

SUMMARY
Several cell populations have been reported to possess intestinal stem cell (ISC) activity during homeostasis and injury-induced regeneration. Here, we explored inter-relationships between putative mouse ISC populations by comparative RNA-sequencing (RNA-seq). The transcriptomes of multiple cycling ISC populations closely resembled Lgr5+ ISCs, the most well-defined ISC pool, but Bmi1-GFP+ cells were distinct and enriched for enteroendocrine (EE) markers including Prox1. Prox1-GFP+ cells exhibited sustained clonogenic growth in vitro, and lineage-tracing of Prox1+ cells revealed long-lived clones during homeostasis and after radiation-induced injury in vivo. Single-cell mRNA-seq revealed two subsets of Prox1-GFP+ cells, one of which resembled mature EE cells while the other displayed low level EE gene expression but co-expressed tuft cell markers, Lgr5 and Ascl2, reminiscent of label-retaining secretory progenitors. Our data suggest that the EE lineage, including mature EE cells, comprise a reservoir of homeostatic and injury-inducible ISCs, extending our understanding of cellular plasticity and stemness.
In Brief
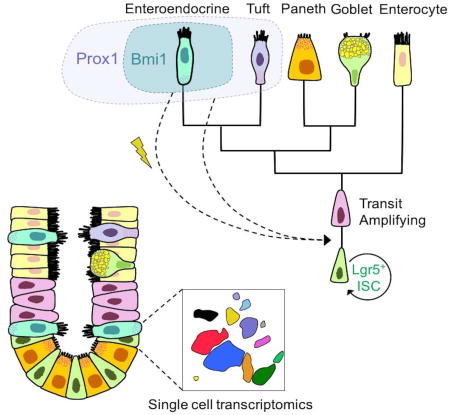
Multiple cell populations, represented by distinct markers including Lgr5 and Bmi1, are capable of reconstituting the intestinal epithelium. Using comparative RNA sequencing and single-cell transcriptomics, Yan et al. define Bmi1-GFP+ and Prox1+ cells as enteroendocrine lineage cells that possess intestinal stem cell activity during homeostasis and injury-induced regeneration.
INTRODUCTION
The intestine exhibits remarkable regenerative potential, with intestinal stem cells (ISCs) residing in proliferative crypts and generating progenitors capable of multi-lineage differentiation and robust homeostatic and regenerative repopulation. The proliferative crypt zone contains an ISC “niche” comprised of epithelial, subepithelial, and luminal components that provide essential paracrine signals (Clevers, 2013). ISCs have been postulated to be either actively cycling crypt-based columnar cells (CBC) or quiescent label-retaining cells (LRC) residing at approximately the +4 position from the crypt base (Cheng and Leblond, 1974; Clevers, 2013; Marshman et al., 2002). Seminal studies defined Lgr5 as a molecular marker of CBC-class ISCs that persist, self-renew, and generate all mature intestinal epithelial lineages in vivo (Barker et al., 2007) and organoids upon clonogenic culture (Sato et al., 2009). CBCs can also be marked by Olfm4, notably Wnt-independent (van der Flier et al., 2009), Ascl2, Prom1, Sox9lo, Cd24lo, Cd166, Grp78− and Lrig1 (Dalerba et al., 2007; Formeister et al., 2009; Levin et al., 2010; von Furstenberg et al., 2011; Wang et al., 2013; Wong et al., 2012; Zhu et al., 2009). Lgr5+ CBC cells undergo homeostatic niche competition resulting in random clone expansion/contraction (“neutral drift dynamics”) and crypt monoclonality (Kozar et al., 2013; Lopez-Garcia et al., 2010; Snippert et al., 2010).
Numerous putative markers of quiescent/slowly-cycling ISC residing in approximately the +4 cell position have also been described. Emerging concepts amongst quiescent populations have included their mobilization upon tissue injury or upon CBC loss and/or possession of secretory progenitor features. The mTert+, Lrig1+ and Bmi1+ ISCs clearly exhibit properties of quiescent but injury-inducible populations (Breault et al., 2008; Montgomery et al., 2011; Powell et al., 2012; Sangiorgi and Capecchi, 2008; Yan et al., 2012).. Dll1+ crypt cells represent short-lived secretory progenitors that regain stemness upon radiation damage (van Es et al., 2012). Lgr5+ or side-population (SP) cells may be subdivided into fractions possessing properties of either actively cycling or quiescent ISCs (Buczacki et al., 2013; von Furstenberg et al., 2014). Indeed, quiescent +4 label-retaining cells (LRCs) are injury-inducible secretory progenitors that co-express Lgr5, Paneth and enteroendocrine (EE) markers (Buczacki et al., 2013). Mex3a+ cells that co-express high levels of Lgr5 are slowly cycling and share common features with LRC(Barriga et al., 2017). Alkaline phosphate-expressing enterocytes repopulate crypts after Lgr5+ ISC ablation but not during homeostasis (Tetteh et al., 2016), suggesting plasticity of the differentiated absorptive enterocyte lineage to support epithelial reconstitution following CBC loss. This diversity of proposed ISC populations and variety of markers has raised numerous questions of ISC inter-relatedness, hierarchy and interconversion. Here, we investigated these issues through systematic transcriptome profiling of diverse ISC marker populations.
RESULTS
To address questions of ISC interrelatedness, we performed comparative bulk cell RNA-seq analysis of diverse FACS-isolated ISC populations. This included Lgr5-eGFPhi cells as a reference for CBC-type ISCs versus cells suggested to exhibit CBC enrichment including Cd166+ (Dalerba et al., 2007; Levin et al., 2010), Cd24lo (von Furstenberg et al., 2011), Grp78− (Wang et al., 2013) and upper side population (SP) (von Furstenberg et al., 2014). Further, as representative +4/quiescent ISC populations, we compared cells expressing Bmi1 (Sangiorgi and Capecchi, 2008), mTert (Breault et al., 2008; Montgomery et al., 2011), Hopx (Takeda et al., 2011), Dclk1 (May et al., 2009) or a lower side population (SP) (von Furstenberg et al., 2014). We previously described an Intestinal Stem Cell Consortium (ISCC) initiative to standardize ISC FACS isolation from primary intestinal epithelium by EDTA-based epithelial isolation, depletion of non-epithelial contaminants and sequential scatter- and antibody-based gating (Magness et al., 2013). Within this common schema, we performed reporter- and non-reporter-based isolation of CBC-like and +4/quiescent-type ISCs. Cd24lo, Cd166+, and GPR78− isolations were antibody-based (Dalerba et al., 2007; Levin et al., 2010). From parental Cd24lo cells, FACS isolation of a Cd24loCd166+ subfraction (designated Cd166+) was facilitated by the crypt-enriching novel monoclonal antibody B6A6 (Wang et al., 2013) to reduce Paneth cell representation. Cells designated as GPR78− were derived from a composite Cd24loCd166+Cd44+Grp78− gate (Wang et al., 2013) and Hoechst dye efflux was utilized for upper and lower SP purification (von Furstenberg et al., 2014). Where pertinent, we utilized intrinsic GFP fluorescence for FACS isolation from well-validated transgenic reporter strains including Lgr5-eGFP-IRES-CreER (Barker et al., 2007; Sato et al., 2009), mTert-GFP (Breault et al., 2008; Montgomery et al., 2011) and Hopx-CreER; Rosa26-mTmG (Takeda et al., 2011). For Bmi1+ ISCs, Bmi1-CreER; Rosa26-YFP mice with short-term tamoxifen-labeling or Bmi1-GFP mice (Hosen et al., 2007), were employed (Figure S1). Notably, Bmi1-GFP enables more specific labeling of Bmi1+ cells than does short-term tamoxifen lineage tracing in Bmi1-GFP; Rosa26-YFP mice (Yan et al., 2012); the latter approach marks Bmi1 downstream progeny in addition to Bmi1+ cells (K.S.Y., N.R.S., M.H.W., and C.J.K., unpublished observations).
For each ISC population of interest, FACS isolation of marker-“positive” and their associated marker-“negative” populations was performed from three independent biological replicate mice. Bulk RNA-seq libraries were generated from polyA+ RNA, followed by deep sequencing, read mapping, calculation of FPKM (Table S1) and determination of transcripts enriched in each marker-positive versus -negative FACS subpopulations. Unsupervised hierarchical pan-population analysis revealed two major clusters (Figure 1A). Cluster 1 showed substantial overlap of Lgr5hi cells with the Cd166+, Cd24lo and Grp78− populations, validating those markers as robust antibody-based methods for CBC enrichment. Interestingly, Cluster 2 was comprised of Bmi1-GFP+ and mTert-GFP+ cells, which share common features of localization to +4 regions and injury-inducibility in vivo. The Hopx, lower SP and Dclk1 entities were intermediate between Clusters 1 and 2. Relative to their respective FACS-isolated marker-negative cell controls, the Lgr5hi, Cd166+, Cd24lo and Bmi1-GFP+ populations all possessed a large number (~200–800) of differentially expressed (DE) transcripts. In contrast, Grp78−, mTert-GFP, Dclk1, and SP had <100 DE genes (Tables S2 and S3), although whether this was attributed to technical factors remains to be determined.
Figure 1. Hierarchical clustering of ISC populations by bulk cell mRNA-seq.

A. Unsupervised hierarchical clustering reveals the association of Lgr5hi, Cd166+, Cd24lo, upper SP and Grp78− cells into Cluster 1. Bmi1-GFP+ and mTert-GFP+ cells are present in Cluster 2, while Hopx+, Dclk1+ and lower SP cells exhibit intermediate features between Clusters 1 and 2. B. Supervised hierarchical analysis demonstrating enrichment of an Lgr5hi/CBC gene signature amongst Cluster 1 populations.
Cluster 1, with the Lgr5hi, Cd166+, Cd24lo and Grp78− populations, exhibited common enrichment of a CBC signature (Koo et al., 2012; Munoz et al., 2012; van der Flier et al., 2009) including Lgr5, Ascl2, Olfm4, Cd44, Sox9, Znrf3 and Tnfrsf19 (Figure 1B; Table S3). Cd24lo and to a lesser extent Lgr5hi exhibited a Paneth signature including Mmp7, Defa21 and Defa26, consistent with the known presence of Paneth cells within non-UEA fractionated Cd24lo cells (von Furstenberg et al., 2011) and the reported expression of Paneth markers in a quiescent label-retaining subfraction of Lgr5+ ISCs (Buczacki et al., 2013). Although Cd166+ and Grp78− are derived from further fractionation of Cd24lo to deplete Paneth cells, Cd24lo itself was sufficient to enrich for the CBC signature (Figure 1B). Unsupervised Gene Set Enrichment Analysis (GSEA) using Gene Ontology (GO) analysis of loci active in Cluster 1 but inactive in Cluster 2 revealed plasma membrane, developmental and ion transport processes (Table S4). Overall, these data indicate strong enrichment for Lgr5+ ISC in the Cd166+, Cd24lo and Grp78− populations, suggesting that corresponding antibody strategies could facilitate ISC isolation from human tissues given the current lack of antibody-based Lgr5+ cell isolation (Wang et al., 2013).
