


Abstract
Herein we describe a formulation of self-encapsulating poly(lactic-co-glycolic acid) (PLGA) micro-spheres for vaccine delivery. Self-healing encapsulation is a novel encapsulation method developed by our group that enables the aqueous loading of large molecules into premade PLGA microspheres. Calcium phosphate (CaHPO4) adjuvant gel was incorporated into the microspheres as a protein-trapping agent for improved encapsulation of antigen. Microspheres were found to have a median size of 7.05 ± 0.31 µm, with a w/w loading of 0.60 ± 0.05% of ovalbumin (OVA) model antigen. The formulation demonstrated continuous release of OVA over a 49-day period. Released OVA maintained its antigenicity over the measured period of >21 days of release. C57BL/6 mice were immunized via the intranasal route with prime and booster doses of OVA (10 µg) loaded into microspheres or coadministered with cholera toxin B (CTB), the gold standard of mucosal adjuvants. Microspheres generated a Th2-type response in both serum and local mucosa, with IgG antibody responses approaching those generated by CTB. The results suggest that this formulation of self-encapsulating microspheres shows promise for further study as a vaccine delivery system.
Keywords: poly(lactic-co-glycolic acid), polymer microspheres, encapsulation, vaccine delivery, calcium phosphate adjuvant, intranasal immunization
Graphical abstract
INTRODUCTION
Vaccination is one of the most successful public health interventions, as evidenced by the eradication of smallpox and the approaching eradication of polio. Despite these successes, there is still much room for improving vaccine coverage and immunity. One major challenge to vaccine coverage is that many vaccines require multiple immunizations for protective immunity.1,2 Noncompliance with vaccination programs can reach 70% in some developing countries.1,2 In addition to coverage, next generation vaccines should address the challenge of inducing potent immunity against complex vaccine targets with a wide spectrum of antigens.3
These long-standing challenges have motivated the field to develop novel approaches to vaccine delivery, with the goal of improving vaccine performance.4 Antigen delivery systems (e.g., microspheres, nanoparticles, and liposomes) and adjuvants (e.g., aluminum and calcium compounds) have been widely explored as approaches to improve the breadth and potency of immune responses. This is especially true for subunit- and recombinant protein antigens, which are generally poorly immunogenic on their own due to a difficulty in activating antigen-presenting cells and the short in vivo half-life of these antigens.5,6 Adjuvants and delivery systems may improve antibody- and cell-mediated immunity, decrease the amount of antigen required per vaccine dose, and decrease the number of necessary immunizations, among other benefits.4
Polymeric microspheres and nanoparticles have been widely examined for their use as vaccine delivery systems. One of the most commonly used polymers for vaccine applications is poly(lactic-co-glycolic acid) (PLGA) due to its biodegradable and biocompatible nature and proven safety record in humans.1,5,7 Depending on polymer molecular weight and the ratio of lactic to glycolic acid components, the release kinetics of antigen from PLGA particles can be tuned from a few days to over a year.8 In addition, studies performed with PLGA microspheres have demonstrated their capacity to induce both B and T cell-mediated immune responses in higher mammals.9
Despite the benefits of PLGA particles, there are a number of challenges facing the development of this delivery system. One of the key challenges is antigen instability, which can occur during microsphere production, lyophilization and storage of the microspheres, and antigen release.7,10 Traditional PLGA microsphere production methods expose the antigen to detrimental conditions, including forces from emulsification procedures and exposure to aqueous/organic solvent interfaces.10,11 Protein instability, which includes unfolding, chemical instability, and aggregation, can result in a significant loss of protein antigenicity.12
In an effort to improve protein stability in PLGA particles, our group has developed a new paradigm for drug encapsulation, termed “self-healing microencapsulation,” that bypasses many of the challenges of traditional PLGA encapsulation techniques.10,11 This method relies on the ability of polymer surface pores to heal (close) spontaneously at temperatures above the glass transition temperature (Tg) of the hydrated polymer under aqueous conditions, without the need to expose antigen to organic solvent, harsh mixing, and micronization.13 The self-encapsulating microspheres contain an interconnecting pore network and a protein-trapping agent within the pores, such as an aluminum- or a calcium-based adjuvant gel, which is accessible to the surface of the microsphere via the percolating pore network. Through gentle mixing of the microspheres in an aqueous solution of antigen, the antigen diffuses into the pores and binds to the trapping agent. Subsequent heating of the system above the Tg of the hydrated polymer results in pore healing and closure of loaded protein within the microsphere. Due to its gentle processing conditions, self-encapsulation is a promising approach for maintaining the antigenicity of encapsulated antigen.
We have previously shown the application of self-healing microencapsulation for subcutaneous delivery of vaccine antigen.14 In this current study, we present more detailed characterization of self-encapsulating microspheres and their application for intranasal vaccine delivery. Formulation characteristics were studied, including the (1) incorporation of protein-trapping agent (CaHPO4 adjuvant gel), (2) encapsulation of ovalbumin (OVA) model antigen, (3) release kinetics, and (4) stability and antigenicity of released OVA. The formulation was then applied to an intranasal immunization study in mice to explore the potency and quality of the immune response generated.
EXPERIMENTAL SECTION
Materials
PLGA 50:50 (i.v. = 0.60 dL/g, Mw = 53.4 kDa, ester terminated) was purchased from Lactel (Durect Corporation, USA). Poly(vinyl alcohol) (PVA) (88% hydrolyzed, Mw = 25 kDa) was purchased from Polysciences Inc. (USA). Alexa Fluor 647-OVA was purchased from Molecular Probes (Thermo Fisher Scientific Inc., USA). Cholera toxin B subunit, OVA grade V, and rhodamine 6G were purchased from Sigma (USA). Endofit OVA (<1 EU/mg) for immunization studies was purchased from Invivogen (USA). Biotinylated goat antimouse IgG, IgG1, IgG2C, and IgA were purchased from Southern Biotech (USA). All other reagents and solvents were purchased from commercial suppliers and were of analytical grade or higher.
