too small to be considered an oligomer building block) and C5H7NO5, which was previously observed in SOA from isoprene photooxidation in the presence of NOx.88 N-Containing oligomers are therefore characterized by a N-containing precursor to which a CxHyOz repeating unit is added (Table S6 and Figure S2). Those oligomers show a similar distribution between different experimental conditions as observed for CHO compounds. The observation of many organonitrogen compounds with nanoESI-HRMS is not in contradiction with HR-TOF-AMS data showing the presence of only one fragment (C4H7N2O3+) containing nitrogen: the former instrument provides qualitative information on the molecular composition of aerosol, while the latter provides quantitative information but uses a hard ionization technique (i.e., EI) that heavily fragments high molecular-weight compounds.
4. Conclusions
In the present study, we have investigated the impact of cloud events on SOA formation from the photooxidation of methacrolein in the presence of NOx and the effect of this phenomenon on SOA composition both in the short term (during cloud events) and in the long term (after cloud dissipation).
We have found that a single and relatively short cloud event triggers immediate formation of aerosol although partly masked by fast evaporative losses. These losses may be caused by hydrolysis of oligomers and re-evaporation of semivolatile compounds, which are enhanced at high RH and acidic pH.45,64 Formation of aqSOA is characterized by the appearance of a second mode at larger diameters, associated with the “droplet mode” previously observed in the atmosphere67−69 and in isoprene SOA experiments conducted in the same conditions.37 The droplet mode is metastable in the simulation chamber, probably as an effect of gas-particles-wall repartitioning of semivolatile compounds.
In all experimental conditions, aerosol mass is dominated by a rather small number of compounds, including 2-methylglyceric acid and its dimers and trimers. Concerning these compounds, no significant changes have been observed between different types of experiments (control vs cloud experiments) and between the two different studied precursors (isoprene vs methacrolein). However, dimers and trimers of 2-methylglyceric acid can make up to 15–20% of the total SOA mass, and therefore the reactivity of this isoprene SOA tracer needs to be taken into account in studies inferring the contribution of isoprene to ambient aerosol.
Detailed chemical characterization at the molecular level reveals a large number of long homologous series of oligomers in all experiments together with a complex co-oligomerized system consisting of monomers with a large variety of different structures. Some compositional changes were observed between control experiments and triphasic (gas-particle-clouds) experiments that are attributed to evaporative losses observed at high RH as well as hydrolysis of oligomers. However, in triphasic experiments performed at high RH for 7–8 h SOA composition is characterized by a wide variety of different and longer oligomers compared to experiments started in dry conditions. On the one hand, this is in contrast to what was observed in previous studies by Zhang et al.45 and by Nguyen et al.15 On the other hand, Rodigast et al.46 observed a dependence of oligomer formation from methylglyoxal not only on RH but also on pH of seed particles, which may partly explain our results, as methylglyoxal is one of the reaction products.
As expected, comparison of oligomer series in SOA from multiphasic (smog-chamber) experiments in the presence of NOx and samples from aqueous-phase oxidation of methacrolein with ·OH radical in NOx-free conditions pointed out different types of oligomerization mechanisms dominating the two different systems. In multiphasic experiments started in dry conditions, reversible reactions, such as esterification and hemiacetal formation, seem to be the dominant processes, while other oligomerization processes occur in bulk aqueous-phase oxidation of methacrolein (Monod et al., in preparation). Aqueous-phase experiments were conducted on oxidation of methacrolein dissolved in water by ·OH produced in situ. In a multiphase environment, as in the smog chamber or in the ambient air, aqueous oxidation is limited by uptake of reagents in the water droplets, which is rather limited in the case of methacrolein89 and especially ·OH.11 However, SOA produced in triphasic experiments starting from 60% RH is characterized by oligomer series observed in both triphasic experiments starting in dry conditions and in aqueous-phase experiments. Previous studies suggested that aqueous chemical processes occur in adsorbed water layers as well as in deliquescent aerosol90 and that isoprene SOA is liquid at 60% RH.62,84 Therefore, long reaction times at high RH create the conditions for more pronounced aqueous-phase processing to occur compared to two relatively short cloud events.
In ambient conditions, the presence of more chemically heterogeneous systems and highly internally and externally mixed particles may dramatically change the reactivity compared with a single-precursor, yet complex, system. The presence of other species (such as photosensitizers, water-soluble inorganic species, and transition metals) as well as different pHs and temperatures are likely to have a significant impact on SOA formation and its composition in the aqueous phase.7,90 For example, it was observed that methacrolein-derived SOA was suppressed in the presence of acidic sulfate seeds in southeastern U.S.25,65 Fundamental laboratory work is necessary to develop the essential framework for understanding the mechanisms of oligomer formation. However, our study points out the need to conduct more experiments with complex chemical systems in a dynamic multiphase environment that can realistically reproduce the complexity of the gas-particle-droplet interaction in atmospheric conditions, as heterogeneous processes, specific to multiphase chemistry (e.g., at the air/water interface) may be important for aerosol properties in the atmosphere.91,92
Acknowledgments
This work was funded by the French National Agency for Research (ANR) through the project CUMULUS ANR-2010-BLAN-617-01. HRMS analysis at the Univ. of Cambridge was supported by the European Research Council (ERC; starting Grant No. 279405). Authors thank the MASSALYA instrumental platform (Aix-Marseille Univ.; lce.univ-amu.fr) for analysis and measurements done during the experiments and Dr. O. Popoola (Univ. of Cambridge) for help with coding required for HRMS data analysis. The authors wish to thank CNRS-INSU for supporting CESAM as a National Facility (http://cesam.cnrs.fr). This work was also supported by the European Commission through the EUROCHAMP-2 project (Contract No. 228335) and through the EUROCHAMP-2020 Integrated Activity (Contract No. 730997). Authors are grateful to Dr. G. Salque-Moreton, K. Buyukispir, C. Andreucci, H. Louati, and H. Louati for their contribution to the aqueous-phase experiments, Dr. F. Siekmann, and M. Della Giustina for help with treatment of HR-TOF-AMS data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpca.7b05933.