Cluster 2 was defined by overlap between the Bmi1-GFP+ and mTert-GFP+ populations, both of which shared common EE enrichment. The mTert-GFP-enriched loci included Chga, Ghrl, Gip, and Reg4 (Figure 2A), but this gene set was too small to perform GSEA. In contrast, 840 genes exhibited significant enrichment in Bmi1-GFP+ versus Bmi1-GFP− populations (Figure 2A and 2B; Figure S1; Table S3). Lgr5+ ISCs express +4 marker transcripts including Bmi1, mTert, Hopx and Dclk1 (Munoz et al., 2012). Accordingly, the Cluster 1 CBC signature genes such as Lgr5, Ascl2 or Olfm4 were detectable at low levels in Bmi1-GFP+ cells, potentially consistent with described interconversion between Lgr5 and Bmi1 or Hopx populations (Takeda et al., 2011; Tian et al., 2011). However, Lgr5, Ascl2 or Olfm4 were de-enriched 5–15 fold in Bmi1-GFP+ versus Bmi1-GFP− cells and were in fact 27–158 fold less enriched than in Lgr5-eGFP+ versus Lgr5-eGFP− cells (Figure 1B and 2B, Table S5).
Figure 2. Characterization of the Bmi1/mTert ISC cluster.

A. Venn diagram of overlapping gene enrichment between Bmi1-GFP+ and mTert-GFP+ cells. B. Enteroendocrine gene signature in Bmi1-GFP+ cells includes numerous EE secretory factors and EE transcription factors (Neurod1, Arx, Prox1) but notably lacks Neurog3. C. Anti-ChgA (white) immunofluorescence co-localization in intestinal crypts is found in 89.9% of crypt Bmi1-GFP cells (anti-GFP, green). Bar = 25 µm. Error bars indicate mean ± s.e.m. D. Overlap between genes enriched in Bmi1-GFP+, LRC and multi-capable EE. E. Clonogenic enteroid culture of FACS-isolated Bmi1-GFP+ cells validating their clonogenic potential ex vivo. D = day, P = passage number. Bar = 100 µm.
Notably, numerous canonical enteroendocrine (EE) secretory products were among the most strongly enriched loci in Bmi1-GFP+ versus Bmi1-GFP− cells (100–3000 fold FPKM) including Cck, Sct, Gip, Glp-1, Pyy and Nts. Multiple EE transcription factor mRNAs were similarly enriched including Prox1, Neurod1, Pax6, Rfx6 and Arx (Figure 2B). mTert-GFP+ cells exhibited hierarchical clustering with Bmi1+ ISCs, and the 36 loci common to Bmi1+ and mTert+ populations included the EE secretory products Chga, Ghrl and Glp (Figure 2A), although mTert+ cells had less pronounced levels of EE gene enrichment. Immunofluorescence co-localization revealed that 90% of Bmi1-GFP+ cells expressed ChgA (Figure 2C). Additionally, Cck was also enriched by bulk RNA-seq of 1d tamoxifen-labeled Bmi1-CreER; Rosa26-YFP cells (Table S3), previously shown to undergo modest homeostatic lineage tracing that is robustly induced by irradiation (Yan et al., 2012). However, the Bmi1-GFP allele allows for more specific Bmi1+ cell isolation compared to Bmi1-CreER, which can additionally label downstream progeny and dilute the stem cell transcriptome.
The EE gene expression within Bmi1+ cells was highly reminiscent of EE marker detection by Dll1+ and label-retaining cell (LRC) injury-inducible ISC populations, which possess secretory progenitor features (Buczacki et al., 2013; van Es et al., 2012). Indeed, the Bmi1+ and LRC transcriptomes shared high-level enrichment of Gip, Pax6, Rfx6, Peg3, Chga and Chgb (Figure 2D). However, Bmi1-GFP+ cells were de-enriched for Paneth and goblet markers ascribed to LRCs including Mmp7 and Muc2. Further, the LRC-specific markers Nfatc3, Cd83 and Nfat5 (Buczacki et al., 2013) were not enriched in Bmi1-GFP+ cells versus Bmi1-GFP− controls. Bmi1-GFP+ cells strongly expressed the differentiated EE marker Neurod1 but had essentially undetectable expression of the EE progenitor marker Neurog3 that is prominent in Dll1+Cd24mid cells (van Es et al., 2012), consistent with Bmi1-GFP+ exhibiting strong EE lineage commitment and differentiation (Figure 2B and Figure 2D). The Bmi1-GFP EE signature resembles a “multi-capable” EE progenitor population that co-expresses Cck, Sct, Gip, Glp-1, Pyy and Nts and Neurod1 but not Ngn3 (Egerod et al., 2012; Engelstoft et al., 2013; Habib et al., 2012) and exhibits developmental requirement for the homeobox gene Arx (Beucher et al., 2012; Du et al., 2012). Bmi1-GFP+ cells were enriched for Cdkn1a but deficient in Cdkn2a, as expected for a quiescent cell. Despite their differentiated transcriptomic features, Bmi1-GFP+ cells exhibited prolonged clonogenic growth in organoid culture (Figure 2E), consistent with stem/progenitor potential. These sorted Bmi1-GFP+ cells did not retain GFP expression upon subsequent enteroid culture (data not shown).
The EE enrichment of the Bmi1 transcriptome suggested that orthogonal EE markers could prospectively identify a Bmi1+ ISC-like injury-inducible stem cell. Prox1 is the mammalian ortholog of Pros, which robustly marks Drosophila intestinal EE cells and is functionally required for ISC commitment to the EE lineage (Guo and Ohlstein, 2015; Zeng and Hou, 2015). Prox1 is a transcription factor expressed in lymphatic endothelial cells and also in quiescent crypt-based EE cells that co-express multiple secretory markers including Pyy, Cck, and Glp-1 (Petrova et al., 2008) reminiscent of the multi-capable EE (Egerod et al., 2012; Engelstoft et al., 2013; Habib et al., 2012). Single-cell RNA-seq has revealed Prox1 expression in a majority of small intestinal EE cells (Grun et al., 2015). We noted enrichment of Prox1 transcripts in Bmi1-GFP+ cells (Figure 2B) and Prox1 protein was detectable in 74% of Bmi1-GFP+ cells by immunostaining (Figure 3A). To functionally characterize Prox1-expressing cells, FACS-isolated Prox1-GFP+ cells from Prox1-GFP mice (Choi et al., 2011) grew clonogenically as enteroids (Figure 3B), similar to sorted Bmi1-GFP+ (Figure 2E) and tamoxifen-labeled Bmi1-CreER; Rosa26-YFP+ cells (Yan et al., 2012). Furthermore, we crossed Prox1-CreER mice (Srinivasan et al., 2007) with a Rosa26-tdTomato reporter strain to facilitate lineage tracing. In Prox1-GFP; Prox1-CreER; Rosa26-tdTomato mice, at 36 hours post-tamoxifen treatment, a subset of crypts (1/20) exhibited 1–2 Prox1+ epithelial cells in a distribution that included mostly crypts at the +3–5 cell positions (Figure 3C) and occasional scattered single cells in the villi (~1 cell/10 villi). Notably, ~10% of Prox1+ cells in Prox1-CreER; Rosa26 tdTomato mice generated long-lived homeostatic lineage stripes persisting to 395 days (Figure 3D) with multi-lineage differentiation (Figure 3E). This suggested rare but detectable clonogenic potential of crypt-based Prox1+ cells during homeostasis. We also confirmed Prox1 expression in lymphatic endothelial cells (Petrova et al., 2008) (Figure 3D). One salient feature of Bmi1+ ISCs is radioresistance and injury-inducibility, in marked contrast to Lgr5+ ISCs (Yan et al., 2012). Thus, we further characterized the regenerative potential of putative Prox1+ ISCs in vivo. Single dose 12 Gy total body irradiation strongly induced lineage tracing from Prox1+ cells in Prox1-CreER; Rosa26 tdTomato mice (Figure 3F), similar to that observed with Bmi1-CreER; Rosa26 YFP mice (Yan et al., 2012). Together, these data support a functional similarity between Prox1+ and Bmi1+ intestinal epithelial cells in vivo and validate our transcriptome findings.
Figure 3. Functional similarities between Bmi1-GFP+ and Prox1+ cells in vivo.

A. Co-localization of Prox1 expression in 74.4% of Bmi1-GFP+ crypt cells by immunofluorescence, Bmi1-GFP (anti-GFP, green) and anti-Prox1 (red). Bar = 25 µm. Error bars indicate mean ± s.e.m. B. Clonogenic enteroid growth ex vivo from Prox1-GFP+ cells isolated from proximal jejunum, day 5. 20× magnification. C. Short-term (36h) in vivo tamoxifen-induced labeling of Prox1+ cells in jejunum of Prox1-GFP; Prox1-CreER; Rosa26 tdTomato mice marks crypt epithelial cells. Bar = 20 µm. D. Persistent lineage tracing in jejunum from a subset of crypt-based Prox1+ cells in tamoxifen-treated Prox1-CreER; Rosa26 tdTomato mice, 9 to 365 days after tamoxifen treatment. Lineage tracing was observed in approximately 1/150 crypts. Labeling was also observed in lymphatic vessels (“lv”, arrow) consistent with the known expression of Prox1 in lacteals and in ~10% of villi. Bar = 50 µm. E. Multi-potency of Prox1-expressing cells. Differentiation markers were detected by IF within Prox1 lineage stripes, with Fabp1 for enterocytes, Muc2 for goblet cells, ChgA for EE cells and Lyz for Paneth cells. Bar = 20 µm. F. Injury-inducibility of Prox1+ lineage traces in tamoxifen-treated Prox1-CreER; Rosa26 tdTomato mice. Tamoxifen was administered 2 days prior to delivery of 12 Gy whole body irradiation, followed by tissue harvest after an additional 7 days. Bar = 50 µm.
As single-cell mRNA-seq (scRNA-seq) can be used to examine cellular identity and heterogeneity at single-cell resolution, we performed scRNA-seq characterization of Prox1+ intestinal epithelial cells. We isolated >1,000 Prox1-GFP+ cells from a Prox1-GFP reporter mouse (Figure 4) using a droplet-based system allowing high cell throughput and >50% single cell capture efficiency (Zheng et al., 2016). These sorted Prox1-GFP+ single cells were lysed followed by in-gel bead cDNA barcoding and next-generation 3’ sequencing. Clustering analysis of 1,051 Prox1-GFP+ cells revealed six heterogeneous populations of cells (designated 0–5) (Figure 4A; Figure S2; Table S6A–C). Despite this heterogeneity, Prox1-GFP+ cells exhibited strong concordance with Prox1 mRNA upon scRNA-seq, validating the Prox1-GFP allele as a faithful reporter (Figure 4B). Cluster 4 expressed high-level secretory EE mRNAs consistent with a mature EE cell population. Additionally, Clusters 0–3 represented tuft cells that co-expressed characteristic markers including Dclk1 and Trpm5 in addition to modest levels of Gip and Sct EE mRNAs (Figure 4B–D). These findings support reports of ChgA expression by microarray analysis in FACS-isolated GFP+ cells from Trpm5-GFP transgenic mice (Bezencon et al., 2008). Notably, the tuft/EE coexpressing population exhibited modest expression of Lgr5 and Ascl2 but not Olfm4 or other canonical CBC signature genes. Furthermore, these cells also exhibited Nfat5 and Nfatc3 (Figure 4B), potentially consistent with the previously described LRC, a secretory progenitor that co-expresses stem cell and secretory lineage markers including EE loci (Buczacki et al., 2013). The Prox1-GFP+ heterogeneity, encompassing both conventional EE cells and tuft cells having low-level EE gene expression, supports the concept of a precursor/progenitor shared between the EE and tuft lineages (Formeister et al., 2009; Kokrashvili et al., 2009).