Preparation of CaHPO4 Adjuvant Gel
CaHPO4 adjuvant gel was prepared as described by Gupta et al.15 Na2HPO4 and CaCl2 salt solutions were rapidly mixed together at equal mole equivalents of HPO4−2 and Ca2+, and the pH immediately adjusted to 6.8–7.0 with NaOH. The resulting precipitate was washed five times with sterile 0.9% NaCl to remove excess phosphate and then was resuspended in sterile 0.9% NaCl to the desired concentration. The mass of the CaHPO4 gel used throughout the manuscript refers to the determined mass of CaHPO4 salt within the aqueous ionomeric gel or dry microspheres.
Preparation of Self-Encapsulating PLGA Micro-spheres
PLGA microspheres without antigen were prepared by the w/o/w double emulsion–solvent evaporation technique. Calcium phosphate adjuvant gel (CaHPO4) was included as a protein-trapping agent and trehalose as a porosigen. The inner water phase contained CaHPO4 gel (28 mg/mL) and trehalose (28 mg/mL) in 25 mM succinate buffer (pH 4.0). One hundred microliters of inner water phase was added to 1 mL of 50 mg/mL PLGA in methylene chloride. The mixture was sonicated at 50% amplitude for 1 min using a Sonics Vibra-Cell VC130 Ultrasonic Processor (Sonics and Materials Inc., USA) to form the first emulsion. Four milliliters of 5% (w/v) PVA solution (used as an emulsion stabilizer) was added to the primary w/o emulsion, and the mixture was vortexed (Genie 2, Scientific Industries Inc., USA) for 30 s to produce the w/o/w double emulsion. The w/o/w emulsion was poured into 100 mL of chilled 0.5% (w/v) PVA under rapid stirring and hardened at room temperature for 6 h. Microspheres were passed through a 10-µm mesh sieve (Newark Wire Cloth Company, USA), and the filtrate was collected and centrifuged (7000 rpm for 5 min). The microspheres in the pellet were washed repeatedly with double-distilled (dd) H2O and then lyophilized (FreeZone 2.5, Labconco, USA) without any additional lyoprotectant for further studies.
Determination of CaHPO4 Loading by ICP-OES
Loading of CaHPO4 in PLGA microspheres was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES). Briefly, approximately 10 mg of microspheres was added to 1 mL of acetone, vortexed, and then centrifuged at 7000 rpm for 5 min. The supernatant polymer solution was removed, and the pellet was washed twice more with 1 mL of acetone. The resulting residue was dried in a fume hood to allow for evaporation of the acetone. One milliliter of 30% HCl was added to each sample to dissolve the residue. The samples were then diluted with ddH2O to bring the theoretical concentration of Ca ion in the samples to between 1 and 15 ppm. The concentration of Ca ion was then analyzed by ICP-OES (PerkinElmer Optima 2000 DV with Winlab software). Each sample was measured three times, and analysis was done in triplicate.
Scanning Electron Microscopy
The surface morphology of microspheres was examined using a Hitachi S3200N scanning electron microscope (SEM) (Hitachi, Japan). Briefly, lyophilized microspheres were fixed on a brass stub using double-sided carbon adhesive tape. The sample was made electrically conductive by coating with a thin layer of gold for 120 s at 40 W under vacuum. Images were taken at an excitation voltage of 8.0 kV. EDAX software was used to obtain the final image.
Microsphere Size and Zeta Potential Analysis
The volume median diameter of the microspheres was measured using a Mastersizer 2000 (Malvern Instruments Ltd., UK). Ten milligrams of lyophilized microspheres was suspended in 5 mL of 0.5% PVA solution and briefly vortexed. Six measurements were performed per sample at a stir speed of 2000 rpm and sampling time of 15 s. Zeta potential was measured with a Zetasizer Nano ZSP (Malvern Instruments Ltd., UK). Lyophilized microspheres were suspended in ddH2O (0.1% mass) and placed in a disposable folded capillary zeta cell (Malvern Instruments Ltd., UK). Each sample was measured six times.
Active Self-Healing Encapsulation of OVA by PLGA Microspheres
Active self-healing encapsulation of OVA by PLGA microspheres was carried out in two phases, as similarly described in Desai et al. and Reinhold et al.10,11 An initial incubation of the porous microspheres in OVA solution at a temperature below the hydrated polymer Tg, for OVA sorption onto the CaHPO4 gel, was followed by incubation at a temperature above the Tg for pore healing. Briefly, approximately 20 mg of lyophilized microspheres were incubated in an OVA solution (0.5 mg/mL OVA in 0.4 mL of 10 mM MOPS buffer, pH 7.4) at 4 °C for 24 h and then 25 °C for 24 h (T < Tg), followed by incubation at 42 °C for 48 h (T > Tg). Incubation was performed under constant agitation using a rigged rotator (Glas-Col, USA).
Determination of Protein Loading and Encapsulation Efficiency
After self-encapsulation of the protein, the microsphere samples were centrifuged at 7000 rpm for 5 min. The supernatant was passed through a low protein-binding Durapore (PVDF) membrane-based syringe filter unit (Millipore Corporation, USA) and the filtrate collected. The microspheres were then washed once with ddH2O and the rinse filtered and collected. A modified Bradford assay was used to determine OVA concentration in the filtrates. Coomassie Plus reagent (Thermo Fisher Scientific, USA) was added to the appropriate volume of standard OVA solution or filtrate sample in a 96-well plate (Nunc, Thermo Scientific, USA). After 10 min, the absorbance was read at 595 nm using a Dynex II MRX microplate reader (Dynex Technology Inc., USA). The mass of OVA encapsulated by the microspheres was calculated by subtracting the mass of OVA in the filtrates from the mass of OVA in the initial loading solution. Percent w/w loading and encapsulation efficiency (EE) were quantified with the following formulas:
Capacity of Unencapsulated CaHPO4 Gel for OVA
To determine the capacity of unencapsulated CaHPO4 gel for OVA, 1 mg of gel was incubated with 200 µg of OVA (0.5 mg/mL in 10 mM MOPS buffer, pH 7.4) at 25 °C for 24 h under mild agitation. Following incubation, the mixture was centrifuged at 7000 rpm for 5 min. The mass of OVA remaining in the supernatant was determined using the Coomassie Plus assay. Based upon the mass of OVA in the original loading solution and the mass of OVA in the supernatant, the amount of OVA loaded onto the gel was calculated. The capacity of the gel for OVA was determined by taking the ratio of OVA loaded and the mass of gel present in the loading mixture.