Details of isoprene experiments, principal component analysis, aqueous phase experiments, evaporative losses at high RH and SOA production during cloud events, additional tabulated results, including three figures (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of The Journal of Physical Chemistry virtual special issue “Veronica Vaida Festschrift”.
Supplementary Material
References
- Roth A.; Schneider J.; Klimach T.; Mertes S.; Van Pinxteren D.; Herrmann H.; Borrmann S. Aerosol Properties, Source Identification, and Cloud Processing in Orographic Clouds Measured by Single Particle Mass Spectrometry on a Central European Mountain Site during HCCT-2010. Atmos. Chem. Phys. 2016, 16 (2), 505–524. 10.5194/acp-16-505-2016. [DOI] [Google Scholar]
- Sareen N.; Carlton A. G.; Surratt J. D.; Gold A.; Lee B.; Lopez-Hilfiker F. D.; Mohr C.; Thornton J. A.; Zhang Z.; Lim Y. B.; et al. Identifying Precursors and Aqueous Organic Aerosol Formation Pathways during the SOAS Campaign. Atmos. Chem. Phys. Discuss. 2016, 1–42. 10.5194/acp-2016-200. [DOI] [Google Scholar]
- Hodas N.; Sullivan A. P.; Skog K.; Keutsch F. N.; Collett J. L.; Decesari S.; Facchini M. C.; Carlton A. G.; Laaksonen A.; Turpin B. J. Aerosol Liquid Water Driven by Anthropogenic Nitrate: Implications for Lifetimes of Water-Soluble Organic Gases and Potential for Secondary Organic Aerosol Formation. Environ. Sci. Technol. 2014, 48 (19), 11127–11136. 10.1021/es5025096. [DOI] [PubMed] [Google Scholar]
- Sandrini S.; Van Pinxteren D.; Giulianelli L.; Herrmann H.; Poulain L.; Facchini M. C.; Gilardoni S.; Rinaldi M.; Paglione M.; Turpin B. J.; et al. Size-Resolved Aerosol Composition at an Urban and a Rural Site in the Po Valley in Summertime: Implications for Secondary Aerosol Formation. Atmos. Chem. Phys. 2016, 16 (17), 10879–10897. 10.5194/acp-16-10879-2016. [DOI] [Google Scholar]
- Sullivan A. P.; Hodas N.; Turpin B. J.; Skog K.; Keutsch F. N.; Gilardoni S.; Paglione M.; Rinaldi M.; Decesari S.; Facchini M. C.; et al. Evidence for Ambient Dark Aqueous SOA Formation in the Po Valley, Italy. Atmos. Chem. Phys. 2016, 16 (13), 8095–8108. 10.5194/acp-16-8095-2016. [DOI] [Google Scholar]
- Hoyle C. R.; Fuchs C.; Järvinen E.; Saathoff H.; Dias A.; El Haddad I.; Gysel M.; Coburn S. C.; Tröstl J.; Bernhammer A.-K.; et al. Aqueous Phase Oxidation of Sulphur Dioxide by Ozone in Cloud Droplets. Atmos. Chem. Phys. 2016, 16 (3), 1693–1712. 10.5194/acp-16-1693-2016. [DOI] [Google Scholar]
- Herrmann H.; Schaefer T.; Tilgner A.; Styler S. A.; Weller C.; Teich M.; Otto T. Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase. Chem. Rev. 2015, 115 (10), 4259–4334. 10.1021/cr500447k. [DOI] [PubMed] [Google Scholar]
- Sorooshian A.; Lu M. L.; Brechtel F. J.; Jonsson H.; Feingold G.; Flagan R. C.; Seinfeld J. H. On the Source of Organic Acid Aerosol Layers above Clouds. Environ. Sci. Technol. 2007, 41 (13), 4647–4654. 10.1021/es0630442. [DOI] [PubMed] [Google Scholar]
- Carlton A. G.; Turpin B. J. Particle Partitioning Potential of Organic Compounds Is Highest in the Eastern US and Driven by Anthropogenic Water. Atmos. Chem. Phys. 2013, 13 (20), 10203–10214. 10.5194/acp-13-10203-2013. [DOI] [Google Scholar]
- Ervens B. Modeling the Processing of Aerosol and Trace Gases in Clouds and Fogs. Chem. Rev. 2015, 115 (10), 4157–4198. 10.1021/cr5005887. [DOI] [PubMed] [Google Scholar]
- Ervens B.; Turpin B. J.; Weber R. J. Secondary Organic Aerosol Formation in Cloud Droplets and Aqueous Particles (aqSOA): A Review of Laboratory, Field and Model Studies. Atmos. Chem. Phys. 2011, 11 (21), 11069–11102. 10.5194/acp-11-11069-2011. [DOI] [Google Scholar]
- Nel A. Toxic Potential of Materials at the Nanolevel. Science (Washington, DC, U. S.) 2006, 311 (5761), 622–627. 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- Hallquist M.; Wenger J. C.; Baltensperger U.; Rudich Y.; Simpson D.; Claeys M.; Dommen J.; Donahue N. M.; George C.; Goldstein A. H.; et al. The Formation, Properties and Impact of Secondary Organic Aerosol: Current and Emerging Issues. Atmos. Chem. Phys. 2009, 9 (14), 5155–5236. 10.5194/acp-9-5155-2009. [DOI] [Google Scholar]