Figure 4. Single-cell transcriptomics of Prox1-GFP+ cells.

A. t-SNE projection of 1,051 Prox1-GFP+ cells colored by clustering. B. Expression of key genes including Prox1 and markers of stem/progenitor cells. Lgr5, Ascl2 and Olfm4 represent canonical CBC markers whereas Nfat5, Nfatc3 and Cd83 are LRC/secretory progenitor makers. C. Tuft cell marker expression among the Prox1-GFP+ clusters. D. EE marker expression among the Prox1-GFP+ clusters. E. Immunofluorescence validation of EE (ChgA) and tuft (Dclk1) marker expression in Prox1+ cells. Bar = 20 µm.
The identity of Prox1+ cells and their degree of overlap with Bmi1- and Lgr5-expressing cells was explored by comparative single-cell RNA-seq. We generated single cell sequencing libraries from sorted cells collected in parallel on the same Chromium Single Cell Chip and sequenced in parallel to minimize experimental variability and any confounding batch effects. Gene-cell matrices from all samples were concatenated, followed by principal component analysis (PCA) to reduce dimensionality before clustering and t-SNE projection. In total, we compared 1,607 FACS-isolated Lgr5-eGFP+, 658 crypt Bmi1-GFP+ cells and 246 Prox1-GFP+ single cells from Lgr5-eGFP-IRES-CreER, Bmi1-GFP, or Prox1-GFP mice, respectively, (Tables S6–S8) versus a fourth control sample of 1,010 Lgr5-eGFP(–) intestinal epithelial cells from an Lgr5-eGFP-IRES-CreER mouse (Tables S6–S8), revealing 13 distinct clusters. The Lgr5-eGFP(−) sample recapitulated clusters corresponding to enterocytes, EE, Paneth, goblet and tuft cells (Figure 5A and Figure S3). Lgr5+ ISCs and Bmi1-GFP+ cells represented small populations relative to these other cell types within the control Lgr5-eGFP(–) sample but were strongly represented within their FACS-enriched samples (Figure 5A and Figure S3). Remarkably, we did not observe significant overlap of Prox1-GFP+ or Bmi1-GFP+ cells with the Lgr5-eGFP+ population at the single-cell level. Consistent with our prior results (Yan et al., 2017), scRNA-seq revealed Lgr5-eGFP+ heterogeneity including non-cycling Lgr5+ ISCs, cycling Lgr5+ ISCs and proliferating Ki67+ cells/transit-amplifying (TA) cells that were distinct from Prox1-GFP+ or Bmi1-GFP+ cells (Figure 5 and Figure S3; Tables S7–8).
Figure 5. Direct comparison of Prox1-GFP+, Bmi1-GFP+ and Lgr5-eGFP+ cells at single-cell resolution.

A. (Top left) Composite t-SNE projection of 1,607 Lgr5-eGFP+, 658 crypt Bmi1-GFP+, 246 crypt Prox1-GFP+ and 1,010 Lgr5-eGFP(−) cells color-coded by FACS-isolated sample. (Top right) Composite t-SNE plot reveals 13 distinct clusters identified by their gene expression. (Bottom) Deconvolution of the cells into their four individual component samples. All five EE clusters visualized by t-SNE represent Bmi1-GFP+ cells (Clusters 3, 6, 7, 10, 12) are globally distinct from Lgr5-eGFP+ cells. B. Bmi1-GFP+ cells express EE genes at the single-cell level. Gene expression heatmaps overlaid on t-SNE plot of crypt Bmi1-GFP+ cells. All five EE clusters representing Bmi1-GFP+ cells express various EE secretory products or EE cell fate determination transcription factors. Bmi1-GFP+ cells (>92%) express EE hormone and transcription factor mRNAs consistent with EE cell identity but lack Lgr5+ ISC signature, secretory progenitor and other lineage marker genes.
In agreement with bulk RNA-Seq data, >92% of individual Bmi1-GFP+ cells expressed EE secretory marker mRNAs such as Chga, Chgb, Cck, Sct, Gip, Glp-1, Pyy and Nts, indicating predominant identity as EE lineage cells (Figure 5 and Figure 6). Bmi1-GFP+ cells existed as 5 distinct EE subpopulations, designated as Clusters 3, 6, 7, 10 and 12, which expressed overlapping but distinct permutations of EE secretory marker mRNAs (Figure 5B and Figure 6). EE secretory mRNAs such as Sct and Chgb were strongly expressed in all five Bmi1-GFP clusters, while others such as Chga, Gip, Tac1, Tph1, Sst, Nts, Reg4 and Cck were variably expressed but typically present in at least low levels, reflecting the known EE cell heterogeneity of secretory product expression (Egerod et al., 2012; Engelstoft et al., 2013; Grun et al., 2015; Habib et al., 2012). The Bmi1-GFP+ clusters typically expressed Neurod1 but not Ngn3 (Figure 5B) in agreement with bulk RNA-seq (Figure 2) and again suggestive that Bmi1-GFP+ cells represent an EE cell with significant differentiation, versus an EE progenitor. Prox1 was expressed in Bmi1-GFP subsets (Figure 5B), corroborating in vivo lineage tracing similarities between Prox1-GFP+ (Figure 3) and Bmi1-GFP+ cells (Yan et al., 2012). Notably, Bmi1 mRNA itself was not exclusively restricted to Bmi1-GFP+ clusters. Bmi1-GFP+ cells likely represent cells with higher relative expression of Bmi1, given their haploinsufficiency for the Bmi1 allele. In addition to Prox1, the EE lineage transcription factors Arx, Pax6 and Neurod1 were also expressed in Bmi1-GFP+ scRNA-seq subpopulations. Interestingly, the five Bmi1-GFP+ EE cell clusters neither overlapped with Lgr5-eGFP+ nor detectably expressed Lgr5. (Figure 5 and Figure 6). Although ~1% of Bmi1-GFP+ cells localized to Lgr5+ ISC cycling and non-cycling clusters, these single Bmi1-GFP+ cells notably lacked expression of signature CBC markers Olfm4, Tnfrsf19, Rnf43 and Znrf3 (Figure 5 and Figure 6).
Figure 6. SPADE single-cell gene expression lineage hierarchy analysis reveals relationship of Prox1-GFP and Bmi1-GFP cell clusters.

A. SPADE lineage hierarchy reconstruction of the intestinal epithelium determined for all cells for the annotated 13 lineages by clustering analysis and t-SNE projection of scRNA-seq data. Cells on the same or neighboring branches are expected to be more hierarchically related compared to cells on different branches in a given tree. A. Hierarchical relationships between the 13 clusters colored by cluster identity. B. Hierarchical relationships colored by sample type. Notably, Bmi1-GFP+ cells mark clusters at the distal branches of the EE lineage whereas Prox1-GFP+ cells mark the tuft lineage and more proximal branches of the EE lineage.
Consistent with the original 1,051 Prox1-GFP+ cells analyzed in isolation (Figure 4), the 246 newly isolated Prox1-GFP+ cells were primarily divided into two major subsets upon direct comparison to the other sorted cell populations (Figure 5A and C). This apparent discrepancy in the number of clusters is attributable to the comparison of Prox1+ cells to all other epithelial subtypes, which overall renders Prox1+ subsets more globally similar in t-SNE analysis. Here, ~52% of Prox1-GFP+ single cells directly overlapped with Bmi1-GFP+ cells with the vast majority of overlap observed in the Cluster 3 EE subset. These Prox1-GFP+ Cluster 3 EE cells exhibited high-level expression of EE marker mRNAs including Cck, Neurod1 and Nts (Figure 5C; Figure S3; Table S6B). The remaining Prox1-GFP+ cells were found in Cluster 8, which exhibited a transcriptional profile consistent with tuft cells, specifically expressing Dclk1, Trpm5, Nrgn, Cox1 and low-level expression of EE secretory mRNAs such as Sct (Figure 5C; Table S6C).
We performed unsupervised placement of Bmi1-GFP+ and Prox1-GFP+ cells along the intestinal differentiation hierarchy using scRNA-seq data and computational analysis with SPADE (Spanning Tree Progression of Density-normalized Events) (Anchang et al., 2016; Qiu et al., 2011), which infers developmental trajectories from single-cell expression data in the form of a minimum spanning tree. We assigned a limited number of genes to identify each major cell type including Lgr5+ ISC, enterocyte, enteroendocrine, goblet, tuft and Paneth cells. These genes included both highly expressed transcripts within individual clusters as well as highly cluster-specific loci. To infer the underlying intestinal differentiation hierarchy, SPADE was optimized on a limited number of 51 genes comprised of a mixture of both enriched cluster-specific genes as well as high variable genes (Table S7). Cells on the same or neighboring branches are expected to be more hierarchically related compared to cells on different branches in a given tree. The SPADE lineage hierarchy was determined for all cells for the annotated 13 lineage subpopulations identified by visualizing median expression profiles (Figure S4) for some key genes on the SPADE tree, concordant with the subgroups previously identified by clustering analysis and t-SNE projection (Figure 5). The SPADE tree displayed a directed hierarchical relationship of the various subgroups conditioned on the root nodes of high density non-cycling and cycling Lgr5+ ISCs (Clusters 0 and 1, respectively), which is consistent with the known biology of intestinal differentiation (Cheng and Leblond, 1974; Clevers, 2013). SPADE analysis revealed that the five Bmi1-GFP+-derived EE clusters (Figure 5) occupy the distal, differentiated branches of the EE lineage (Figure 6). Interestingly, our SPADE analysis revealed a detailed lineage hierarchy among the EE subpopulations with Cluster 3 EE cells serving as a common precursor for the other four EE Clusters 6, 7, 10 and 12; furthermore, the tuft lineage was closely related to that of EE cells (Figure 6A). In our analysis, Bmi1-GFP+ and Prox1+ cells overlap in identity and are related by lineage. Importantly, Prox1 labeled both tuft and EE cells localized in the more truncal branches of the hierarchy tree where these lineages diverge, whereas Bmi1-GFP additionally labeled terminal branches representing mature EE cells (Figure 6B). The Prox1+ tuft marker expression while still simultaneously retaining low-level EE expression is suggestive of recent tuft cell commitment and concomitant ongoing EE marker loss.