Distribution of Encapsulated OVA within Micro-spheres
To observe the distribution of self-encapsulated OVA within microspheres, fluorescently labeled OVA was loaded into microspheres containing a rhodamine dye in the polymer phase and observed by confocal microscopy. Briefly, rhodamine 6G was added to the oil phase during microsphere production to dye the polymer. The microspheres were then used to load Alexa Fluor 647-OVA using the self-healing encapsulation process. Directly following encapsulation, the loaded microspheres were centrifuged at 7000 rpm for 5 min and the supernatant removed. Next, the microspheres were rinsed once with ddH2O. Following centrifugation and removal of the supernatant, the microspheres were resuspended in ddH2O, placed on a glass side, and covered with a coverslip. The sample was viewed using a Nikon A-1 spectral confocal microscope with NIS Elements software (Nikon Instruments).
Size Exclusion-High Performance Liquid Chromatography of OVA
Size exclusion-high performance liquid chromatography (SE-HPLC) was performed using a TSKgel G3000SWxl column (Tosoh Bioscience, USA) on a Waters HPLC system (Waters, USA). The mobile phase consisted of PBS, pH 7.4, at a flow rate of 0.7 mL/min and injection volume of 50 µL. Protein detection by UV was done at 210 and 280 nm.
In Vitro Release of OVA from Self-Encapsulating Microspheres
Directly after antigen loading, the OVA-encapsulated microspheres (~20 mg) were rinsed with ddH2O, as described above, to remove any unencapsulated OVA. The microspheres were then immediately resuspended in 0.5 mL of PBS, pH 7.4, and incubated at 37 °C under constant agitation (240 rpm/min). At different incubation times (1, 3, 5, and 7 days, and then every 7 days until day 49), the mixture was centrifuged at 7000 rpm for 5 min, and the supernatant was collected through a low protein-binding Durapore (PVDF) membrane-based syringe filter unit (Millipore Corporation, USA). Fresh PBS release media (0.5 mL) was then added to the sample, and the microspheres were resuspended to continue the release study. The OVA content in the supernatants was analyzed by SE-HPLC.
ELISA for Quantifying Release of Antigenic OVA
In vitro release samples collected and analyzed by SE-HPLC were further analyzed by enzyme-linked immunosorbent assay (ELISA) to quantify the amount of antigenic OVA in each sample. Samples were collected at time points of 1, 3, 5, 7, 14, and 21 days. Antigenic OVA was detected using a commercial chicken egg OVA ELISA kit (Alpha Diagnostic International, USA), which was used according to the manufacturer’s instructions. Samples were diluted with the sample diluent provided with the kit to fall within the working range of the assay and then assayed in duplicate. The dilution was based upon the calculated concentration of OVA in the samples determined by SE-HPLC. The ELISA plate was read at 405 nm using a Synergy Neo plate reader with Gen5 software (Biotek Instruments Inc., USA).
Extraction of Unreleased Protein
At the end of release, microspheres were dissolved in acetone (three washes at 1 mL of acetone per wash) to remove the polymer. The CaHPO4 and OVA pellet was allowed to dry before resuspension in 1 mL of 10% w/v sodium citrate to elute soluble OVA from the gel. The samples were analyzed by SE-HPLC to determine remaining soluble OVA. The residue was then dissolved in 0.1 mL of a denaturing-reducing agent (6 M urea, 1 mM EDTA, and 10 mM dithiothreitol) to dissolve any noncovalent and disulfide-bonded aggregates. The aggregated OVA was quantified using the Coomassie Plus assay using the denaturing-reducing solvent as the standard solution matrix for the standard curve.
Immunization Study
Female C57BL/6 mice, 6–7 weeks old, were purchased from Harlan Laboratories, Inc., and handled according to the University of Michigan Institutional Animal Care guidelines. Mice (10 mice per group) were immunized intranasally (i.n.) with 15 µL (~7.5 µL per nare) of sterile PBS, cholera toxin B (CTB) coadministered with OVA (10 µg CTB + 10 µg OVA), or microspheres loaded with OVA (10 µg OVA). Microspheres were loaded with the antigen, rinsed once with sterile ddH2O after loading, and immediately resuspended in sterile PBS for administration. A booster dose was given 3 weeks later on day 21 after primary immunization. On days 20 and 41, blood samples were collected by submandibular bleed for analysis of the serum antibody titers. Blood was collected into Microvette 500 Z-Gel serum collection tubes (Sarstedt, Germany) and centrifuged at 10,000g for 5 min to separate the serum. Serum was stored at −80 °C until analysis.
Bronchial Alveolar Lavage
Mice were euthanized on day 42, and bronchial alveolar lavage (BAL) was performed for analysis of mucosal antibody titers. Briefly, the trachea was cannulated, and lavage was carried out with approximately 0.75 mL of PBS. The bronchial alveolar lavage fluid (BALF) was centrifuged, and the supernatant collected and stored at −80 °C until ELISA analysis.
Measurement of Antibody Titers
Serum and BALF samples were sent to the Immunology Core at the University of Michigan Cancer Center for ELISA analysis. The assays were performed using a standard ELISA protocol. Briefly, OVA (10 µg/mL in 0.05 M carbonate/bicarbonate buffer, pH 9.6) was used to coat 384-well plates overnight at 4 °C. Plates were washed with 0.1× PBS + 0.05% Tween-20 and residual binding sites blocked with 0.2% casein in PBS, pH 7.4, for at least 1 h at room temperature. Samples were diluted using the PBS–casein buffer and added to the plates at a working volume of 25 µL in duplicate wells. Plates were incubated overnight at 4 °C and then washed. Ig detection antibodies were minimally diluted in PBS to the manufacturer’s suggested working range and incubated in the plates for 2.5 h at room temperature. IgG, IgG1, and IgG2C response was measured for both serum and BALF samples, while IgA response was also analyzed for the BALF. After washing, plates were incubated with Streptavidin-HRP (R&D Systems, USA) for 1 h at room temperature, followed by further washing and incubation with TMB substrate (Surmodics, USA); development was stopped after 4–10 min with 2 N H2SO4 solution. Plates were read on a microplate reader at 450 nm with a correction at 650 nm.
Statistical Analysis
Statistical analyses were carried out using GraphPad Prism 6.0 g software. Two-way ANOVA with Bonferroni’s post-test was used to compare multiple groups. Values are reported as mean ± SEM (standard error of the mean).
