

Abstract
Long-distance biological electron transfer occurs through a hopping mechanism and often involves tyrosine as a high potential intermediate, for example in the early charge separation steps during photosynthesis. Protein design allows for the development of minimal systems to study the underlying principles of complex systems. Herein, we report the development of the first ruthenium-linked designed protein for the photogeneration of a tyrosine radical by intramolecular electron transfer.
Keywords: electron transfer, photochemistry, protein design, radicals, tyrosine
Electron transfer (ET) is an important process that plays key roles in sustaining life through photosynthesis and respiration as well as in the development of new technologies for green energy. Biological ET must often occur over large distances (≈ 30 Å) with a sufficiently high rate to sustain life.[1] Long-distance electron transfer proceeds by a series of hops, which reduces the distance dependence of the rate of ET.[2–4] The protein matrix itself can be involved in mediating ET directly through the participation of redox-active amino acids, such as tyrosine and tryptophan.[5]
Stable amino acid based radicals are recognized as essential components of both electron transfer and catalytic processes in many proteins that can play both oxidative and reductive roles, as their reduction potentials are highly sensitive to their protonation state and chemical environment.[6] In particular, tyrosine radicals are vital to long-distance ET in many important biological systems, such as photosystem II (PSII), ribonucleotide reductase (RNR), and cytochrome c oxidase.[1] Tyrosine radical chemistry in biological processes is complicated by the fact that acid-base chemistry is coupled to electron transfer, which results from the difference in pKa values between neutral tyrosine (pKa = 10) and the tyrosyl radical (pKa = −2).[7] Thus, over the physiologically relevant pH range, proton-coupled electron transfer (PCET) occurs. Since tyrosine undergoes a PCET process, tyrosines in native proteins are often hydrogen bonded to nearby amino acids and water molecules, facilitating fast reactions by providing nearby proton acceptors.[8] Although the importance of tyrosine radicals in natural processes is well known, the details of their behavior is challenging to study because of the complexity of the systems of which they are a part. Moreover, understanding their role in vectorial electron transfer may help chemists to employ similar functionalities in the design of bioinspired materials.
Previously, amino acid based radicals had been studied by spectroscopic characterization of mutants and perturbed forms of native proteins.[5,9,10] Engineered forms of azurin and RNR have been used to study tyrosine radicals in native proteins.[11–13] More recently, scientists have turned to protein design as a method to create systems with sufficient complexity to understand the basics of functionality in native systems, while retaining a high degree of control over the system. A single tyrosine in a de novo designed α-helical bundle was recently reported and was characterized by electrochemistry[14] and transient absorption spectroscopy.[15] This tyrosine radical was extremely stable and exhibited long lifetimes as a result of the fact that it is buried in the hydrophobic interior of the protein. As radicals are typically intermediate electron relays in intramolecular electron-transfer reactions, some synthetic systems have been developed to mimic tyrosine-based relays.[16,17] However, further work is needed to better understand the roles and mechanisms inherent to protein-based tyrosines.
By using de novo protein design methods, we sought to develop a system to generate and study tyrosine radical involvement in intramolecular electron transfer. Our lab has previously reported de novo designed α-helical bundles for modeling metal binding sites, hydrolytic and redox catalysis, and electron transfer.[18–24] These proteins are designed to mimic globular proteins and exhibit a well-folded and characterized tertiary structure. We hypothesized that a variant form of the peptide α3DH3, which was previously characterized for hydrolytic activity,[21] would be an excellent scaffold for controlled studies on intramolecular electron transfer in proteins via tyrosine radicals. Replacing the terminal residue with a cysteine generated a site useful for the covalent attachment of chromophores within the electron-transfer distance of Tyr70, which is located at the interface between two helices (Figure 1 A). In this study, we have not optimised and studied the chemical environment of the tyrosine motif that can be tuned by its position on the protein scaffold. Such a systematic study is beyond the scope of this preliminary communication and will be addressed in future work.
Figure 1.