DISCUSSION
The numerous proposed markers of both actively cycling and quiescent ISC raise questions of their overlap, interrelatedness and plasticity. Here, we performed a comprehensive RNA-seq-based transcriptome survey of diverse ISC populations with unsupervised analyses revealing the existence of two hierarchical clusters. Cluster 1 contains Lgr5hi and Cd166+, Cd24lo, Grp78− and upper SP populations, unified by common enrichment for a CBC gene signature containing Lgr5 itself, Ascl2 and Olfm4 amongst others. Interestingly, although Cd166+ and Grp78− cells are obtained by Cd24lo sub-fractionation (Levin et al., 2010; Wang et al., 2013), the single marker Cd24 alone appears sufficient for prominent enrichment for an Lgr5-like transcriptome. On the other hand, the additional marker fractionation present in Cd166+ and Grp78− likely assists in the removal of Paneth cells from Cd24lo, which can be alternatively achieved by superimposed UEA sorting. The strong degree of interrelatedness of the Cluster 1 populations strongly support the use of Cd166+, Cd24lo, Grp78−, and upper SP for prospective FACS-based isolation of CBC-type ISCs, for instance, in primary human tissues where knock-in mouse alleles are not applicable.
The presence of both Bmi1+ and mTert+ populations in Cluster 2 parallels reported common features of these ISCs including baseline quiescence, crypt localization and injury-inducible lineage tracing (Montgomery et al., 2011; Sangiorgi and Capecchi, 2008; Yan et al., 2012). Certainly, our analyses are not comprehensive and other unevaluated populations such as LRC, Dll1+ or Lrig1+ (Buczacki et al., 2013; Powell et al., 2012; van Es et al., 2012; Wong et al., 2012) could either exhibit similarities to Clusters 1 or 2, or demonstrate more unique features such as for Hopx, SP or Dclk1 cells. Further, these studies suggest that despite marker overlap and plasticity (Sonoshita et al., 2011; Tian et al., 2011; van Es et al., 2012), distinct ISC species can exhibit distinguishing patterns of global gene expression. Indeed, in agreement with prior studies (Itzkovitz et al., 2012; Munoz et al., 2012), we detected transcripts representing Cluster 2 markers at low levels in Cluster 1 populations, although these “+4” transcripts are generally de-enriched in Cluster 1 versus Cluster 2. Detection of Bmi1+ ISC has typically utilized tamoxifen labeling of Bmi1-CreER mice bred to Rosa26 reporter strains (Ootani et al., 2009; Sangiorgi and Capecchi, 2008; Tian et al., 2011; Yan et al., 2012). However, this potentially detects both Bmi1+ ISCs and their downstream progeny despite short-term (<24h) tamoxifen exposure. In contrast, the present studies performed FACS isolation from Bmi1-GFP knock-in mice, allowing direct fluorescence detection of Bmi1+ cells and not their progeny (Hosen et al., 2007; Tian et al., 2011), thus minimizing associated transcriptome dilution. In our hands, the Bmi1-GFP strategy appears superior as it allows removal of differentiation markers that are otherwise observed in RNA-seq from short-term (<24h) tamoxifen-traced Bmi1-CreER; Rosa26-YFP mice. Furthermore, Bmi1-GFP+ cells retain the clonogenic culture activity that we previously described for short-term tamoxifen-labeled cells from Bmi1-CreER; Rosa26-YFP mice (Yan et al., 2012).
The Bmi1-GFP transgene defines a cell population demonstrating prominent EE lineage commitment (>92%) upon bulk and single-cell RNA-seq. Here, scRNA-seq of 658 individual Bmi1-GFP+ cells revealed significant Bmi1-GFP heterogeneity within distinct subpopulations expressing overlapping subsets of EE secretory and EE transcription factor mRNAs. We currently cannot specifically attribute the clonogenic organoid-forming activity of Bmi1-GFP+ cells to any individual subpopulation(s), which will be a subject for further investigation. Thus, it remains entirely possible that stem cell activity resides in individual Bmi1-GFP+ subset(s). Bmi1-GFP may mark a subset of mature EE cells as our Bmi1-GFP+ bulk and single-cell RNA-seq analyses were confined to crypt as opposed to villus fractions. Crucially, computational lineage inference from scRNA-seq reveal Bmi1-GFP+ cells as mature EE cells, consistent with absent Neurog3 mRNA. The scRNA-Seq analyses indicate that Bmi1-GFP selectively and fortuitously marks a subset of total Bmi1 mRNA-expressing cells, consistent with the companion paper by Jadhav et al.
The Bmi1-GFP EE signature further resembles a recently-described “multi-capable” EE population that co-expresses a common module of Cck, Sct, Gip, Glp-1, Pyy and Nts (Egerod et al., 2012; Engelstoft et al., 2013; Habib et al., 2012), express Prox1 and Neurod1 but not Neurog3, and that is ablated by homeobox gene Arx deletion (Beucher et al., 2012; Du et al., 2012); Arx is highly enriched in Bmi1+ ISCs. Bmi1-GFP+ cells also resemble an EE cell subset that migrates to the crypt base to co-express Cck, Secretin, Gip, Glp-1, Pyy, Nts, Ghrl, Lgr5 and Dclk1(Sei et al., 2011) (Bjerknes and Cheng, 1981). (Rindi et al., 1999; Roth et al., 1992). The current data for Bmi1-GFP, along with published single-cell analysis of Reg4+ EE cells (Grun et al., 2015), support established notions of significant heterogeneity within multi-capable and conventionally defined EE populations (Egerod et al., 2012; Engelstoft et al., 2013; Habib et al., 2012; Rindi et al., 1999; Roth et al., 1992).
The EE transcription factor Prox1 is enriched in the Bmi1-GFP+ transcriptome and Prox1 independently marks injury-inducible ISCs, similar to Bmi1+ ISC (Yan et al., 2012). In contrast to constitutive lineage tracing from Neurog3-Cre (Schonhoff et al., 2004), the current Prox1-CreER strain allows inducible tracing from adult cells. Indeed, Prox1 marks quiescent mouse crypt EE cells that express Pyy, Cck, and Glp1 (Petrova et al., 2008), while in Drosophila, the Prox1 ortholog Pros robustly marks and is functionally required for direct generation of midgut intestinal EE cells via symmetric and asymmetric ISC divisions (Guo and Ohlstein, 2015; Zeng and Hou, 2015). Our data suggest overlap between Bmi1+ (Yan et al., 2012) and Prox1+ cells and implicate Prox1 as a novel marker for injury-inducible ISCs that exhibit extensive similarities with Bmi1+ ISCs including crypt localization, EE gene expression and radio-resistance. However, distinct from Bmi1-GFP+ cells, intestinal epithelial Prox1+ cells possess dual EE and tuft identity with either high level EE gene expression, or high levels of tuft mRNAs associated with low levels of EE mRNAs without Neurog3 mRNA. The common subset of EE cells marked by Prox1-GFP and Bmi1-GFP could conceivably possess the regenerative capacity seen upon clonogenic culture and in vivo lineage tracing. However, we cannot exclude that this activity arises from non-EE-based lineage tracing from Prox1+ or Bmi1-GFP+ populations, albeit quite rare in the latter. Alternatively, multiple populations, including differentiated EE and tuft cells could possess injury-induced regenerative capacity. Previous studies have failed to show Dclk1+ tuft cell lineage tracing during intestinal homeostasis and suggest that tuft cells are quite radiosensitive (Nakanishi et al., 2013; Westphalen et al., 2014). In contrast, Prox1-CreER; Rosa26-tdTomato-derived Prox1+ cells, comprised of both Tufthi/EElo and EEhi subsets, produce both homeostatic and radiation injury-induced lineage tracing.
Several precedents for the stem cell potential of intestinal secretory progenitors have been recently described. Dll1+ crypt cells are short-lived secretory progenitors that express the EE marker Neurog3 and produce long-lived lineage traces upon damage (van Es et al., 2012). Further, quiescent +4 label-retaining cells (LRC) are secretory progenitors co-expressing Lgr5, Paneth and EE markers under homeostasis, but robustly lineage trace after injury (Buczacki et al., 2013). Additionally, differentiated intestinal enterocytes exhibit injury-inducible stem cell activity (Tetteh et al., 2016). Adult EE cells have been proposed to have in vivo stem cell potential, although lineage tracing from EE marker genes was not employed (Sei et al., 2011; Van Landeghem et al., 2012). Intriguingly, both Neurog3-Cre (Schonhoff et al., 2004), and Nkx2.2-Cre (Gross et al., 2015) robustly mark adult small intestine EE cells and reveal epithelial lineage traces that include all four small intestinal lineages; however, the use of constitutively active Cre strains did not allow inducible lineage tracing in adult animals so cell interconversion events originating in embryogenesis could not be excluded. Interestingly, other studies have suggested that Neurog3+ cells exclusively contribute to EE lineages without multi-lineage potential (Bjerknes and Cheng, 2006). There may be substantial overlap between Nkx2.2+, Prox1+ and Bmi1+ cells, and indeed Nkx2.2 is strongly enriched in our Bmi1-GFP transcriptome. However, Nkx2.2+ ISCs do not appear to be mobilized by injury (Gross et al., 2015), in contrast to that of Prox1+ and Bmi1+ cells.
Overall, our unsupervised hierarchical clustering of diverse ISC populations reveals CBC-like and quiescent/reserve-like subsets. Our findings are consonant with an emerging concept that secretory lineage precursors or enterocytes can serve as injury-inducible ISCs that facilitate epithelial regeneration and indicate that such plasticity in fact extends to highly differentiated intestinal EE cells marked by Bmi1-GFP. The companion paper by Jadhav et al. also ascribes prominent EE characteristics to Bmi1-GFP cells, which furthermore undergo conversion to a Lgr5-like chromatin state upon injury. Our studies highlight the utility of single-cell transcriptomics to explore questions of stem cell heterogeneity, lineage hierarchy and prospective stem cell marker discovery. These results, combined with a growing body of evidence suggesting the stem cell potential of secretory progenitors, should catalyze further investigation into the plasticity of the EE lineage during intestinal regeneration.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, Dr. Calvin Kuo (cjkuo@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Participating Institutions and Bulk Cell Data
This study includes mRNA sequencing data from 10 bulk cell populations isolated in laboratories across six different institutions. Adult male and female mice between 6 to 12 weeks of age were used for our bulk cell RNA-seq studies. A total of n=3 mice (i.e. biological replicates) were used per population. For each population, 3 samples were prepared per mouse, including a marker positive, marker negative, and total epithelial fraction. The populations and laboratories include Lgr5-eGFPhi, mTert-GFP, Bmi1-GFP, Bmi1-CreER; Rosa-YFP (Stanford University); Cd44+Cd24loCd166+Grp78− (termed Grp78−, Stowers Institute); Dclk1-GFP (University of Oklahoma), Cd166+Cd24− (termed Cd166+, Oregon Health Sciences University); side population (SP, lower and upper) and Cd24lo (University of North Carolina); and Hopx-GFP (University of Pennsylvania/Stanford). Our single-cell studies utilized adult male and female mice between 8–20 weeks of age. The specific genotypes and number of replicates used in our analyses are described below in Methods Details.