RESULTS AND DISCUSSION
Preparation of Self-Encapsulating Microspheres
Microspheres were prepared by a standard double emulsion–solvent evaporation technique, with trehalose in the inner water phase to act as a porosigen to create an interconnected pore network. This pore network is necessary for the protein in the loading solution to diffuse through the pores into the microspheres prior to healing the pores shut.11 As shown in Figure 1A, scanning electron microscopy was used to verify that the formulation produced porous particles with a spherical morphology.
Figure 1.

(A) SEM image of porous, self-encapsulating PLGA microspheres prior to encapsulation of protein. (B) Healed microspheres after self-encapsulation of OVA. Scale bars represent 5 µm.
Incorporation of CaHPO4 Adjuvant Gel
Calcium phosphate (CaHPO4) adjuvant gel was present in the inner water phase to be incorporated into the microspheres. The purpose of the gel is to act as a protein-trapping agent within the microsphere pores. In previous work done by our lab, use of a protein-trapping agent, such as Al(OH)3 or CaHPO4 adjuvant gels, improved the encapsulation efficiency of the microspheres, compared with microspheres without an inner trapping agent.10,11 To distinguish the two strategies, use of a protein-trapping agent is termed “active” self-encapsulation.
Calcium phosphate is an alternative to the common aluminum-based adjuvants. Being a natural constituent of the body, it is well-tolerated and readily resorbed.15 CaHPO4 has been used in France for many years as an adjuvant with diphtheria–tetanus–pertussis (DTP) vaccines.16 Unlike aluminum adjuvants, calcium phosphate does not lead to enhanced IgE production in animals and humans.15 The adjuvant is thought to act as an antigen depot and improve uptake of the antigen by antigen-presenting cells (APCs).1,15,17
The CaHPO4 gel was prepared by rapid mixing (<10 s) of equimolar solutions of disodium hydrogen phosphate and calcium chloride. Gel prepared by rapid mixing was found to have much better adsorption of diphtheria toxoid, compared with gel made by slower mixing (10 min).15 The adjuvant was then washed thoroughly with 0.9% sodium chloride to remove excess phosphate ions that would interfere with antigen adsorption.
Successful incorporation of the adjuvant in the microspheres was confirmed by ICP-OES (Table 1). In addition to measurement of the % w/w loading of the adjuvant (2.2 ± 0.1%), the distribution of the gel within the microspheres was viewed by confocal microscopy (Figure S1). The CaHPO4 gel was preloaded with Alexa Fluor 647-OVA and used in the inner water phase to be incorporated into microspheres. The gel was observed to be loaded within both smaller and larger microspheres and to be homogeneously distributed.
Table 1.
Composition, Size, and Zeta Potential of the Self-Encapsulating PLGA Microsphere Formulationa
| PLGA concentration (mg in 1 mL) |
volume of inner water phase (mL) |
trehalose loading (wt %)b |
Measured CaHPO4 loading (wt %)c |
volume median diameter (µm) |
zeta potential (mV) |
|---|---|---|---|---|---|
| 50 | 0.1 | 5 | 2.2 ± 0.1 | 7.05 ± 0.31 | −21.9 ± 2.1 |
Data represent mean ± SEM, n = 3.
Theoretical loading.
Theoretical loading = 5 wt %.
Microsphere Size and Zeta Potential
Based upon their size, microspheres may carry out a number of functions. For example, microspheres may (1) act as a depot for sustained release of antigen, prolonging exposure of the immune system to the antigen; and (2) facilitate uptake of the antigen by APCs, improving delivery of the antigen to the lymphoid organs.18 Consequently, microsphere size is an important consideration for antigen delivery.
The formulation parameters were designed to produce microspheres of a size range smaller than 10 µm. In particular, sonication was used to form the primary emulsion since greater agitation is known to generate smaller particles.19 A smaller microsphere size was desired for improved immune response as prior research has shown that particles smaller than 10 µm are preferentially phagocytosed by APCs, in comparison with larger particles, with smaller particles eliciting stronger immune responses.20–24 The median microsphere diameter was found to be within the desired size range (7.05 ± 0.31 µm), with a zeta potential of −21.9 ± 2.1 mV (Table 1). We have also previously shown that the self-healing process did not typically change the overall size distribution of the microspheres.10,11
Self-Encapsulation of OVA Model Antigen
OVA was loaded into microspheres by the active self-healing encapsulation process. Lyophilized microspheres were initially incubated in a solution of OVA at lower temperatures, with mild mixing, to allow the protein to diffuse through the pores and sorb onto the CaHPO4 gel. Following this stage, the mixture was then incubated at a temperature above the Tg of the hydrated polymer in order for the microsphere pores to heal, thereby trapping the antigen within the particle. Scanning electron microscopy was used to verify pore closure after encapsulation (Figure 1B).
Results of the self-encapsulation are shown in Table 2. The mass of OVA loaded into the microspheres was assessed by measuring the OVA mass loss from the original loading solution. The measured % w/w loading of OVA was found to be 0.60 ± 0.05%. The OVA capacity of CaHPO4 gel in either the microsphere form or unencapsulated form was comparable (Table 2). These results suggest that incorporation of the gel within the microspheres did not adversely affect the ability of the gel to sorb the protein and that the loading of OVA into PLGA microspheres was likely to be limited by the binding capacity of the CaHPO4 gel for the antigen within the microspheres. CaHPO4 may adsorb antigen by electrostatic interaction, hydrophobic interaction, and ligand exchange.17 The latter occurs by exchange of hydroxyl groups in the gel with phosphate groups in the protein. Commercial OVA, such as the type used in this study, contains 0–2 mol of PO4 covalently bound per mol of protein.25 Cargo loading of 0.60 ± 0.05% w/w may be low for common pharmaceutical proteins but is sufficient for many antigens (e.g., the tetanus toxoid human dose of 10 Lf or 30–40 µg of antigen would require only a very modest ~5–7 mg of microspheres to accommodate each dose).
Table 2.