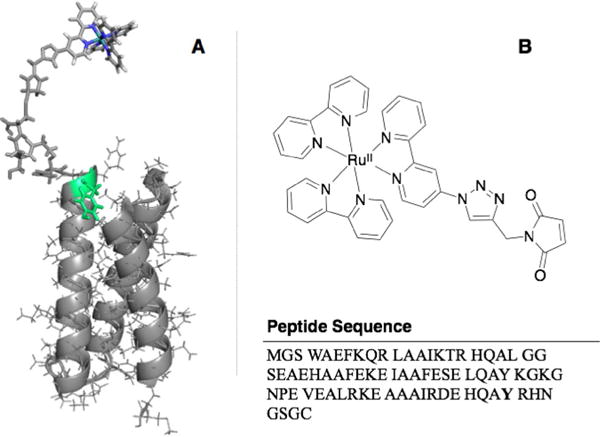
A) Schematic representation of the structure of α3DH3-Rubpymal based on the solution NMR structure of a closely related scaffold (PDB: 2MTQ). The key tyrosine residue is marked in green. B) Chemical structure of Rubpymal (top) and sequence of α3DH3 (bottom).
Ruthenium trisbipyridine [RuII(bpy)3]2+ compounds and their derivatives are well-characterized and widely used photosensitizers with a long-lived triplet excited state that has the capacity to either accept or donate one electron.[25,26] These characteristics have led to the widespread use of [RuII(bpy)3]2+ analogues to trigger and understand photoinduced catalytic and electron-transfer reactions.[25] Previous studies on [RuII(bpy)3]2+ derivatives showed that functionalization of the bipyridines with a triazole ring resulting from the copper-catalyzed azide-alkyne addition reaction does not significantly alter the characteristics of this excited state.[27,28] Further photophysical studies showed that the triazole linkage can efficiently mediate electron transfer from the [RuII*(bpy)3]2+ either to an electron acceptor or an electron donor.[28] Hence, we reasoned that modifying the bipyridine ligand through click chemistry to introduce a maleimide moiety should not affect the photophysical properties of the lumophore. Inspired by this prior work, we have developed a [RuII(bpy)3]2+ derivative (Rubpymal) that can be appended to free cysteine residues (Figure 1 A; see also Scheme S1 in the Supporting Information). This was used to modify α3DH3 selectively at position 75 to give α3DH3-Rubpymal. These studies provide a proof-of-principle for being able to design complex systems involving separate redox sites that must communicate over long distances. We present a designed system that examines ET properties of a solvent-exposed tyrosine. The combination of a new chromophore for protein labelling with a scaffold for the systematic alteration of protein- or metal-based redox co-factors provides an attractive platform to generate and characterize tyrosine radicals produced by photoinduced electron transfer. More importantly, this protein system represents a foundation for the development of artificial electron-transfer conduits based on α-helical bundles.
Rubpymal was synthesized (see the Supporting Information) and conjugated to α3DH3 by reaction of the thiol of Cys75 with the maleimide moiety. After purification, the successful formation of α3DH3-Rubpymal was confirmed by QTOF-MS. The estimated distance between the ruthenium and the tyrosine is about 16 Å, based on the solution structure of a related pep- tide[22] and the sizes of the triazole and ruthenium trisbipyridine units.[29] The ground-state absorption spectrum of α3DH3-Rubpymal exhibits a typical metal-to-ligand charge transfer (MLCT) band for ruthenium trisbipyridine-based chromophores (Figure S1). The emission spectrum band maximum is located at λ= 640 nm, in agreement with Ru(bpy)3-triazole derivatives previously investigated.[28] The excited-state lifetime in α3DH3-Rubpymal was halved (τ ≈400 ns) compared to Ru-triazole, suggesting that some interaction between the peptide and the chromophore may take place in the excited state. However, the nanosecond transient absorption spectrum shows features typical of triazole functionalization (Figure S2).