METHOD DETAILS
Mice
Adult male and female mice between 6 to 12 weeks of age were used for FACS isolation of all cell populations for bulk cell RNA-seq analysis. Wild type C57BL/6J mice (The Jackson Laboratory; Bar Harbor, ME) were used to isolate Cd166+, Cd24lo, and SP cells. The following transgenic reporter mice were utilized: Lgr5-eGFP-IRES-CreER (Hans Clevers, JAX) (Barker et al., 2007; Sato et al., 2009), mTert-GFP (David Breault) (Breault et al., 2008; Montgomery et al., 2011) and Hopx-CreER; Rosa26-mTmG (Jonathan Epstein) (Takeda et al., 2011). Bmi1 ISC isolation utilized either acute tamoxifenlabeled Bmi1-CreER; Rosa26-YFP mice (Sangiorgi and Capecchi, 2008; Yan et al., 2012) or Bmi1-GFP mice containing a GFP knock-in at the Bmi1 start codon (Irv Weissman) (Hosen et al., 2007). The Bmi1-GFP reporter strain allows direct detection of Bmi1+ ISC (Tian et al., 2011) and removes contamination by Paneth cells and potentially other Bmi1 progeny that have been associated with Bmi1+ detection by short-term lineage tracing detection in Bmi1-CreER; Rosa26-YFP mice (Sangiorgi and Capecchi, 2008; Yan et al., 2012). Hopx-CreER; Rosa26-mTmG and Bmi1-CreER; Rosa26-YFP mice were treated with a single dose of intraperitoneal tamoxifen (2 mg/head) to label cells and the intestine was harvested 20 hours later for cell isolation. Prox1+ cells were analyzed using Prox1-GFP (Choi et al., 2011) or Prox1-CreER mice (Srinivasan et al., 2007), which were a kind gift of Guillermo Oliver. Lineage tracing experiments were conducted in Prox1-CreER; Rosa26-tdTomato mice using a single dose of intraperitoneal tamoxifen (2 mg/head) and intestinal tissue was analyzed at various time points after administration. All animal procedures were performed with the approval and authorization of the Institutional Review Board and/or Institutional Animal Care and Use Committee at each participating institution. Animals were housed and bred under according to institutional guidelines. Specifically, animals were housed in a controlled environment with 12 hour light/dark cycles and standard diet and water ad libitum. No blinding, randomization or sample-size estimates were performed. All data were analyzed, thus no exclusion criteria were used.
Immunofluorescence (IF) co-localization
Proximal jejunum from Bmi1-GFP mice was used for IF colocalization of Bmi1-GFP with ChgA or serotonin. Five µm frozen mouse intestinal tissue sections were used for immunostaining. Primary antibodies were used to stain against GFP (rabbit anti-GFP, 1:500, Life Technologies; chicken anti-GFP, 1:250; Aves Labs) and enteroendocrine (EE) markers (rat antiserotonin, 1:500, Abcam; rabbit anti-chromogranin A, 1:500, Abcam), and Prox1 (rabbit anti-Prox1, 1:200, RELIATech; or goat anti-Prox1, 1:250, R&D). The staining was then visualized by fluorescence microscopy with fluorescent-conjugated species-specific secondary antibodies (Alexa Fluor 488, 1:500, Life Technologies; indocarbocyanine5 [Cy5], 1:500, Jackson ImmunoResearch), (Davies et al. BMC Gastroenterology 2008; 8:57). Nuclei were counterstained with Hoechst 33258 dye (0.1 µg/mL; Sigma-Aldrich; St Louis, MO). Tissue sections were analyzed on a Leica DM6000 microscope. Quantification of single Bm1-GFP expressing versus Bmi1-GFP/EE cells was determined by analyzing 500 crypts per mouse from Bmi1-GFP reporter mice (n = 5). Quantification of single Bmi1-GFP expressing versus Bmi1-Prox1 cells was determined by analyzing 200–500 crypts/mouse (n=4).
10 µm frozen sections from proximal jejunum of Prox1-CreER; Rosa26-tdTomato mice were used for IF localization of enterocytes (rabbit anti-FABP1, 1:100, Novus Biologicals), goblet (rabbit anti-Mucin 2, 1:100, Santa Cruz), EE (chromogranin A, 1:200, Abcam), and Paneth cells (Lysozyme, 1:200, Dako) within Prox1-tdTomato lineage stripes. Staining was visualized by fluorescent microscopy (Leica DMi8 microscope) with fluorescent conjugated species-specific secondary antibody (Donkey anti-Rabbit Alexa Fluor 488, 1:500, Invitrogen).
Five µm paraffin embedded sections from proximal jejunum were used for IF colocalization of Prox1 (goat anti-Prox1, 1:100, R&D Systems) with either Dclk1 (rabbit anti-Dclk1, 1:200, Abcam) or ChgA (rabbit anti-chromogranin A, 1:200, Abcam). Staining was visualized through fluorescence microscopy (Leica DMi8 microscope) using species-specific secondary antibodies (donkey anti-goat Alexa Fluor 568 and donkey anti-rabbit Alexa Fluor 488, respectively).
Intestinal Epithelial Cell (IEC) Preparation
For all bulk cell and single cell mRNA-seq analysis of different cell populations, a 10cm segment of proximal jejunum (designated as cm 2–12 past the gastroduodenal junction, following discard of the first 2 cm of duodenum) was dissected and used to obtain dissociated epithelial cells, as previously described (Magness et al., 2013). The intestinal segment was flushed with cold PBS and the tissue was opened longitudinally and rinsed extensively with cold PBS to remove debris. The tissue was then incubated with 30 mM EDTA on ice for 20 minutes. The epithelial layer was liberated from the stroma/muscle using vigorous manual shaking and the stroma/muscle was then discarded. The remaining epithelial cells were pelleted by centrifugation, resuspended in cold PBS and subsequently treated with dispase/collagenase 30 units/ml (Roche) at 37°C for 10 minutes with gentle agitation for enzymatic dissociation into a single-cell suspension. Cells were then placed on ice and FBS was added to 10% of the final volume. This epithelial cell suspension was then passed through a 40 µm cell strainer to generate a single-cell suspension.
Flow Cytometry and Sample Collection
Mouse intestinal epithelial cells (IECs) were dissociated to single cells as described above (Magness et al., 2013) and all populations were purified by FACS using sorters with large bore (100–130 µm) nozzles at the respective institutions. After scatter discrimination to remove doublets and enrich for epithelium, the IEC preparation was negatively selected with propidium iodide, anti-CD45, and anti-CD31 to remove dead, hematopoietic and endothelial cells, respectively. The cells were then positively selected with anti-EpCAM to enrich for IEC as described (Magness et al., 2013). Conditions for cell sorting and sample collection have been previously described for Cd24lo (von Furstenberg et al., 2011), SP (von Furstenberg et al., 2014), Dclk1+ (Chandrakesan et al., 2015), Grp78− (Wang et al., 2013) and Cd166+. For Cd166mid/Cd24mid cells, additional pre-enrichment with the cryptenriching novel monoclonal antibody B6A6 (ref. (Wang et al., 2013)) was performed. GPR78− cells were derived from a composite Cd166hi, Cd24lo Cd44+ gate (Wang et al., 2013). SP purification utilized Hoechst dye efflux with FACS UV laser (von Furstenberg et al., 2014). Antibodies for FACS purification included: anti-rat Alexa-488 secondary (Jackson Immuno Research, 1:200), Anti-mouse CD31-PE/Cy7 (Biolegend, clone 390, 1:200), anti-mouse CD45-PE/Cy7 (Biolegend, clone 30-F11, 1:200), anti-mouse CD24-APC (Biolegend, clone M1/69, 1:200) and anti-mouse CD166-PE (R&D Systems, 1:200), anti-GPR78 and rat mAb B6A6 (Melissa Wong, undiluted(Wang et al., 2013)). In general, samples were collected from n=3 biological replicates (i.e. from independent mice). These consisted of a markerpositive population, a marker-negative population and a total epithelial population, each consisting of up to 10,000 cells sorted directly into RNA lysis buffer (Lysis Buffer RLT; Qiagen, Valencia, CA). RNA was extracted and RNA-seq libraries were constructed and submitted for RNA-sequencing analysis at the Stanford Genome Center.
The Lgr5hi, Bmi1-GFP, mTert-GFP and Hopx+ populations were obtained by jejunal IEC isolation, with negative selection for dead/stromal components and positive EpCAM selection for epithelium as described above. This was followed by FACS purification using intrinsic GFP fluorescence from Lgr5-eGFP-IRES-CreER (Barker et al., 2007; Sato et al., 2009), crypt Bmi1-GFP (Tian et al., 2011) using crypt-enrichment with the monoclonal antibody B6A6, mTert-GFP (Breault et al., 2008; Montgomery et al., 2011) and Hopx-CreER; Rosa26-mTmG (Takeda et al., 2011), respectively.
Clonogenic intestinal enteroid culture
Bmi1-GFP+ cells or Prox1-GFP+ cells were isolated using the endogenous GFP signal to sort cells on an FACSAria II cell sorter (BD) from single-cell suspensions of proximal jejunal epithelium from Bmi1-GFP or Prox1-GFP mice. Sorted cells were grown in Matrigel with Advanced DMEM/F12 supplemented with Glutamax, HEPES, B27, N2, Y27632 (10 µM), EGF (50 ng/ml), Noggin (100 ng/ml) and R-spondin1 (500 ng/ml) as previously described(Yan et al., 2012).