- Carlton A. G.; Wiedinmyer C.; Kroll J. H. A Review of Secondary Organic Aerosol (SOA) Formation from Isoprene. Atmos. Chem. Phys. 2009, 9 (14), 4987–5005. 10.5194/acp-9-4987-2009. [DOI] [Google Scholar]
- Nguyen T. B.; Roach P. J.; Laskin J.; Laskin A.; Nizkorodov S. A. Effect of Humidity on the Composition of Isoprene Photooxidation Secondary Organic Aerosol. Atmos. Chem. Phys. 2011, 11 (14), 6931–6944. 10.5194/acp-11-6931-2011. [DOI] [Google Scholar]
- Nguyen T. B.; Laskin J.; Laskin A.; Nizkorodov S. A. Nitrogen-Containing Organic Compounds and Oligomers in Secondary Organic Aerosol Formed by Photooxidation of Isoprene. Environ. Sci. Technol. 2011, 45 (16), 6908–6918. 10.1021/es201611n. [DOI] [PubMed] [Google Scholar]
- Dommen J.; Metzger A.; Duplissy J.; Kalberer M.; Alfarra M. R.; Gascho A.; Weingartner E.; Prévôt A. S. H.; Verheggen B.; Baltensperger U. Laboratory Observation of Oligomers in the Aerosol from isoprene/NOX Photooxidation. Geophys. Res. Lett. 2006, 33 (13), L13805. 10.1029/2006GL026523. [DOI] [Google Scholar]
- Surratt J. D.; Murphy S. M.; Kroll J. H.; Ng N. L.; Hildebrandt L.; Sorooshian A.; Szmigielski R.; Vermeylen R.; Maenhaut W.; Claeys M.; et al. Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene. J. Phys. Chem. A 2006, 110 (31), 9665–9690. 10.1021/jp061734m. [DOI] [PubMed] [Google Scholar]
- Edney E. O.; Kleindienst T. E.; Jaoui M.; Lewandowski M.; Offenberg J. H.; Wang W.; Claeys M. Formation of 2-Methyl Tetrols and 2-Methylglyceric Acid in Secondary Organic Aerosol from Laboratory Irradiated isoprene/NOX/SO2/air Mixtures and Their Detection in Ambient PM2.5 Samples Collected in the Eastern United States. Atmos. Environ. 2005, 39 (29), 5281–5289. 10.1016/j.atmosenv.2005.05.031. [DOI] [Google Scholar]
- Claeys M.; Graham B.; Vas G.; Wang W.; Vermeylen R.; Pashynska V.; Cafmeyer J.; Guyon P.; Andreae M. O.; Artaxo P.; et al. Formation of Secondary Organic Aerosols through Photooxidation of Isoprene. Science 2004, 303 (5661), 1173–1176. 10.1126/science.1092805. [DOI] [PubMed] [Google Scholar]
- Taraborrelli D.; Lawrence M. G.; Crowley J. N.; Dillon T. J.; Gromov S.; Groß C. B. M.; Vereecken L.; Lelieveld J. Hydroxyl Radical Buffered by Isoprene Oxidation over Tropical Forests. Nat. Geosci. 2012, 5 (3), 190–193. 10.1038/ngeo1405. [DOI] [Google Scholar]
- Kiendler-Scharr A.; Andres S.; Bachner M.; Behnke K.; Broch S.; Hofzumahaus A.; Holland F.; Kleist E.; Mentel T. F.; Rubach F.; et al. Isoprene in Poplar Emissions: Effects on New Particle Formation and OH Concentrations. Atmos. Chem. Phys. 2012, 12 (2), 1021–1030. 10.5194/acp-12-1021-2012. [DOI] [Google Scholar]
- Hatch L. E.; Creamean J. M.; Ault A. P.; Surratt J. D.; Chan M. N.; Seinfeld J. H.; Edgerton E. S.; Su Y.; Prather K. A. Measurements of Isoprene-Derived Organosulfates in Ambient Aerosols by Aerosol Time-of-Flight Mass Spectrometry-Part 2: Temporal Variability and Formation Mechanisms. Environ. Sci. Technol. 2011, 45 (20), 8648–8655. 10.1021/es2011836. [DOI] [PubMed] [Google Scholar]
- Fu P.; Kawamura K.; Chen J.; Barrie L. A. Isoprene, Monoterpene, and Sesquiterpene Oxidation Products in the High Arctic Aerosols during Late Winter to Early Summer. Environ. Sci. Technol. 2009, 43 (11), 4022–4028. 10.1021/es803669a. [DOI] [PubMed] [Google Scholar]
- Budisulistiorini S. H.; Li X.; Bairai S. T.; Renfro J.; Liu Y.; Liu Y. J.; McKinney K. A.; Martin S. T.; McNeill V. F.; Pye H. O. T.; et al. Examining the Effects of Anthropogenic Emissions on Isoprene-Derived Secondary Organic Aerosol Formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee Ground Site. Atmos. Chem. Phys. 2015, 15 (15), 8871–8888. 10.5194/acp-15-8871-2015. [DOI] [Google Scholar]