Active Self-healing Encapsulation of Ovalbumin (OVA) by PLGA Microspheresa
| theoretical % (w/w) loadingb |
measured % (w/w) loading |
% encapsulation efficiencyc |
OVA capacity of CaHPO4 within microspheresd,e |
OVA capacity of unencapsulated CaHPO4 geld,f |
|---|---|---|---|---|
| 1.0 | 0.60 ± 0.05 | 56 ± 6 | 0.27 ± 0.03 | 0.21 ± 0.01 |
Encapsulation was performed using 20 mg of microspheres in 0.4 mL of a 0.5 mg/mL OVA solution. Data represent mean ± SEM, n = 3.
mg of total OVA in loading solution/(mg of microspheres in sample + mg of total OVA in solution).
Efficiency relative to the original mass of OVA in the loading solution.
mg OVA loaded/mg CaHPO4 gel.
Based upon the determined mass of CaHPO4 gel loaded within the microspheres.
Determined for OVA loaded onto unencapsulated CaHPO4 gel.
Self-encapsulation of OVA was also monitored by confocal microscopy. Microspheres containing CaHPO4 gel were made fluorescent by the addition of rhodamine 6G into the polymer phase during microsphere preparation, and the self-encapsulation process was carried out with Alexa Fluor 647-OVA. Incorporation of the protein into the microspheres was confirmed by confocal microscopy (Figure 2). Orthogonal images verified that protein was internal to the microspheres and not on the particle surface.
Figure 2.

Confocal microscopy image of the distribution of Alexa Fluor 647-OVA (violet) loaded within rhodamine-labeled PLGA microspheres (cyan) by self-encapsulation. Orthogonal images are shown in the right and bottom panels. Scale bar represents 10 µm.
Release of Encapsulated OVA
Sustained release of OVA from the microspheres containing CaHPO4 gel was observed over a 49-day period, with a moderate burst release of 28% (Figure 3). OVA in the PBS release media was quantified by SE-HPLC, and the % cumulative release over time was calculated based on the mass of OVA loaded into the microspheres. PLGA-encapsulated CaHPO4 gel allowed slightly more gradual release of OVA (50% at day 21), compared with the unencapsulated CaHPO4 gel (61% at day 21, p < 0.0001, Figure S2) but was highly similar. By the end of the study at day 49, 70% of the PLGA-encapsulated OVA was observed in the release media, and the monomer fraction remained stable (88–98%) relative to the commercial OVA received (88–90%) (data not shown). It is important to note that because of the way OVA incorporates in the CaHPO4 gel with a ligand exchange component, slow release from the microspheres may in fact be strongly controlled by this interaction.
Figure 3.

Cumulative OVA released as a function of time by self-encapsulating PLGA microspheres. In vitro release was conducted in PBS (pH 7.4) at 37 °C. Data represent mean ± SEM (n = 3).
At the conclusion of the release study, the remaining unreleased OVA was extracted from the polymer and desorbed from the CaHPO4 gel. The fraction of soluble and insoluble (covalent and noncovalent aggregates) remaining was determined. No unreleased soluble OVA was detected; however, a small fraction of insoluble aggregates (3%) was recovered after extraction (data not shown). Following mass balance, a total of 73% of the encapsulated OVA was accounted for after the study. Protein loss may have occurred during the wash cycles for extraction from the polymer and/or because of potential incomplete desorption from the CaHPO4 gel.
The stability of OVA at the temperatures of self-healing encapsulation was studied by incubating the protein in loading solution buffer at the different process temperatures for the specified periods (Figure S3). Samples of OVA were treated separately at (1) 4 °C for 24 h, (2) 25 °C for 24 h, (3) 42 °C for 48 h, and (4) 4 °C (24 h) + 25 °C (24 h) + 42 °C (48 h) (the full self-encapsulation process). A separate sample of protein was also incubated at 80 °C for 24 h to purposefully induce denaturation and aggregation.26 Aggregation of the protein was not observed during the self-encapsulation process, as compared to the protein sample denatured at 80 °C.
Antigenicity of OVA Released from Microspheres
The stability of released OVA was analyzed by measuring the antigenicity of the protein by ELISA, using antibodies monospecific for OVA. Previous work from our lab has shown improved stability of protein loaded by self-encapsulation compared to traditional encapsulation methods. Self-encapsulated tetanus toxoid maintained >96% of its antigenicity over 28 days of release, compared with only 27% for tetanus toxoid loaded into PLGA particles by a traditional w/o/w method.10
The antigenicity of released OVA was measured over 21 days. The protein was analyzed by HP-SEC to measure total protein released and also by ELISA to determine the fraction of released protein that was antigenic. As shown in Figure 4, there was excellent agreement between the total protein released and its antigenicity as measured by HP-SEC and ELISA, respectively. This suggests that the model antigen loaded within the microspheres via the self-healing encapsulation process maintained its stability and antigenicity for at least 21 days.
Figure 4.

Plot of OVA released over a 21-day period from microspheres as measured by HP-SEC (○) and ELISA (□). ELISA analysis was performed to specifically measure the antigenic OVA present in the release samples.
Serum and Mucosal Antibody Response in Mice Following Intranasal Immunization with Microspheres
Intranasal immunization is an attractive route for vaccine administration since the nasal cavity is easily accessible and has a high density of dendritic cells.27 Intranasal vaccination has shown the ability to produce an antibody response in the serum and in both local and distal mucosal secretions.28,29 Since many infections occur at mucosal surfaces, induction of a mucosal immune response is important for protection against the infectious agent.30
Female C57BL/6 mice were immunized via intranasal administration of microspheres, which were used immediately following loading with OVA (10 µg OVA per dose). A booster dose was given 3 weeks later after primary immunization. The immune response generated by immunization with microspheres was compared with that for mice immunized with OVA and cholera toxin B (CTB) (10 µg CTB + 10 µg OVA) or PBS. We chose cholera toxin as the positive control in our studies because, despite its reported toxicity in humans, it is widely used as an experimental, gold-standard adjuvant for mucosal immunization in preclinical studies.31,32 The B subunit of cholera toxin used in our studies maintains intranasal adjuvanticity but lacks the toxicity of the whole toxin.33
Following prime and booster intranasal immunization of mice, the serum anti-OVA antibody response was measured. The total IgG response was analyzed (Figure 5A), as well as titers for IgG1 and IgG2C subclasses (Figures 5B,C, respectively). Subclass titers provide information about the polarization of the Th response, with IgG1 associated with a Th2-type response and IgG2C associated with a Th1 response.34,35 Despite high variability among the groups, prime vaccination with the CTB- and microsphere-vaccines elicited similar levels of anti-OVA antibody responses (Figure 5). Poor response of some mice to immunization may be attributed to inadequate intranasal deposition of the micro-spheres, as intranasal administration is known to be a challenging route of vaccination.36–38
Figure 5.