Nanosecond laser flash photolysis was used to investigate the electron-transfer reactions and kinetics of the covalently linked ruthenium-peptide adduct (α3DH3-Rubpymal). Ruthenium hexaamine [(RuIII(NH3)6)]3+ was used as a reversible electron acceptor to generate the oxidized form of the photoactive unit, for example, [RuIIIbpymal]3+, which could then, in turn, oxidize Tyr70. Upon irradiation into the MLCT of [RuIIbpymal]2+, traces were collected at wavelengths of interest to characterize the kinetics of the observed process. Experiments on α3DH3-Rubpymal resulted in traces with three kinetic regimes (Figure 2A). By comparing control experiments with only [RuII(bpy)3]2+ and [RuIII(NH3)6]3+ in solution, the fastest kinetic step (τ = 40 ns, kq = 1.1 × 109m−1s−1; where kq is the quenching rate constant) was identified as an intermolecular electron transfer from the excited state of the chromophore to [RuIII(NH3)6]3+ to generate α3DH3-[RuIIIbpymal]3+ (Figure 2B, inset). This step is independent of the subsequent reactions involving the generated RuIII species and is present in all spectra collected. The photogenerated [RuIIIbpymal]3+ was characterized by the depletion of the MLCT band at 450 nm and possesses a high oxidizing power with a reduction potential (Eo) for the Ru3+/2+ couple measuring circa 1.2 V vs. NHE.[25,27,28] The second kinetic phase was associated with an increase in absorption between 360 nm and 420 nm, with maxima at 390 nm and 410 nm, which is typical of a tyrosine radical (Figure 2B). Traces at multiple wavelengths were used to reconstruct the transient absorption spectrum of the tyrosine radical (Figure 2B), which agrees with previously observed tyrosyl radical spectra[13,15,30] and supports the model that reduction of [RuIIIbpymal]3+ occurs by means of electron transfer from Tyr70. A global fit (Scheme 1; see also the Supporting Information) at wavelengths associated with the tyrosine radical found that the intramolecular electron-transfer rate (kiet) from tyrosine to [RuIIIbpymal]3+ is circa 3.3 × 105s−1. This kiet value is consistent with the expected rate constant for electron transfer at 16 Å, based on a simplified model of electron tunneling in proteins.[31] Using Δε410= 3000 m−1 cm−1[30] and Δε410 ≈4500 m−1 cm−1 for RuIII (where ε is the molar extinction coefficient),26 about 0.9 μm tyrosine radical is produced from 1.4 μM α3DH3-[RuIIIbpymal]3+, corresponding to a yield of approximately 60% for the electron transfer from tyrosine to α3DH3-[RuIIIbpymal]3+. The third kinetic phase observed corresponds to recombination of the tyrosine radical and [RuIII(NH3)6]3+ (τ = 210 μs) to return the system to the ground state.
Figure 2.
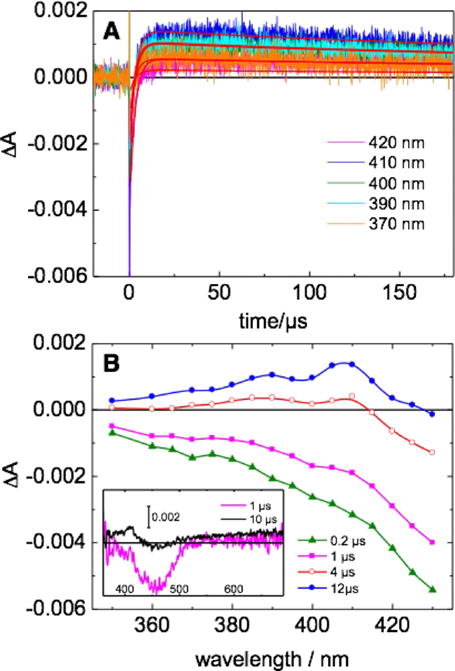
A) Kinetic traces at different wavelengths for an argon-purged aqueous solution of α3DH3-Rubpymal in the presence of 20 mM [RuIII(NH3)6]3+, 20 mM acetate-phosphate-borate (APB), 140 mM KCl, pH 5.0. Excitation at λ = 460 nm, laser energy 4 mJ. Red curves correspond to global fitting analysis. B) Differential absorption spectrum calculated from kinetic traces in (A). The spectrum of the tyrosine radical is observed at 12 μs post-laser flash. Inset in (B): transient absorption spectra from 350–700nm.