Bulk cell RNA library prep and sequencing
A 1.8× volume of AMPure beads (Beckman Coulter, Brea, CA) was added to the thawed cell lysates (~10,000 cells sorted into 500 µl Lysis Buffer RLT (Qiagen, Valencia, CA)). After a 30 minute incubation at room temperature, the samples were washed 2× with 70% ethanol and eluted in 22 µl water. The samples were then digested with 0.6 mAU Proteinase K (Qiagen, Valencia, CA) in the presence of 1× NEB Buffer 1 (New England Biolabs, Ipswitch, MA) at 50°C for 20 minutes, followed by a heat inactivation step at 65°C for 10 minutes. A DNase digestion was performed using the RNase-Free DNase Set (Qiagen, Valencia, CA) at 37°C for 30 minutes. The samples were cleaned with a 1.8× volume of AMPure XP beads (Beckman Coulter, Brea, CA). 1 ng of purified total RNA, as determined by Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA), was processed with the mRNA direct micro kit (Life Technologies, Foster City, CA) to select for poly A RNA. Each entire sample was input into the Ambion WT Expression Kit (Life Technologies, Foster City, CA) to perform double stranded cDNA synthesis followed by in vitro transcription to generate amplified cRNA. The cRNA was purified following the manufacturer’s instructions and the concentration was determined by a NanoDrop instrument (Thermo Fisher Scientific, Waltham, MA). 1 µg of cRNA was fragmented in 1× Fragmentation Buffer (mRNA-seq Sample Prep Kit, Illumina, Hayward, CA) at 94°C for 5 minutes, then placed on ice and the reaction was stopped by the addition of 20 mM EDTA. The fragments were precipitated with 70 mM sodium acetate (Life Technologies, Foster City, CA), 40 µg glycogen (Life Technologies, Foster City, CA) and 70% ethanol at −80°C for 1 hour followed by centrifugation and washing with 70% ethanol. 3 µg of random hexamer (Life Technologies, Foster City, CA) was added to the fragmented, purified cRNA and incubated at 70°C for 10 minutes to anneal the primer. The first strand reaction was performed with 200 units of SuperScript II (Life Technologies, Foster City, CA) with 0.625 mM dNTP’s (NEB, Ipswitch, MA) and 8 units SUPERase RNase Inhibitor (Life Technologies, Foster City, CA) at 25°C for 10 minutes, then 42°C for 50 minutes, then 75°C for 15 minutes and cooled to 4°C. In second strand synthesis, 1× second strand buffer (Illumina, Hayward, CA) and 0.3 mM dNTP’s (Illumina, Hayward, CA) were added and the samples were incubated at 4°C for 5 minutes before adding 50 units of DNA Polymerase (NEB, Ipswitch, MA) and 5 units of Rnase H (NEB, Ipswitch, MA). The samples were mixed well and incubated at 16°C for 2.5 hours, followed by purification with the MinElute Kit (Qiagen, Valencia, CA).
To perform library prep, the samples were end repaired using a Quick Blunting Kit (NEB, Ipswitch, MA) and incubated at 20°C for 1 hour, then 75°C for 30 minutes to inactivate the enzyme. A’ bases were added to the 3’ ends of each fragment with 2 mM dATP and 5 units of Klenow fragment 3’–5’ exo-DNA Polymerase (NEB, Ipswitch, MA) at 37°C for 45 minutes, followed by 75°C for 30 minutes to inactivate the enzyme. Using a quick ligase kit (NEB, Ipswitch, MA), 0.5 uM of adaptors were ligated to the cDNA fragments at 12°C for 75 minutes, then 80°C for 20 minutes and cooled to 4°C. These adaptors contain barcodes to facilitate sample multiplexing during sequencing. The adaptor sequence is preceded by four random nucleotides to add diversity to the pooled library. The samples were pooled by combining 5 µl of each library. After AMPure XP cleanup, one half of the pooled library was run on the Pippin Size Selection Instrument (Sage Sciences, Beverly, MA) to select for 200 bp fragments. Library amplification was performed on one half of the Pippin eluate in 1× Phusion GC buffer with 0.2 mM dNTP’s, 0.1 µM forward primer (IDT, Coralville, IA), 0.1 µM reverse primer, 1 unit of Phusion Hot Start II Polymerase (Thermo Fisher Scientific, Waltham, MA). The reaction was run with the following program: 98°C for 30s, then 15 cycles of 98°C for 10s, 65°C for 30s, 72°C for 30s, then 72°C for 4 minutes and cooled to 4°C. The amplified library was cleaned using a 1× volume of AMPure XP beads and QC was run with the Agilent Bioanalyzer DNA 1000 kit, followed by concentration determination by qPCR using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA). To qualify for sequencing, the library must have a minimum concentration of 4 nM and have no trace of low MW (<150 bp) primer/dimer product. To perform sequencing, the library was diluted to 4 nM and denatured with 0.1N NaOH. Following denaturation, the library was further diluted to 4 pM and run on the Illumina HiSeq 2000 or HiSeq 2500 (Illumina, Hayward, CA) in paired-end, 100×100 bp format to a minimum depth of 100M reads per population.
-
Forward adaptor sequence 5’ /5Phos/ATCGCACNNNNAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG 3’
Reverse adaptor sequence 5’ ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNGTGCGATT 3’
-
Red=sample specific barcode sequence
Blue=T overhang to enable TA ligation
-
Reverse library amplification primer (PAGE ultramer): 5’ CAAGCAGAAGACGGCATACGAGATC 3’
Forward library amplification primer (PAGE ultramer): 5’ AATGATACGGCGACCACCGAGATCTA 3’
Bulk cell RNA-seq bioinformatics analysis
Paired-end RNA-seq reads were mapped to the mm9 reference using TopHat 2.08(Kim et al., 2013) which internally uses the Bowtie 2.1 aligner(Langmead et al., 2009) and samtools 0.1.19(Langmead et al., 2009), and additionally finds splice junctions. The resulting transcripts were assembled using cufflinks 2.0(Trapnell et al., 2013). This sequence of steps generated Fragments Per Kilobase Million (FPKM) calls, which is a read count metric normalized for sequencing depth and gene length. In preparation for differential expression analysis, the cuffmerge module of cufflinks 2.0 was run to merge transcripts from each set of biological replicates. Differentially expressed genes were then identified using cuffdiff 2.0(Trapnell et al., 2013). Hierarchical clustering of all populations and external datasets (Lrig1, LRC) (Buczacki et al., 2013) was performed and dendrogram and p-values for interrelationships generated. p-values underwent Benjamini-Hochberg correction for multiple testing to control the FDR. The most differentially expressed genes (FDR<0.05) were used for heatmaps. Gene Ontology (GO) pathway analysis of populations was performed with Bioconductor/GOSeq to remove RNA-seq gene length biases. Gene set enrichment analysis (GSEA) was performed against the Molecular Signatures Database (MSigDB) and ISC marker-positive gene lists.
Single-cell mRNA-seq
Individual FACS-sorted Prox1-GFP+, Bmi1-GFP+, Lgr5-eGFP+, and Lgr5-eGFP(−) cells were FACS isolated from jejunum of Prox1-GFP, Bmi1-GFP and Lgr5-eGFP-IRES-CreER mice, respectively. All mice used in our single-cell analysis were adults (8–20 weeks of age) and male, except for the repeat collection of Prox1-GFP+ cells, which was collected from a female mouse. Prox1-GFP and Bmi1-GFP epithelial cells were isolated from intestinal crypts (enriched using mAB B6A6 to exclude villus cells) as described above. Additionally, Lgr5-eGFP(−) from CD31−CD45−EpCAM+ epithelial cells were also sorted and used in the single cell analysis as a control. Single-cell RNA-seq libraries were generated, sequenced and analyzed as described below.
GemCode platform
The Prox1-GFP+ libraries from a total of 1,051 cells separated into two technical replicates of 521 and 537 cells were made using the GemCode platform. Cellular suspensions were loaded on a GemCode Single Cell Instrument (10× Genomics, Pleasanton, CA) to generate single cell GEMs. Approximately ~1000 cells were loaded per channel. Two technical replicates were generated per sample. Single cell RNA-Seq libraries were prepared using GemCode Single Cell 3’ Gel Bead and Library Kit (now sold as P/N 120230, 120231, 120232, 10× Genomics) following previously described protocols (Zheng et al., 2017). Sequencing libraries were loaded at 2.1 pM on an Illumina NextSeq500 with 2 × 75 paired-end kits using the following read length: 98 bp Read1, 14 bp I7 Index, 8 bp I5 Index and 5 bp Read2. Note that these libraries were generated before the official launch of GemCode Single Cell 3’ Gel Bead and Library Kit. Thus, 5bp UMI was used (the official GemCode Single Cell 3’ Gel Bead contains 10 bp UMI).
Chromium platform
The remainder of the libraries (Bmi1-GFP+, repeat collection of Prox1-GFP+, Lgr5-eGFP+, and Lgr5-eGFP− cells were made using the Chromium platform and Chromium Single Cell 3’ v2 chemistry. Cellular suspensions were loaded on a Chromium Single Cell Instrument (10× Genomics, Pleasanton, CA) to generate single cell GEMs. Approximately ~100–2000 cells were loaded per channel. Two technical replicates were generated per sample. Single cell RNA-Seq libraries were prepared using the Chromium Single Cell 3’ Gel Bead and Library Kit (P/N 120235, 120234, 120236, 120262, 10× Genomics). Sequencing libraries were loaded at 240 pM on an Illumina HiSeq4000 with 2 × 75 pairedend kits using the following read length: 26 bp Read1, 8 bp I7 Index and 98 bp Read2.
Alignment, barcoding and UMI counting
The Cell Ranger Single Cell Software Suite 1.3 was used to perform sample demultiplexing, barcode processing, and single cell 3’ gene counting (http://software.10xgenomics.com/single-cell/overview/welcome). For the old Prox1+ samples, UMI=5 was used for the analysis.
PCA and t-SNE analysis
For the Prox1-GFP+ analysis in Figure 4, 1,058 cells were recovered from 2 technical replicates of the Prox1-GFP+ sample. The gene-cell barcode matrix was filtered based on number of genes detected per cell (any cells with less than 500 or more than 4000 genes per cell were filtered) and percentage of mitochondrial UMI counts (any cells with more than 10% of mitochondrial UMI counts were filtered). Altogether, 1,051 cells and 12,963 genes were kept for analysis by Seurat(Macosko et al., 2015). 2,013 variable genes were selected based on their expression and dispersion (expression cutoff=0.1, and dispersion cutoff=0.5). The first 10 principal components were used for the t-SNE projection(van der Maaten and Hinton, 2008) and clustering analysis (resolution=0.4, k.seed=100). For the samples of Figures 5–6 (Bmi1-GFP+, repeat collection of Prox1-GFP+, Lgr5-eGFP+, and Lgr5-eGFP−) we recovered 4,512 cells from 4 samples, each with two technical replicates (the number of cells recovered per sample ranges from ~40 to 1,000 cells.). The mean raw reads per cell varies from ~43k to 90k. Each sample was downsampled to ~25k confidently mapped reads per cell. Then the gene-cell barcode matrix from each sample was concatenated.
For the analysis of four single-cell samples collected in parallel (Figure 5), the gene-cell barcode matrix was filtered based on number of genes detected per cell (any cells with less than 100 or more than 5000 genes per cell were filtered) and percentage of mitochondrial UMI counts (any cells with more than 10% of mitochondrial UMI counts were filtered). Altogether, 3,521 cells, and 16,025 genes were kept for analysis by Seurat(Macosko et al., 2015). 2,419 variable genes were selected based on their expression and dispersion (expression cutoff=0. 5, and dispersion cutoff=0.5). The first 20 principal components were used for the t-SNE projection and clustering analysis (resolution=0.4, k.seed=100).
For all the sample collections for construction of libraries, we applied sSeq (Yu et al., 2013) to identify genes that are enriched in a specific cluster (the specific cluster is assigned as group a, and the rest of clusters is assigned as group b) respectively to each dataset. There are a few differences between our implementation and Yu et al. 1) we used the ratio of total UMI counts and median of total UMI counts across all cells) as the size factors. 2) The quantile rule of thumb was used to estimate the shrinkage target. 3) For genes with large counts, an asymptotic approximation from the edgeR package (McCarthy et al., 2012) was used instead of the negative binomial exact test to speed up the computation.