Serum anti-OVA antibody titers for groups on days 20 (prime response) and 41 (boost response): (A) IgG, (B) IgG1, and (C) IgG2C. Data were fit using a four-parameter curve, and titers were calculated by solving for the inverse dilution factor resulting in an absorbance value of 0.5. Data represent mean ± SEM (n = 10). All groups were compared using two-way ANOVA followed by Bonferroni’s post-test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). N.B. = no binding detected.
Boost immunization resulted in >80% of mice achieving seroconversion, with significant total IgG and IgG1 responses observed for both CTB and microsphere groups. Importantly, the IgG and IgG1 responses following the boost immunization with microspheres approached those for the mice immunized with CTB, a strong mucosal adjuvant, although the CTB group was statistically superior to the microsphere group. Neither group displayed a significant IgG2C response after prime or boost vaccination, suggesting that the response for both groups was Th2-biased. This is confirmed in the literature as CTB is known to trigger a more Th2-type response.39,40
The immune response was further assessed by studying the cytokine profiles (IL-2, IL-6, IL-10, and IFN-γ) of splenocytes restimulated ex vivo. IL-2 and IFN-γ are secreted by Th1 cells, while IL-6 and IL-10 are indicative of a Th2-skewed response.41,42 Splenocytes were collected from immunized mice 3 weeks after booster administration and restimulated with whole OVA. Splenocytes from microsphereimmunized mice produced IL-6 and IL-10 as well as a robust level of IFN-γ comparable to those from CTB-immunized mice (Figure S3).
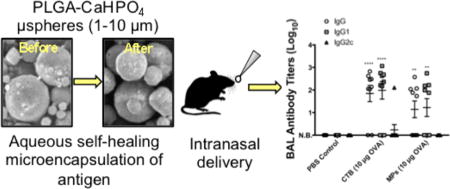
Apart from systemic immunity, local mucosal responses were also analyzed by determining anti-OVA antibody titers in bronchial alveolar lavage fluid collected after booster administration (Figure 6). The mucosal antibody responses reflected the Th2-biased serum responses as we observed predominant anti-OVA IgG1 titers with no detectable levels of IgG2C. In addition, the IgG and IgG1 antibody titers for the microsphere-immunized group once again approached the responses generated in the CTB-immunized mice. No detectable levels of anti-OVA IgA were found in the lavage fluid (data not shown).
Figure 6.

Anti-OVA antibody titers from bronchial alveolar lavage (BAL) samples collected from groups on day 42. Data were fit using a four-parameter curve, and titers were calculated by solving for the inverse dilution factor resulting in an absorbance value of 0.5. Data represent mean ± SEM (n = 9). All groups were compared using two-way ANOVA followed by Bonferroni’s post-test. Statistical significance shown is in relation to the corresponding PBS control (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). N.B. = no binding detected.
Taken together, the data presented in this article suggest that our PLGA microspheres loaded with OVA via the “active” self-healing encapsulation process have the ability to maintain the antigenicity of the protein and to generate a robust Th2-type antibody response in both serum and local mucosa, approaching that of a gold-standard intranasal adjuvant. This work builds upon our previous research to develop a formulation of self-encapsulating microspheres for vaccine delivery, which showed unprecedented stability of tetanus toxoid in PLGA10 and superior induction of T and B cell immune responses compared with plain CaHPO4 geladjuvanted OVA after SC vaccination.14 As the current study on IN vaccination employed the same formulation of PLGA-OVA microspheres in the same rodent species as in our earlier SC study,14 we summarized the antibody responses from the two studies (Table 3). SC vaccination with PLGA-OVA microspheres generated significantly higher levels of anti-OVA serum IgG, IgG1, and IgG2C responses, compared with IN vaccination with PLGA-OVA microspheres or the CTB-OVA positive control. While these results demonstrate the potency of self-healing PLGA microspheres to elicit antibody responses after SC and IN routes of vaccination, they also underscore the need to further optimize our strategy for IN vaccination.
Table 3.
Comparison of Serum Antibody Titers after Intranasal (IN) or Subcutaneous (SC) Route of Vaccinationa
| after prime | after boost | |
|---|---|---|
| Anti-OVA Serum IgG Titer (Log10) | ||
| PBS control-IN | 0.49 ± 0.33 | 0.47 ± 0.31 |
| CTB (10 µg OVA)-IN | 1.70 ± 0.49 | 4.11 ± 0.31#### |
| MPs (10 µg OVA)-IN | 0.90 ± 0.46 | 2.55 ± 0.47##, † |
| MPs (10 µg OVA)-SC | 5.85 ± 0.21**** | 7.32 ± 0.14**** |
| Anti-OVA Serum IgG1 Titer (Log10) | ||
| PBS control-IN | N.B. | N.B. |
| CTB (10 µg OVA)-IN | 0.77 ± 0.51 | 4.41 ± 0.39#### |
| MPs (10 µg OVA)-IN | 0.934 ± 0.34 | 2.48 ± 0.58###,†† |
| MPs (10 µg OVA)-SC | 4.84 ± 1.22**** | 7.74 ± 0.11####,††††,‡‡‡ |
| Anti-OVA Serum IgG2C Titer (Log10) | ||
| PBS control-IN | N.B. | 0.63 ± 0.43 |
| CTB (10 µg OVA)-IN | N.B. | 0.63 ± 0.43 |
| MPs (10 µg OVA)-IN | N.B. | 0.50 ± 0.33 |
| MPs (10 µg OVA)-SC | N.B. | 4.80 ± 0.22**** |
The data for MPs (10 µg OVA)-SC is from Bailey et al.14 Data represent mean ± SEM (n = 10 for PBS-IN, CTB-IN, and MPs-IN; and n = 5 for MPs-SC).
p < 0.0001 against all other groups.
p ≤ 0.01;
p ≤ 0.001;
p ≤ 0.0001 against PBS control-IN.
p ≤ 0.05;
p ≤ 0.01;
p ≤ 0.0001 against CTB-IN.
p ≤ 0.001 against MPs-IN. N.B. = no binding detected.