Scheme 1.
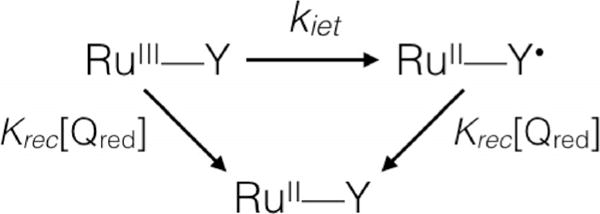
Reaction model for global fitting analysis.
The generation of a tyrosyl radical is tightly linked to intricate electron- and proton-coupled processes. In α3DH3-Rubpymal, the kiet value is four times faster at pH 9.5 as compared to pH 5.0 (Figure 3), which is consistent with PCET mechanisms in which proton transfer forms part of the rate-limiting step. The yield (≈60%) of tyrosyl radical formation did not change over the range of pH values tested, although the decay became four times faster with increasing pH values. Although the pH dependence of the rate of tyrosine oxidation is not in and of itself enough to assign a mechanism to this process, this system can form the basis of a series of studies to understand protein-based PCET processes through the introduction of hydrogen-bond donors. By studying the effects of these modifications and the extent to which they may perturb the PCET process, we may be able to understand how one mechanism is “chosen” over another.
Figure 3.
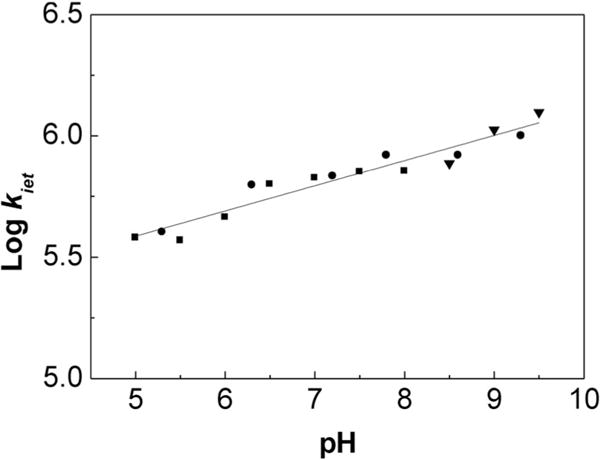
The pH dependence of kiet values from pH 5 to 9.5 in 20 mM APB buffer with 140 mM KCl.
We performed X-band EPR experiments to confirm the production of a tyrosine radical. In the presence of the nonreversible electron acceptor [CoIII(NH3)5Cl]2+, illumination for 1 min at λ= 460 nm produces a characteristic spectrum of high-spin [CoII(H2O)6]2+ with a g value of 4.44 and an organic radical (Figure 4A). Closer examination of the organic radical reveals a broad signal centered at giso = 2.0052, which is typical of a deprotonated tyrosine radical. The spectrum of the radical species between 10 and 60 K is shown in Figure 4B. The EPR spectrum of tyrosine is very sensitive to the conformation and environment of the radical; depending on the angle of rotation, up to six hydrogen atoms can contribute to the hyperfine structure. The radical observed in this system is consistent with other previously observed tyrosine radicals in native proteins[32–34] and thus confirms that this construct is a good model for tyrosine radicals in native proteins.
Figure 4.