- Fu P.; Aggarwal S. G.; Chen J.; Li J.; Sun Y.; Wang Z.; Chen H.; Liao H.; Ding A.; Umarji G. S.; et al. Molecular Markers of Secondary Organic Aerosol in Mumbai, India. Environ. Sci. Technol. 2016, 50 (9), 4659–4667. 10.1021/acs.est.6b00372. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Parikh H. M.; Bapat J.; Lin Y.-H.; Surratt J. D.; Kamens R. M. Modelling of Secondary Organic Aerosol Formation from Isoprene Photooxidation Chamber Studies Using Different Approaches. Environ. Chem. 2013, 10 (3), 194. 10.1071/EN13029. [DOI] [Google Scholar]
- Riedel T. P.; Lin Y. H.; Zhang Z.; Chu K.; Thornton J. A.; Vizuete W.; Gold A.; Surratt J. D. Constraining Condensed-Phase Formation Kinetics of Secondary Organic Aerosol Components from Isoprene Epoxydiols. Atmos. Chem. Phys. 2016, 16 (3), 1245–1254. 10.5194/acp-16-1245-2016. [DOI] [Google Scholar]
- Fisher J. A.; Jacob D. J.; Travis K. R.; Kim P. S.; Marais E. A.; Chan Miller C.; Yu K.; Zhu L.; Yantosca R. M.; Sulprizio M. P.; et al. Organic Nitrate Chemistry and Its Implications for Nitrogen Budgets in an Isoprene- and Monoterpene-Rich Atmosphere: Constraints from Aircraft (SEAC4RS) and Ground-Based (SOAS) Observations in the Southeast US. Atmos. Chem. Phys. Discuss. 2016, 1–38. 10.5194/acp-2016-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais E. A.; Jacob D. J.; Jimenez J. L.; Campuzano-Jost P.; Day D. A.; Hu W.; Krechmer J.; Zhu L.; Kim P. S.; Miller C. C.; et al. Aqueous-Phase Mechanism for Secondary Organic Aerosol Formation from Isoprene: Application to the Southeast United States and Co-Benefit of SO2 Emission Controls. Atmos. Chem. Phys. 2016, 16 (3), 1603–1618. 10.5194/acp-16-1603-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surratt J. D.; Lewandowski M.; Offenberg J. H.; Jaoui M.; Kleindienst T. E.; Edney E. O.; Seinfeld J. H. Effect of Acidity on Secondary Organic Aerosol Formation from Isoprene. Environ. Sci. Technol. 2007, 41 (15), 5363–5369. 10.1021/es0704176. [DOI] [PubMed] [Google Scholar]
- Gaston C. J.; Riedel T. P.; Zhang Z.; Gold A.; Surratt J. D.; Thornton J. A. Reactive Uptake of an Isoprene-Derived Epoxydiol to Submicron Aerosol Particles. Environ. Sci. Technol. 2014, 48 (19), 11178–11186. 10.1021/es5034266. [DOI] [PubMed] [Google Scholar]
- Riedel T. P.; Lin Y. H.; Budisulistiorini S. H.; Gaston C. J.; Thornton J. A.; Zhang Z.; Vizuete W.; Gold A.; Surratt J. D. Heterogeneous Reactions of Isoprene-Derived Epoxides: Reaction Probabilities and Molar Secondary Organic Aerosol Yield Estimates. Environ. Sci. Technol. Lett. 2015, 2 (2), 38–42. 10.1021/ez500406f. [DOI] [Google Scholar]
- Budisulistiorini S. H.; Baumann K.; Edgerton E. S.; Bairai S. T.; Mueller S.; Shaw S. L.; Knipping E. M.; Gold A.; Surratt J. D. Seasonal Characterization of Submicron Aerosol Chemical Composition and Organic Aerosol Sources in the Southeastern United States: Atlanta, Georgia,and Look Rock, Tennessee. Atmos. Chem. Phys. 2016, 16 (8), 5171–5189. 10.5194/acp-16-5171-2016. [DOI] [Google Scholar]
- Surratt J. D.; Chan A. W.; Eddingsaas N. C.; Chan M.; Loza C. L.; Kwan A. J.; Hersey S. P.; Flagan R. C.; Wennberg P. O.; Seinfeld J. H. Reactive Intermediates Revealed in Secondary Organic Aerosol Formation from Isoprene. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6640–6645. 10.1073/pnas.0911114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Monod A.; Tritscher T.; Praplan A. P.; Decarlo P. F.; Temime-Roussel B.; Quivet E.; Marchand N.; Dommen J.; Baltensperger U. Aqueous Phase Processing of Secondary Organic Aerosol from Isoprene Photooxidation. Atmos. Chem. Phys. 2012, 12 (13), 5879–5895. 10.5194/acp-12-5879-2012. [DOI] [Google Scholar]
- Brégonzio-Rozier L.; Giorio C.; Siekmann F.; Pangui E.; Morales S. B.; Temime-Roussel B.; Gratien A.; Michoud V.; Cazaunau M.; DeWitt H. L.; et al. Secondary Organic Aerosol Formation from Isoprene Photooxidation during Cloud Condensation–evaporation Cycles. Atmos. Chem. Phys. 2016, 16 (3), 1747–1760. 10.5194/acp-16-1747-2016. [DOI] [Google Scholar]