Overall, the combination of slow release of antigens and targeting of antigen-presenting cells from the novel microsphere formulation provides a unique vaccine delivery platform potentially suitable for numerous antigens. The results of this work motivate further investigation of this formulation for delivery of clinically relevant antigen as well as direct comparison to other adjuvants and vaccine delivery systems. We also wish to develop means to increase antigen loading, reduce the time for the self-encapsulation process, and improve the IN vaccination, methods for which are under current development.
Supplementary Material
Acknowledgments
The authors thank the following staff from the University of Michigan for their expertise: Dr. Gordon Moore from the Electron Beam Analysis Laboratory (EMAL), Linda Barthel from the Microscopy and Image Analysis Laboratory (MIL), and Joel Whitfield from the Cancer Center Immunology Core. This work was supported in part by the NIH (R21 EB 08873, to S.P.S; R01EB022563, to J.J.M.; R01AI127070, to J.J.M.; R01CA210273, to J.J.M.), DoD/CDMRP (W81XWH-16-1-0369, to J.J.M.), and NSF (1553831, to J.J.M.). B.B. acknowledges financial support from the University of Michigan Rackham Merit Fellowship, the American Foundation for Pharmaceutical Education (AFPE), and NIGMS (GM007767). L.O. is supported by predoctoral fellowships from UM Rackham and AFPE. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS.
ABBREVIATIONS
- APC
antigen-presenting cell
- BAL
bronchial alveolar lavage
- BALF
bronchial alveolar lavage fluid
- CTB
cholera toxin B
- EE
encapsulation efficiency
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- ICP-OES
inductively coupled plasma-optical emission spectrometry
- OVA
ovalbumin
- PLGA
poly(lactic-co-glycolic acid)
- SE-HPLC
size exclusion-high performance liquid chromatography
Footnotes
ASSOCIATED CONTENT
Methods for analyzing the distribution of CaHPO4 gel within microspheres, evaluation of OVA stability during the self-healing encapsulation process, and measuring cytokine response from splenocytes; confocal image of CaHPO4 gel distribution within microspheres; plot of OVA release kinetics from microspheres relative to unencapsulated CaHPO4 gel; HP-SEC chromatograms of OVA stability at different temperatures; and plots of cytokine production by mouse splenocytes restimulated ex vivo (PDF)
The authors declare no competing financial interest.
References
- 1.Wilson-Welder JH, Torres MP, Kipper MJ, Mallapragada SK, Wannemuehler MJ, Narasimhan B. Vaccine adjuvants: current challenges and future approaches. J. Pharm. Sci. 2009;98(4):1278–316. doi: 10.1002/jps.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friede M, Aguado MT. Need for new vaccine formulations and potential of particulate antigen and DNA delivery systems. Adv. Drug Delivery Rev. 2005;57(3):325–31. doi: 10.1016/j.addr.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 3.O’Hagan DT, De Gregorio E. The path to a successful vaccine adjuvant-’the long and winding road’. Drug Discovery Today. 2009;14(11–12):541–51. doi: 10.1016/j.drudis.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 4.O’Hagan DT, Rappuoli R. Novel approaches to vaccine delivery. Pharm. Res. 2004;21(9):1519–30. doi: 10.1023/B:PHAM.0000041443.17935.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akagi T, Baba M, Akashi M. Biodegradable nanoparticles as vaccine adjuvants and delivery systems: regulation of immune responses by nanoparticle-based vaccine. Adv. Polym. Sci. 2011;247:31–64. [Google Scholar]
- 6.Storni T, Kundig TM, Senti G, Johansen P. Immunity in response to particulate antigen-delivery systems. Adv. Drug Delivery Rev. 2005;57(3):333–55. doi: 10.1016/j.addr.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Kersten G, Hirschberg H. Antigen delivery systems. Expert Rev. Vaccines. 2004;3(4):453–62. doi: 10.1586/14760584.3.4.453. [DOI] [PubMed] [Google Scholar]
- 8.O’Hagan DT, Rahman D, McGee JP, Jeffery H, Davies MC, Williams P, Davis SS, Challacombe SJ. Biodegradable microparticles as controlled release antigen delivery systems. Immunology. 1991;73(2):239–42. [PMC free article] [PubMed] [Google Scholar]
- 9.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470(7335):543–7. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desai KG, Schwendeman SP. Active self-healing encapsulation of vaccine antigens in PLGA microspheres. J. Controlled Release. 2013;165(1):62–74. doi: 10.1016/j.jconrel.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reinhold SE, Desai KG, Zhang L, Olsen KF, Schwendeman SP. Self-healing microencapsulation of biomacromolecules without organic solvents. Angew. Chem., Int. Ed. 2012;51(43):10800–3. doi: 10.1002/anie.201206387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit. Rev. Ther. Drug Carrier Syst. 2002;19(1):73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Wang BM, Schwendeman SP. Characterization of the initial burst release of a model peptide from poly(D,L-lactide-co-glycolide) microspheres. J. Controlled Release. 2002;82(2–3):289–307. doi: 10.1016/s0168-3659(02)00137-2. [DOI] [PubMed] [Google Scholar]
- 14.Bailey BA, Ochyl LJ, Schwendeman SP, Moon JJ. Toward a Single-Dose Vaccination Strategy with Self-Encapsulating PLGA Microspheres. Adv. Healthcare Mater. 2017;6:12. doi: 10.1002/adhm.201601418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta RK, Rost BE, Relyveld E, Siber GR. Adjuvant properties of aluminum and calcium compounds. In: Powell MF, Newman MJ, editors. Vaccine Design: The Subunit and Adjuvant Approach. Vol. 6. Plenus Press; New York: 1995. pp. 229–249. [DOI] [PubMed] [Google Scholar]
- 16.Gupta RK, Siber GR. Comparison of adjuvant activities of aluminium phosphate, calcium phosphate and stearyl tyrosine for tetanus toxoid. Biologicals. 1994;22(1):53–63. doi: 10.1006/biol.1994.1008. [DOI] [PubMed] [Google Scholar]