For the heatmap in Figure S3b, the gene list was furthered filtered requiring minimum UMI counts of 5 in each group, with a positive log2 fold change of mean expression between the 2 groups, and an adjusted p value of less than 0.01. The top 10 genes specific to each cluster was picked, and their mean expression was center scaled before heatmap generation.
For all datasets, classification of cells was inferred from the annotation of cluster-specific genes. For example, the Lgr5-eGFP+ stem cell clusters (Clusters 0 and 1) were marked by enrichment of Lgr5, Olfm4, and Ascl2. Non-cycling and cycling stem cells were distinguished by the enrichment of cell cycle markers such as Mki67. Enterocytes (Clusters 2, 4 and 9) were annotated based on the enrichment of markers such as Reg1. EE cells (Clusters 3, 6, 7, 10, and 12) were annotated based on the enrichment of markers such as Chga and Chgb. Goblet cells (Cluster 5) were annotated based on the enrichment of markers such as Muc2 and Guca2a. Tuft cells (Cluster 8) were annotated based on the enrichment of markers such as Dclk1. Paneth cells (Cluster 11) were annotated based on the enrichment of Defa genes.
SPADE gene set selection
Since SPADE can produce suboptimal results when poor lineage markers are chosen to build the tree, we summarize the process of selecting the optimal gene set for building the tree. Based on the t-SNE clusters, our gene selection approach combines the top 2–3 significantly enriched genes in one cluster over the rest of clusters with the top high variable genes in each cluster derived using the robust measure of variability, median absolute deviation (MAD) scores (Rousseeuw and Croux, 1993) defined as median of the absolute deviations from the data's median. For each gene, the MAD score is determined from the non-zero count data, minimizing the bias from the high zero density data points.
SPADE computation
Using the R implementation of SPADE (Anchang et al., 2016), results were generated with R version 3.2.3 that runs on Mac OS X. Gene expression counts were asinh column-transformed to minimize the high variability between transcript estimates while keeping the zero counts constant. The minimum spanning tree (MST) layout involves running the usual 4 major steps of SPADE: (1) density-dependent down-sampling of single-cell data, (2) agglomerative clustering, (3) joining clusters with a minimum spanning tree and (4) up-sampling to map all cells onto the final output tree. Each node and node size in the MST output represents a median marker expression and the number of cells within that node respectively. For this particular analysis, SPADE was run on an initial number of 200 clusters with no downsampling by disabling the downsampling parameter setting it to a value of 1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental design
For mouse experiments, all groups consisted of age-matched mice and n represents the number of independent biological replicates. All data were included in the analysis and thus no exclusion criteria were used. Experiments were not blinded. Sample-size estimates were not performed beforehand. Details are provided in the detailed methods section.
Statistical analysis
General statistical analysis for Figures 2 and 3 were performed using n biological replicates as indicated in the detailed methods. Error bars indicated mean ± s.e.m. and statistical differences were assessed by a t-test. Statistical analysis for single-cell RNA-seq and SPADE analysis are described in the detailed methods.
DATA AND SOFTWARE AVAILABILITY
All data generated in these studies were deposited in the Gene Expression Omnibus (GEO) database (using accession GSE99457 and GSE99815) and on the NIH ISCC Consortium website (https://iscconsortium.org) upon publication.
Supplementary Material
(Related to Figure 1 and Figure 2)
FACS-isolated samples were collected across six institutions for RNA-seq library preparation, sequencing and bioinformatics analysis. Samples from three biological replicates were used to examine each marker population of interest, as well as their marker-negative control and unfractionated epithelium control. Sample sequencing and read mapping metrics are indicated for each individual sample.
(Related to Figure 1 and Figure 2)
For each marker-positive population of interest, the number of differentially expressed genes relative to each of their marker-negative control population is shown. Only Bmi1-GFP+, CD166+, CD24lo and Lgr5-eGFP+ populations exhibit >200 differentially expressed genes relative to their marker-negative controls.
(Related to Figure 1 and Figure 2). Detailed differentially expressed genes are shown for each marker-positive population analyzed, including FPKMs, log2 (fold-change), p-values, and q-values.
(Related to Figure 1). Top genes common to populations and other genes of interest are shown in Cluster 1.
(Related to Figure 4 and Figure 5)
Table S6A. Single cell RNA sequencing metrics summary. Single-cell libraries were constructed from sorted Prox1-GFP+, Bmi1-GFP+ and Lgr5-eGFP+ cells, as well as from a sample of unfractionated intestinal epithelial cells. Two technical replicate libraries were constructed from each biological samle. Sequencing metrics for each library are provided. Table S6B. Proportion of individual cell clusters by sample. Table S6C. Cluster-specific genes for 1,051 Prox1-GFP+ cells and upon direct comparison of Bmi1-GFP+, Prox1-GFP+, Lgr5-eGFP+ and Lgr5-eGFP− cells.
(Related to Figure 6).
Highlights.
Bmi1-GFP+ cells express mature enteroendocrine (EE) genes, including Prox1
Prox1-GFP+ and Bmi1-GFP+ cells exhibit ISC properties in clonogenic cultures
Prox1+ cells co-express EE and tuft cell genes in vivo
Lineage tracing shows Prox1+ cells contribute to crypt homeostasis and regeneration
Acknowledgments
We are indebted to Timothy Wang, Moritz Middelhoff, Michael Quante, Kari Alitalo, the members of the NIDDK Intestinal Stem Cell Consortium, and the Kuo lab for extensive discussions. Prox1-CreER and Bmi1-CreER mice were kind gifts of Guillermo Oliver and Mario Capecchi, respectively. K.S.Y. was supported by a CIRM MD Scholars Fellowship, NIH K08DK096048 and Burroughs Wellcome Fund Career Award for Medical Scientists. A.H. was supported by NIH K08DK100739. C.S.P. was supported by the NSF Graduate Research Fellowship DGE-114747, R.J. by BWF CAMS and WW Smith Foundation and D.T.B. by R01DK084056, the Timothy Murphy Fund, IDDRC P30HD18655, and HDDC P30DK034854. This work was also funded by the NIH Intestinal Stem Cell Consortium supported by NIDDK and NIAID UO1DK085547 (S.J.H), U01DK085525 (M.H.W.), U01DK085532 (J.N.), U01DK085508 (C.H.), U01DK085535 (M.M.), U01DK085551 (J.L.) and U01DK085507 (L.L.) and U01DK085527 (C.J.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
K.S.Y. designed and executed experiments, analyzed data and wrote the manuscript. O.G., G.X.Y.Z., B.A., C.S.P., S.G., D.C., C.C. and H.P.J. performed bioinformatics and computational analysis. K.A.L., P.S.D., Z.C., J.S.K., A.H., K.R., R.I.C., E.C., X.Y., K.M., J.W., S.C.B., J.M.T, P.B, S.B.Z., T.S.M., F.W., R.J.vF., N.R.S., P.C., R.M., M.C., and R.J. conducted experiments and analyzed data. C.A.C., J.C.N., Y.H., J.C., D.B., J.E., C.W.H., J.P.L., M.G.M., S.K.P., C.C., H.P.J., L.L., S.J.H., M.H.W., and C.J.K. supervised experiments/analysis. C.J.K. designed experiments and wrote the manuscript.
References
- Anchang B, Hart TD, Bendall SC, Qiu P, Bjornson Z, Linderman M, Nolan GP, Plevritis SK. Visualization and cellular hierarchy inference of single-cell data using SPADE. Nature protocols. 2016;11:1264–1279. doi: 10.1038/nprot.2016.066. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;7165;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Barriga FM, Montagni E, Mana M, Mendez-Lago M, Hernando-Momblona X, Sevillano M, Guillaumet-Adkins A, Rodriguez-Esteban G, Buczacki SJ, Gut M, et al. Mex3a Marks a Slowly Dividing Subpopulation of Lgr5+ Intestinal Stem Cells. Cell stem cell. 2017 doi: 10.1016/j.stem.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beucher A, Gjernes E, Collin C, Courtney M, Meunier A, Collombat P, Gradwohl G. The homeodomain-containing transcription factors Arx and Pax4 control enteroendocrine subtype specification in mice. PLoS One. 2012;7:e36449. doi: 10.1371/journal.pone.0036449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezencon C, Furholz A, Raymond F, Mansourian R, Metairon S, Le Coutre J, Damak S. Murine intestinal cells expressing Trpm5 are mostly brush cells and express markers of neuronal and inflammatory cells. The Journal of comparative neurology. 2008;509:514–525. doi: 10.1002/cne.21768. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H. The stem-cell zone of the small intestinal epithelium. III. Evidence from columnar, enteroendocrine, and mucous cells in the adult mouse. The American journal of anatomy. 1981;160:77–91. doi: 10.1002/aja.1001600107. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H. Neurogenin 3 and the enteroendocrine cell lineage in the adult mouse small intestinal epithelium. Dev Biol. 2006;300:722–735. doi: 10.1016/j.ydbio.2006.07.040. [DOI] [PubMed] [Google Scholar]
- Breault DT, Min IM, Carlone DL, Farilla LG, Ambruzs DM, Henderson DE, Algra S, Montgomery RK, Wagers AJ, Hole N. Generation of mTert-GFP mice as a model to identify and study tissue progenitor cells. Proc Natl Acad Sci U S A. 2008;105:10420–10425. doi: 10.1073/pnas.0804800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. 2013;495:65–69. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- Chandrakesan P, May R, Qu D, Weygant N, Taylor VE, Li JD, Ali N, Sureban SM, Qante M, Wang TC, et al. Dclk1+ small intestinal epithelial tuft cells display the hallmarks of quiescence and self-renewal. Oncotarget. 2015;6:30876–30886. doi: 10.18632/oncotarget.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. I. Columnar cell. The American journal of anatomy. 1974;141:461–479. doi: 10.1002/aja.1001410403. [DOI] [PubMed] [Google Scholar]
- Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, Yoo J, Choi D, Lee YS, Aguilar B, et al. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood. 2011;117:362–365. doi: 10.1182/blood-2010-07-298562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154:274–284. doi: 10.1016/j.cell.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du A, McCracken KW, Walp ER, Terry NA, Klein TJ, Han A, Wells JM, May CL. Arx is required for normal enteroendocrine cell development in mice and humans. Dev Biol. 2012;365:175–188. doi: 10.1016/j.ydbio.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerod KL, Engelstoft MS, Grunddal KV, Nohr MK, Secher A, Sakata I, Pedersen J, Windelov JA, Fuchtbauer EM, Olsen J, et al. A major lineage of enteroendocrine cells coexpress CCK, secretin, GIP, GLP-1, PYY, and neurotensin but not somatostatin. Endocrinology. 2012;153:5782–5795. doi: 10.1210/en.2012-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelstoft MS, Egerod KL, Lund ML, Schwartz TW. Enteroendocrine cell types revisited. Curr Opin Pharmacol. 2013 doi: 10.1016/j.coph.2013.09.018. [DOI] [PubMed] [Google Scholar]