- Nguyen T. B.; Laskin A.; Laskin J.; Nizkorodov S. A. Direct Aqueous Photochemistry of Isoprene High-NOx Secondary Organic Aerosol. Phys. Chem. Chem. Phys. 2012, 14 (27), 9702. 10.1039/c2cp40944e. [DOI] [PubMed] [Google Scholar]
- Kalberer M.; Paulsen D.; Sax M.; Steinbacher M.; Dommen J.; Prévôt A. S. H.; Fisseha R.; Weingartner E.; Frankevich V.; Zenobi R.; et al. Identification of Polymers as Major Components of Atmospheric Organic Aerosols. Science 2004, 303 (5664), 1659–1662. 10.1126/science.1092185. [DOI] [PubMed] [Google Scholar]
- Yasmeen F.; Vermeylen R.; Szmigielski R.; Iinuma Y.; Böge O.; Herrmann H.; Maenhaut W.; Claeys M. Terpenylic Acid and Related Compounds: Precursors for Dimers in Secondary Organic Aerosol from the Ozonolysis of α- and β-Pinene. Atmos. Chem. Phys. 2010, 10 (19), 9383–9392. 10.5194/acp-10-9383-2010. [DOI] [Google Scholar]
- Kourtchev I.; Giorio C.; Manninen A.; Wilson E.; Mahon B.; Aalto J.; Kajos M.; Venables D.; Ruuskanen T.; Levula J.; et al. Enhanced Volatile Organic Compounds Emissions and Organic Aerosol Mass Increase the Oligomer Content of Atmospheric Aerosols. Sci. Rep. 2016, 6, 35038. 10.1038/srep35038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall W. A. IV; Johnston M. V. Oligomer Formation Pathways in Secondary Organic Aerosol from MS and MS/MS Measurements with High Mass Accuracy and Resolving Power. J. Am. Soc. Mass Spectrom. 2012, 23 (6), 1097–1108. 10.1007/s13361-012-0362-6. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y.; Inomata S.; Hirokawa J. Oligomerization Reaction of the Criegee Intermediate Leads to Secondary Organic Aerosol Formation in Ethylene Ozonolysis. J. Phys. Chem. A 2013, 117 (48), 12912–12921. 10.1021/jp408672m. [DOI] [PubMed] [Google Scholar]
- Riva M.; Budisulistiorini S. H.; Zhang Z.; Gold A.; Thornton J. A.; Turpin B. J.; Surratt J. D. Multiphase Reactivity of Gaseous Hydroperoxide Oligomers Produced from Isoprene Ozonolysis in the Presence of Acidified Aerosols. Atmos. Environ. 2017, 152, 314–322. 10.1016/j.atmosenv.2016.12.040. [DOI] [Google Scholar]
- Zhang H.; Surratt J. D.; Lin Y. H.; Bapat J.; Kamens R. M. Effect of Relative Humidity on SOA Formation from isoprene/NO Photooxidation: Enhancement of 2-Methylglyceric Acid and Its Corresponding Oligoesters under Dry Conditions. Atmos. Chem. Phys. 2011, 11 (13), 6411–6424. 10.5194/acp-11-6411-2011. [DOI] [Google Scholar]
- Rodigast M.; Mutzel A.; Herrmann H. A Quantification Method for Heat-Decomposable Methylglyoxal Oligomers and Its Application on 1,3,5-Trimethylbenzene SOA. Atmos. Chem. Phys. 2017, 17 (6), 3929–3943. 10.5194/acp-17-3929-2017. [DOI] [Google Scholar]
- Lin Y. H.; Zhang Z.; Docherty K. S.; Zhang H.; Budisulistiorini S. H.; Rubitschun C. L.; Shaw S. L.; Knipping E. M.; Edgerton E. S.; Kleindienst T. E.; et al. Isoprene Epoxydiols as Precursors to Secondary Organic Aerosol Formation: Acid-Catalyzed Reactive Uptake Studies with Authentic Compounds. Environ. Sci. Technol. 2012, 46 (1), 250–258. 10.1021/es202554c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard P.; Siekmann F.; Gandolfo A.; Socorro J.; Salque G.; Ravier S.; Quivet E.; Clément J.-L.; Traikia M.; Delort A.-M.; et al. Radical Mechanisms of Methyl Vinyl Ketone Oligomerization through Aqueous Phase OH-Oxidation: On the Paradoxical Role of Dissolved Molecular Oxygen. Atmos. Chem. Phys. 2013, 13 (13), 6473–6491. 10.5194/acp-13-6473-2013. [DOI] [Google Scholar]
- Ervens B.; Renard P.; Tlili S.; Ravier S.; Clément J.-L.; Monod A. Aqueous-Phase Oligomerization of Methyl Vinyl Ketone through Photooxidation – Part 2: Development of the Chemical Mechanism and Atmospheric Implications. Atmos. Chem. Phys. 2015, 15 (16), 9109–9127. 10.5194/acp-15-9109-2015. [DOI] [Google Scholar]
- Liu Y.; Siekmann F.; Renard P.; El Zein A.; Salque G.; El Haddad I.; Temime-Roussel B.; Voisin D.; Thissen R.; Monod A. Oligomer and SOA Formation through Aqueous Phase Photooxidation of Methacrolein and Methyl Vinyl Ketone. Atmos. Environ. 2012, 49, 123–129. 10.1016/j.atmosenv.2011.12.012. [DOI] [Google Scholar]