- 17.Jiang D, Premachandra GS, Johnston C, Hem SL. Structure and adsorption properties of commercial calcium phosphate adjuvant. Vaccine. 2004;23(5):693–8. doi: 10.1016/j.vaccine.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 18.Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Rev. Vaccines. 2010;9(9):1095–107. doi: 10.1586/erv.10.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M, Rouaud O, Poncelet D. Microencapsulation by solvent evaporation: state of the art for process engineering approaches. Int. J. Pharm. 2008;363(1–2):26–39. doi: 10.1016/j.ijpharm.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm. Res. 2008;25(8):1815–21. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabata Y, Ikada Y. New Polymer Materials. 1. Springer-Verlag; Berlin Heidelberg: 1990. Phgocytosis of Polymer Microspheres by Macrophages; pp. 107–141. [Google Scholar]
- 22.Yoshida M, Babensee JE. Molecular aspects of microparticle phagocytosis by dendritic cells. J. Biomater. Sci., Polym. Ed. 2006;17(8):893–907. doi: 10.1163/156856206777996844. [DOI] [PubMed] [Google Scholar]
- 23.Joshi VB, Geary SM, Salem AK. Biodegradable particles as vaccine delivery systems: size matters. AAPS J. 2013;15(1):85–94. doi: 10.1208/s12248-012-9418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Audran R, Peter K, Dannull J, Men Y, Scandella E, Groettrup M, Gander B, Corradin G. Encapsulation of peptides in biodegradable microspheres prolongs their MHC class-I presentation by dendritic cells and macrophages in vitro. Vaccine. 2003;21(11–12):1250–5. doi: 10.1016/s0264-410x(02)00521-2. [DOI] [PubMed] [Google Scholar]
- 25.Morefield GL, HogenEsch H, Robinson JP, Hem SL. Distribution of adsorbed antigen in mono-valent and combination vaccines. Vaccine. 2004;22(15–16):1973–84. doi: 10.1016/j.vaccine.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 26.Rumbo M, Chirdo FG, Fossati CA, Anon MC. Analysis of structural properties and immunochemical reactivity of heat-treated ovalbumin. J. Agric. Food Chem. 1996;44(12):3793–3798. [Google Scholar]
- 27.Pawar D, Mangal S, Goswami R, Jaganathan KS. Development and characterization of surface modified PLGA nanoparticles for nasal vaccine delivery: effect of mucoadhesive coating on antigen uptake and immune adjuvant activity. Eur. J. Pharm. Biopharm. 2013;85(3 PtA):550–9. doi: 10.1016/j.ejpb.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Alpar HO, Somavarapu S, Atuah KN, Bramwell VW. Biodegradable mucoadhesive particulates for nasal and pulmonary antigen and DNA delivery. Adv. Drug Delivery Rev. 2005;57(3):411–30. doi: 10.1016/j.addr.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Carcaboso AM, Hernandez RM, Igartua M, Rosas JE, Patarroyo ME, Pedraz JL. Potent, long lasting systemic antibody levels and mixed Th1/Th2 immune response after nasal immunization with malaria antigen loaded PLGA microparticles. Vaccine. 2004;22(11–12):1423–32. doi: 10.1016/j.vaccine.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 30.Holmgren J, Czerkinsky C, Eriksson K, Mharandi A. Mucosal immunisation and adjuvants: a brief overview of recent advances and challenges. Vaccine. 2003;21(Suppl 2):S89–95. doi: 10.1016/s0264-410x(03)00206-8. [DOI] [PubMed] [Google Scholar]
- 31.de Geus B, Dol-Bosman M, Scholten JW, Stok W, Bianchi A. A comparison of natural and recombinant cholera toxin B subunit as stimulatory factors in intranasal immunization. Vaccine. 1997;15(10):1110–3. doi: 10.1016/s0264-410x(97)00007-8. [DOI] [PubMed] [Google Scholar]
- 32.Kim D, Kim YG, Seo SU, Kim DJ, Kamada N, Prescott D, Philpott DJ, Rosenstiel P, Inohara N, Nunez G. Nod2-mediated recognition of the microbiota is critical for mucosal adjuvant activity of cholera toxin. Nat. Med. 2016;22(5):524–30. doi: 10.1038/nm.4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moschos SA, Bramwell VW, Somavarapu S, Alpar HO. Adjuvant synergy: the effects of nasal coadministration of adjuvants. Immunol. Cell Biol. 2004;82(6):628–37. doi: 10.1111/j.0818-9641.2004.01280.x. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Ostroff GR, Lee CK, Specht CA, Levitz SM. Robust stimulation of humoral and cellular immune responses following vaccination with antigen-loaded beta-glucan particles. mBio. 2010;1(3):e00164–10. doi: 10.1128/mBio.00164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin RM, Brady JL, Lew AM. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J. Immunol. Methods. 1998;212(2):187–92. doi: 10.1016/s0022-1759(98)00015-5. [DOI] [PubMed] [Google Scholar]
- 36.Kaufman DR, Bivas-Benita M, Simmons NL, Miller D, Barouch DH. Route of adenovirus-based HIV-1 vaccine delivery impacts the phenotype and trafficking of vaccine-elicited CD8+ T lymphocytes. J. Virol. 2010;84(12):5986–96. doi: 10.1128/JVI.02563-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buonaguro L, Tagliamonte M, Visciano ML, Andersen H, Lewis M, Pal R, Tornesello ML, Schroeder U, Hinkula J, Wahren B, Buonaguro FM. Immunogenicity of HIV virus-like particles in rhesus macaques by intranasal administration. Clin Vaccine Immunol. 2012;19(6):970–3. doi: 10.1128/CVI.00068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lycke N. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol. 2012;12(8):592–605. doi: 10.1038/nri3251. [DOI] [PubMed] [Google Scholar]
- 39.Eriksson K, Fredriksson M, Nordstrom I, Holmgren J. Cholera toxin and its B subunit promote dendritic cell vaccination with different influences on Th1 and Th2 development. Infect. Immun. 2003;71(4):1740–7. doi: 10.1128/IAI.71.4.1740-1747.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhee JH, Lee SE, Kim SY. Mucosal vaccine adjuvants update. Clin. Exp. Vaccine Res. 2012;1(1):50–63. doi: 10.7774/cevr.2012.1.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002;39(9):531–6. doi: 10.1016/s0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 42.Borish L, Rosenwasser LJ. Update on cytokines. J. Allergy Clin. Immunol. 1996;97(3):719–34. doi: 10.1016/s0091-6749(96)80146-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.