- Formeister EJ, Sionas AL, Lorance DK, Barkley CL, Lee GH, Magness ST. Distinct SOX9 Levels Differentially Mark Stem/Progenitor Populations and Enteroendocrine Cells of the Small Intestine Epithelium. Am J Physiol Gastrointest Liver Physiol. 2009 doi: 10.1152/ajpgi.00004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Balderes D, Liu J, Asfaha S, Gu G, Wang TC, Sussel L. Nkx2.2 is expressed in a subset of enteroendocrine cells with expanded lineage potential. Am J Physiol Gastrointest Liver Physiol. 2015;309:G975–987. doi: 10.1152/ajpgi.00244.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grun D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- Guo Z, Ohlstein B. Stem cell regulation. Bidirectional Notch signaling regulates Drosophila intestinal stem cell multipotency. Science. 2015;350 doi: 10.1126/science.aab0988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CA, Parker HE, Morley TC, Yeo GS, Reimann F, Gribble FM. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology. 2012;153:3054–3065. doi: 10.1210/en.2011-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosen N, Yamane T, Muijtjens M, Pham K, Clarke MF, Weissman IL. Bmi-1-green fluorescent protein-knock-in mice reveal the dynamic regulation of bmi-1 expression in normal and leukemic hematopoietic cells. Stem Cells. 2007;25:1635–1644. doi: 10.1634/stemcells.2006-0229. [DOI] [PubMed] [Google Scholar]
- Itzkovitz S, Lyubimova A, Blat IC, Maynard M, van Es J, Lees J, Jacks T, Clevers H, van Oudenaarden A. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat Cell Biol. 2012;14:106–114. doi: 10.1038/ncb2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokrashvili Z, Rodriguez D, Yevshayeva V, Zhou H, Margolskee RF, Mosinger B. Release of endogenous opioids from duodenal enteroendocrine cells requires Trpm5. Gastroenterology. 2009;137:598–606. 606 e591–592. doi: 10.1053/j.gastro.2009.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488:665–669. doi: 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- Kozar S, Morrissey E, Nicholson AM, van der Heijden M, Zecchini HI, Kemp R, Tavare S, Vermeulen L, Winton DJ. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell stem cell. 2013;13:626–633. doi: 10.1016/j.stem.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin TG, Powell AE, Davies PS, Silk AD, Dismuke AD, Anderson EC, Swain JR, Wong MH. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology. 2010;139:2072–2082 e2075. doi: 10.1053/j.gastro.2010.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–825. doi: 10.1126/science.1196236. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magness ST, Puthoff BJ, Crissey MA, Dunn J, Henning SJ, Houchen C, Kaddis JS, Kuo CJ, Li L, Lynch J, et al. A multicenter study to standardize reporting and analyses of fluorescence-activated cell-sorted murine intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2013;305:G542–551. doi: 10.1152/ajpgi.00481.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. Bioessays. 2002;24:91–98. doi: 10.1002/bies.10028. [DOI] [PubMed] [Google Scholar]
- May R, Sureban SM, Hoang N, Riehl TE, Lightfoot SA, Ramanujam R, Wyche JH, Anant S, Houchen CW. Doublecortin and CaM kinase-like-1 and leucine-rich-repeat-containing G-protein-coupled receptor mark quiescent and cycling intestinal stem cells, respectively. Stem Cells. 2009;27:2571–2579. doi: 10.1002/stem.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic acids research. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk ME, Henderson DE, Baffour-Awuah NY, Ambruzs DM, Fogli LK, Algra S, et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A. 2011;108:179–184. doi: 10.1073/pnas.1013004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent '+4' cell markers. Embo J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, Nakanishi N, Kanda K, Komekado H, Kawada M, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet. 2013;45:98–103. doi: 10.1038/ng.2481. [DOI] [PubMed] [Google Scholar]
- Ootani A, Li X, Sangiorgi E, Ho QT, Ueno H, Toda S, Sugihara H, Fujimoto K, Weissman IL, Capecchi MR, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009;15:701–706. doi: 10.1038/nm.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova TV, Nykanen A, Norrmen C, Ivanov KI, Andersson LC, Haglund C, Puolakkainen P, Wempe F, von Melchner H, Gradwohl G, et al. Transcription factor PROX1 induces colon cancer progression by promoting the transition from benign to highly dysplastic phenotype. Cancer Cell. 2008;13:407–419. doi: 10.1016/j.ccr.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 2012;149:146–158. doi: 10.1016/j.cell.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nature biotechnology. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindi G, Ratineau C, Ronco A, Candusso ME, Tsai M, Leiter AB. Targeted ablation of secretin-producing cells in transgenic mice reveals a common differentiation pathway with multiple enteroendocrine cell lineages in the small intestine. Development. 1999;126:4149–4156. doi: 10.1242/dev.126.18.4149. [DOI] [PubMed] [Google Scholar]
- Roth KA, Kim S, Gordon JI. Immunocytochemical studies suggest two pathways for enteroendocrine cell differentiation in the colon. Am J Physiol. 1992;263:G174–180. doi: 10.1152/ajpgi.1992.263.2.G174. [DOI] [PubMed] [Google Scholar]
- Rousseeuw PJ, Croux C. Alternatives to the Median Absolute Deviation. Journal of the American Statistical Association. 1993;88:1273–1283. [Google Scholar]
- Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Schonhoff SE, Giel-Moloney M, Leiter AB. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol. 2004;270:443–454. doi: 10.1016/j.ydbio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Sei Y, Lu X, Liou A, Zhao X, Wank SA. A stem cell marker-expressing subset of enteroendocrine cells resides at the crypt base in the small intestine. Am J Physiol Gastrointest Liver Physiol. 2011;300:G345–356. doi: 10.1152/ajpgi.00278.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Sonoshita M, Aoki M, Fuwa H, Aoki K, Hosogi H, Sakai Y, Hashida H, Takabayashi A, Sasaki M, Robine S, et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell. 2011;19:125–137. doi: 10.1016/j.ccr.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Srinivasan RS, Dillard ME, Lagutin OV, Lin FJ, Tsai S, Tsai MJ, Samokhvalov IM, Oliver G. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 2007;21:2422–2432. doi: 10.1101/gad.1588407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420–1424. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetteh PW, Basak O, Farin HF, Wiebrands K, Kretzschmar K, Begthel H, van den Born M, Korving J, de Sauvage F, van Es JH, et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell stem cell. 2016;18:203–213. doi: 10.1016/j.stem.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature biotechnology. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, van Gijn ME, Hatzis P, Kujala P, Haegebarth A, Stange DE, Begthel H, van den Born M, Guryev V, Oving I, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–912. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- van der Maaten LJP, Hinton GE. Visualizing High-Dimensional Data Using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- van Es JH, Sato T, van de Wetering M, Lyubimova A, Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–1104. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Landeghem L, Santoro MA, Krebs AE, Mah AT, Dehmer JJ, Gracz AD, Scull BP, McNaughton K, Magness ST, Lund PK. Activation of two distinct Sox9-EGFP-expressing intestinal stem cell populations during crypt regeneration after irradiation. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1111–1132. doi: 10.1152/ajpgi.00519.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Furstenberg RJ, Buczacki SJ, Smith BJ, Seiler KM, Winton DJ, Henning SJ. Side population sorting separates subfractions of cycling and non-cycling intestinal stem cells. Stem Cell Res. 2014;12:364–375. doi: 10.1016/j.scr.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Furstenberg RJ, Gulati AS, Baxi A, Doherty JM, Stappenbeck TS, Gracz AD, Magness ST, Henning SJ. Sorting mouse jejunal epithelial cells with CD24 yields a population with characteristics of intestinal stem cells. Am J Physiol Gastrointest Liver Physiol. 2011;300:G409–417. doi: 10.1152/ajpgi.00453.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Scoville D, He XC, Mahe MM, Box A, Perry JM, Smith NR, Lei NY, Davies PS, Fuller MK, et al. Isolation and characterization of intestinal stem cells based on surface marker combinations and colony-formation assay. Gastroenterology. 2013;145:383–395. e381–321. doi: 10.1053/j.gastro.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen CB, Asfaha S, Hayakawa Y, Takemoto Y, Lukin DJ, Nuber AH, Brandtner A, Setlik W, Remotti H, Muley A, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. The Journal of clinical investigation. 2014;124:1283–1295. doi: 10.1172/JCI73434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VW, Stange DE, Page ME, Buczacki S, Wabik A, Itami S, van de Wetering M, Poulsom R, Wright NA, Trotter MW, et al. Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nat Cell Biol. 2012;14:401–408. doi: 10.1038/ncb2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci U S A. 2012;109:466–471. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Janda CY, Chang J, Zheng GXY, Larkin KA, Luca VC, Chia LA, Mah AT, Han A, Terry JM, et al. Non-equivalence of Wnt and R-spondin ligands during Lgr5+ intestinal stem-cell self-renewal. Nature. 2017;545:238–242. doi: 10.1038/nature22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Huber W, Vitek O. Shrinkage estimation of dispersion in Negative Binomial models for RNA-seq experiments with small sample size. Bioinformatics. 2013;29:1275–1282. doi: 10.1093/bioinformatics/btt143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Hou SX. Enteroendocrine cells are generated from stem cells through a distinct progenitor in the adult Drosophila posterior midgut. Development. 2015;142:644–653. doi: 10.1242/dev.113357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. Massively parallel digital transcriptional profiling of single cells. Nature communications. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. Massively parallel digital transcriptional profiling of single cells. bioRxiv. 2016 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(Related to Figure 1 and Figure 2)
FACS-isolated samples were collected across six institutions for RNA-seq library preparation, sequencing and bioinformatics analysis. Samples from three biological replicates were used to examine each marker population of interest, as well as their marker-negative control and unfractionated epithelium control. Sample sequencing and read mapping metrics are indicated for each individual sample.
(Related to Figure 1 and Figure 2)
For each marker-positive population of interest, the number of differentially expressed genes relative to each of their marker-negative control population is shown. Only Bmi1-GFP+, CD166+, CD24lo and Lgr5-eGFP+ populations exhibit >200 differentially expressed genes relative to their marker-negative controls.
(Related to Figure 1 and Figure 2). Detailed differentially expressed genes are shown for each marker-positive population analyzed, including FPKMs, log2 (fold-change), p-values, and q-values.
(Related to Figure 1). Top genes common to populations and other genes of interest are shown in Cluster 1.
(Related to Figure 4 and Figure 5)
Table S6A. Single cell RNA sequencing metrics summary. Single-cell libraries were constructed from sorted Prox1-GFP+, Bmi1-GFP+ and Lgr5-eGFP+ cells, as well as from a sample of unfractionated intestinal epithelial cells. Two technical replicate libraries were constructed from each biological samle. Sequencing metrics for each library are provided. Table S6B. Proportion of individual cell clusters by sample. Table S6C. Cluster-specific genes for 1,051 Prox1-GFP+ cells and upon direct comparison of Bmi1-GFP+, Prox1-GFP+, Lgr5-eGFP+ and Lgr5-eGFP− cells.
(Related to Figure 6).