- Renard P.; Tlili S.; Ravier S.; Quivet E.; Monod A. Aqueous Phase Oligomerization of A,β-Unsaturated Carbonyls and Acids Investigated Using Ion Mobility Spectrometry Coupled to Mass Spectrometry (IMS-MS). Atmos. Environ. 2016, 130, 153–162. 10.1016/j.atmosenv.2015.10.060. [DOI] [Google Scholar]
- Wang J.; Doussin J. F.; Perrier S.; Perraudin E.; Katrib Y.; Pangui E.; Picquet-Varrault B. Design of a New Multi-Phase Experimental Simulation Chamber for Atmospheric Photosmog, Aerosol and Cloud Chemistry Research. Atmos. Meas. Tech. 2011, 4 (11), 2465–2494. 10.5194/amt-4-2465-2011. [DOI] [Google Scholar]
- Brégonzio-Rozier L.; Siekmann F.; Giorio C.; Pangui E.; Morales S. B.; Temime-Roussel B.; Gratien A.; Michoud V.; Ravier S.; Cazaunau M.; et al. Gaseous Products and Secondary Organic Aerosol Formation during Long Term Oxidation of Isoprene and Methacrolein. Atmos. Chem. Phys. 2015, 15, 2953–2968. 10.5194/acp-15-2953-2015. [DOI] [Google Scholar]
- Afif C.; Jambert C.; Michoud V.; Colomb A.; Eyglunent G.; Borbon A.; Daële V.; Doussin J.-F.; Perros P. NitroMAC: An Instrument for the Measurement of HONO and Intercomparison with a Long-Path Absorption Photometer. J. Environ. Sci. 2016, 40, 105–113. 10.1016/j.jes.2015.10.024. [DOI] [PubMed] [Google Scholar]
- El-Sayed M. M. H.; Amenumey D.; Hennigan C. J. Drying-Induced Evaporation of Secondary Organic Aerosol during Summer. Environ. Sci. Technol. 2016, 50 (7), 3626–3633. 10.1021/acs.est.5b06002. [DOI] [PubMed] [Google Scholar]
- Kourtchev I.; Doussin J.-F.; Giorio C.; Mahon B.; Wilson E. M.; Maurin N.; Pangui E.; Venables D. S.; Wenger J. C.; Kalberer M. Molecular Composition of Fresh and Aged Secondary Organic Aerosol from a Mixture of Biogenic Volatile Compounds: A High-Resolution Mass Spectrometry Study. Atmos. Chem. Phys. 2015, 15 (10), 5683–5695. 10.5194/acp-15-5683-2015. [DOI] [Google Scholar]
- Zielinski A.; Kourtchev I.; Bortolini C.; Fuller S. J.; Giorio C.; Popoola O.; Bogialli S.; Tapparo A.; Jones R. L.; Kalberer M.. A New Processing Routine for Ultra-High Resolution Direct Infusion Mass Spectrometry Data. Atmos. Environ. 2017, submitted
- Kind T.; Fiehn O. Seven Golden Rules for Heuristic Filtering of Molecular Formulas Obtained by Accurate Mass Spectrometry. BMC Bioinf. 2007, 8 (1), 105. 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson A. C.; Marshall A. G.; Cooper W. T. Exact Masses and Chemical Formulas of Individual Suwannee River Fulvic Acids from Ultrahigh Resolution Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectra. Anal. Chem. 2003, 75 (6), 1275–1284. 10.1021/ac026106p. [DOI] [PubMed] [Google Scholar]
- Reinhardt A.; Emmenegger C.; Gerrits B.; Panse C.; Dommen J.; Baltensperger U.; Zenobi R.; Kalberer M. Ultrahigh Mass Resolution and Accurate Mass Measurements as a Tool to Characterize Oligomers in Secondary Organic Aerosols. Anal. Chem. 2007, 79 (11), 4074–4082. 10.1021/ac062425v. [DOI] [PubMed] [Google Scholar]
- Wilson J.; Imre D.; Beránek J.; Shrivastava M.; Zelenyuk A. Evaporation Kinetics of Laboratory-Generated Secondary Organic Aerosols at Elevated Relative Humidity. Environ. Sci. Technol. 2015, 49 (1), 243–249. 10.1021/es505331d. [DOI] [PubMed] [Google Scholar]
- Song M.; Liu P. F.; Hanna S. J.; Li Y. J.; Martin S. T.; Bertram A. K. Relative Humidity-Dependent Viscosities of Isoprene-Derived Secondary Organic Material and Atmospheric Implications for Isoprene-Dominant Forests. Atmos. Chem. Phys. 2015, 15 (9), 5145–5159. 10.5194/acp-15-5145-2015. [DOI] [Google Scholar]
- Chan A. W. H.; Chan M. N.; Surratt J. D.; Chhabra P. S.; Loza C. L.; Crounse J. D.; Yee L. D.; Flagan R. C.; Wennberg P. O.; Seinfeld J. H. Role of Aldehyde Chemistry and NOx Concentrations in Secondary Organic Aerosol Formation. Atmos. Chem. Phys. 2010, 10 (15), 7169–7188. 10.5194/acp-10-7169-2010. [DOI] [Google Scholar]
- Nguyen T. B.; Bates K. H.; Crounse J. D.; Schwantes R. H.; Zhang X.; Kjaergaard H. G.; Surratt J. D.; Lin P.; Laskin A.; Seinfeld J. H.; et al. Mechanism of the Hydroxyl Radical Oxidation of Methacryloyl Peroxynitrate (MPAN) and Its Pathway toward Secondary Organic Aerosol Formation in the Atmosphere. Phys. Chem. Chem. Phys. 2015, 17 (27), 17914–17926. 10.1039/C5CP02001H. [DOI] [PubMed] [Google Scholar]
- Rattanavaraha W.; Chu K.; Budisulistiorini S. H.; Riva M.; Lin Y. H.; Edgerton E. S.; Baumann K.; Shaw S. L.; Guo H.; King L.; et al. Assessing the Impact of Anthropogenic Pollution on Isoprene-Derived Secondary Organic Aerosol Formation in PM2.5 Collected from the Birmingham, Alabama, Ground Site during the 2013 Southern Oxidant and Aerosol Study. Atmos. Chem. Phys. 2016, 16 (8), 4897–4914. 10.5194/acp-16-4897-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Schwantes R. H.; McVay R. C.; Lignell H.; Coggon M. M.; Flagan R. C.; Seinfeld J. H. Vapor Wall Deposition in Teflon Chambers. Atmos. Chem. Phys. 2015, 15 (8), 4197–4214. 10.5194/acp-15-4197-2015. [DOI] [Google Scholar]
- Hering S. V.; Friedlander S. K. Origins of Aerosol Sulfur Size Distributions in the Los Angeles Basin. Atmos. Environ. 1982, 16 (11), 2647–2656. 10.1016/0004-6981(82)90346-8. [DOI] [Google Scholar]
- Meng Z.; Seinfeld J. H. On the Source of the Submicrometer Droplet Mode of Urban and Regional Aerosols. Aerosol Sci. Technol. 1994, 20 (3), 253–265. 10.1080/02786829408959681. [DOI] [Google Scholar]
- John W.; Wall S. M.; Ondo J. L.; Winklmayr W. Modes in the Size Distributions of Atmospheric Inorganic Aerosol. Atmos. Environ., Part A 1990, 24 (9), 2349–2359. 10.1016/0960-1686(90)90327-J. [DOI] [Google Scholar]
- Bianchi F.; Dommen J.; Mathot S.; Baltensperger U. On-Line Determination of Ammonia at Low Pptv Mixing Ratios in the CLOUD Chamber. Atmos. Meas. Tech. 2012, 5 (7), 1719–1725. 10.5194/amt-5-1719-2012. [DOI] [Google Scholar]
- Xu L.; Kollman M. S.; Song C.; Shilling J. E.; Ng N. L. Effects of NOx on the Volatility of Secondary Organic Aerosol from Isoprene Photooxidation. Environ. Sci. Technol. 2014, 48 (4), 2253–2262. 10.1021/es404842g. [DOI] [PubMed] [Google Scholar]
- Ng N. L.; Canagaratna M. R.; Zhang Q.; Jimenez J. L.; Tian J.; Ulbrich I. M.; Kroll J. H.; Docherty K. S.; Chhabra P. S.; Bahreini R.; et al. Organic Aerosol Components Observed in Northern Hemispheric Datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2010, 10 (10), 4625–4641. 10.5194/acp-10-4625-2010. [DOI] [Google Scholar]
- Ng N. L.; Canagaratna M. R.; Jimenez J. L.; Chhabra P. S.; Seinfeld J. H.; Worsnop D. R. Changes in Organic Aerosol Composition with Aging Inferred from Aerosol Mass Spectra. Atmos. Chem. Phys. 2011, 11 (13), 6465–6474. 10.5194/acp-11-6465-2011. [DOI] [Google Scholar]
- Szmigielski R.; Surratt J. D.; Vermeylen R.; Szmigielska K.; Kroll J. H.; Ng N. L.; Murphy S. M.; Sorooshian A.; Seinfeld J. H.; Claeys M. Characterization of 2-Methylglyceric Acid Oligomers in Secondary Organic Aerosol Formed from the Photooxidation of Isoprene Using Trimethylsilylation and Gas Chromatography/ion Trap Mass Spectrometry. J. Mass Spectrom. 2007, 42 (1), 101–116. 10.1002/jms.1146. [DOI] [PubMed] [Google Scholar]
- Hu Q.-H.; Xie Z.-Q.; Wang X.-M.; Kang H.; He Q.-F.; Zhang P. Secondary Organic Aerosols over Oceans via Oxidation of Isoprene and Monoterpenes from Arctic to Antarctic. Sci. Rep. 2013, 3, 2280. 10.1038/srep02280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleindienst T. E.; Jaoui M.; Lewandowski M.; Offenberg J. H.; Lewis C. W.; Bhave P. V.; Edney E. O. Estimates of the Contributions of Biogenic and Anthropogenic Hydrocarbons to Secondary Organic Aerosol at a Southeastern US Location. Atmos. Environ. 2007, 41 (37), 8288–8300. 10.1016/j.atmosenv.2007.06.045. [DOI] [Google Scholar]
- Docherty K. S.; Jaoui M.; Corse E.; Jimenez J. L.; Offenberg J. H.; Lewandowski M.; Kleindienst T. E. Collection Efficiency of the Aerosol Mass Spectrometer for Chamber-Generated Secondary Organic Aerosols. Aerosol Sci. Technol. 2013, 47 (3), 294–309. 10.1080/02786826.2012.752572. [DOI] [Google Scholar]
- Nguyen T. B.; Bateman A. P.; Bones D. L.; Nizkorodov S. A.; Laskin J.; Laskin A. High-Resolution Mass Spectrometry Analysis of Secondary Organic Aerosol Generated by Ozonolysis of Isoprene. Atmos. Environ. 2010, 44 (8), 1032–1042. 10.1016/j.atmosenv.2009.12.019. [DOI] [Google Scholar]
- Renard P.; Siekmann F.; Salque G.; Demelas C.; Coulomb B.; Vassalo L.; Ravier S.; Temime-Roussel B.; Voisin D.; Monod A. Aqueous-Phase Oligomerization of Methyl Vinyl Ketone through Photooxidation -- Part 1: Aging Processes of Oligomers. Atmos. Chem. Phys. 2015, 15 (1), 21–35. 10.5194/acp-15-21-2015. [DOI] [Google Scholar]
- Nguyen T. B.; Laskin A.; Laskin J.; Nizkorodov S. A. Direct Aqueous Photochemistry of Isoprene High-NOx Secondary Organic Aerosol. Phys. Chem. Chem. Phys. 2012, 14 (27), 9702. 10.1039/c2cp40944e. [DOI] [PubMed] [Google Scholar]
- Renard P.; Tlili S.; Ravier S.; Quivet E.; Monod A. Aqueous Phase Oligomerization of A,β-Unsaturated Carbonyls and Acids Investigated Using Ion Mobility Spectrometry Coupled to Mass Spectrometry (IMS-MS). Atmos. Environ. 2016, 130, 153–162. 10.1016/j.atmosenv.2015.10.060. [DOI] [Google Scholar]
- Renard P.; Reed Harris A. E.; Rapf R. J.; Ravier S.; Demelas C.; Coulomb B.; Quivet E.; Vaida V.; Monod A. Aqueous Phase Oligomerization of Methyl Vinyl Ketone by Atmospheric Radical Reactions. J. Phys. Chem. C 2014, 118 (50), 29421–29430. 10.1021/jp5065598. [DOI] [Google Scholar]
- Slade J. H.; Knopf D. A. Multiphase OH Oxidation Kinetics of Organic Aerosol: The Role of Particle Phase State and Relative Humidity. Geophys. Res. Lett. 2014, 41 (14), 5297–5306. 10.1002/2014GL060582. [DOI] [Google Scholar]
- Smith M. L.; Bertram A. K.; Martin S. T. Deliquescence, Efflorescence, and Phase Miscibility of Mixed Particles of Ammonium Sulfate and Isoprene-Derived Secondary Organic Material. Atmos. Chem. Phys. 2012, 12 (20), 9613–9628. 10.5194/acp-12-9613-2012. [DOI] [PubMed] [Google Scholar]
- Duplissy J.; De Carlo P. F.; Dommen J.; Alfarra M. R.; Metzger A.; Barmpadimos I.; Prevot A. S. H.; Weingartner E.; Tritscher T.; Gysel M.; et al. Relating Hygroscopicity and Composition of Organic Aerosol Particulate Matter. Atmos. Chem. Phys. 2011, 11 (3), 1155–1165. 10.5194/acp-11-1155-2011. [DOI] [Google Scholar]
- Guo H.; Xu L.; Bougiatioti A.; Cerully K. M.; Capps S. L.; Hite J. R.; Carlton A. G.; Lee S. H.; Bergin M. H.; Ng N. L.; et al. Fine-Particle Water and pH in the Southeastern United States. Atmos. Chem. Phys. 2015, 15 (9), 5211–5228. 10.5194/acp-15-5211-2015. [DOI] [Google Scholar]
- Nguyen T. K. V; Zhang Q.; Jimenez J. L.; Pike M.; Carlton A. G. Liquid Water: Ubiquitous Contributor to Aerosol Mass. Environ. Sci. Technol. Lett. 2016, 3 (7), 257–263. 10.1021/acs.estlett.6b00167. [DOI] [Google Scholar]
- Lee B. H.; Mohr C.; Lopez-Hilfiker F. D.; Lutz A.; Hallquist M.; Lee L.; Romer P.; Cohen R. C.; Iyer S.; Kurtén T.; et al. Highly Functionalized Organic Nitrates in the Southeast United States: Contribution to Secondary Organic Aerosol and Reactive Nitrogen Budgets. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (6), 1516–1521. 10.1073/pnas.1508108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander R. Compilation of Henry’s Law Constants (Version 4.0) for Water as Solvent. Atmos. Chem. Phys. 2015, 15 (8), 4399–4981. 10.5194/acp-15-4399-2015. [DOI] [Google Scholar]
- McNeill V. F. Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols. Environ. Sci. Technol. 2015, 49 (3), 1237–1244. 10.1021/es5043707. [DOI] [PubMed] [Google Scholar]
- Reed Harris A. E.; Pajunoja A.; Cazaunau M.; Gratien A.; Pangui E.; Monod A.; Griffith E. C.; Virtanen A.; Doussin J.-F.; Vaida V. Multiphase Photochemistry of Pyruvic Acid Under Atmospheric Conditions. J. Phys. Chem. A 2017, 121, 3327. 10.1021/acs.jpca.7b01107. [DOI] [PubMed] [Google Scholar]
- Vaida V. Atmospheric Radical Chemistry Revisited. Science 2016, 353 (6300), 650–650. 10.1126/science.aah4111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.