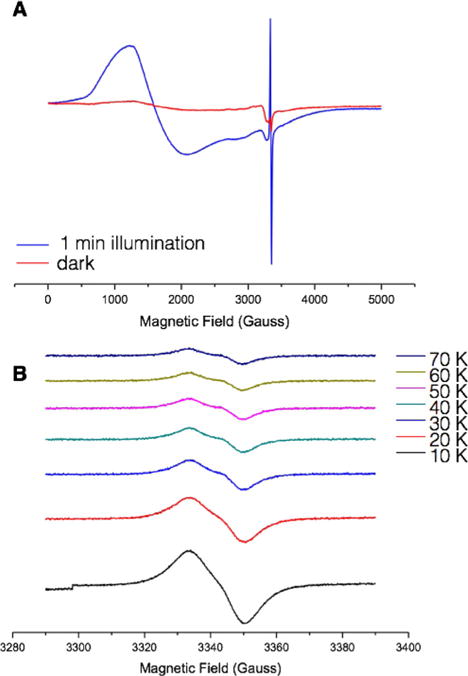
A) EPR spectra of α3DH3-Rubpymal in the dark (red trace) and after exposure to light at 460 nm for 1 min (blue trace). Conditions: 0.17 mM α3DH3-Rubpymal, 7.2 mM [CoIIII(NH3)5Cl]2+, pH 5.5, temperature 10 K, microwave frequency 9.38 GHz, microwave power 0.04 mW, modulation amplitude 10 G, modulation frequency 100 kHz. B) EPR spectra of tyrosyl radical in α3DH3-Rubpymal from 10 K to 70 K. Conditions: as for (A) but with a modulation amplitude of 2 G.
Given the importance of protein radicals and the complexity of the processes in which they function, simplified model systems can help elucidate the key aspects for function. This system represents the first de novo designed system for phototriggered intramolecular tyrosine radical formation and is an important tool for exploring the behavior of tyrosine radicals. The similarity to fundamental processes occurring in proteins such as PSII and the ability to selectively modify the protein suggest a departure point for systematic studies. Such a system may be used to explore the requirements for PCET mechanisms, the effect of nearby hydrogen-bonding residues, and the distance dependence of electron-transfer relays. The insertion of a coordinating site to bind a redox-active transition metal ion will provide an alternative way to study the role of redox-active amino acids in charge accumulation at a catalytic site. Furthermore, this study also stands as a first incursion in the de novo design of redox-active proteins and also lays the groundwork for the development of bio-inspired, fully artificial energy-harvesting systems.
Supplementary Material
Acknowledgments
A.G.T. acknowledges training grant support from the University of Michigan Chemistry-Biology Interface (CBI) training program (NIH grant 5T32GM008597) as well as support from the Chateaubriand Fellowship. V.L.P would like to thank the National Institutes of Health (NIH) for financial support for this research (ES012236). This work was supported in part by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01 and Labex CHARMMAT
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: http ://dx.doi.org/10.1002/cptc.201600044.
References
- 1.Dempsey JL, Winkler JR, Gray HB. Chem Rev. 2010;110:7024–7039. doi: 10.1021/cr100182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray HB, Winkler JR. Chem Phys Lett. 2009;483:1–9. doi: 10.1016/j.cplett.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gray HB, Winkler JR. Quart Rev Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 4.Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 5.Barry BA. J Photochem Photobiol B. 2011;104:60–71. doi: 10.1016/j.jphotobiol.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rappaport F, Diner BA. Coord Chem Rev. 2008;252:259–272. [Google Scholar]
- 7.Dixon WT, Murphy D. J Chem Soc Faraday Trans 2. 1976;72:1221–1230. [Google Scholar]
- 8.Umena Y, Kawakami K, Shen J-R, Kamiya N. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 9.Barry BA. BBA—Bioenergetics. 2015;1847:46–54. doi: 10.1016/j.bbabio.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Migliore A, Polizzi NF, Therien MJ, Beratan DN. Chem Rev. 2014;114:3381–3465. doi: 10.1021/cr4006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Bilio AJ, Crane BR, Wehbi WA, Kiser CN, Abu-Omar MM, Carlos RM, Richards JH, Winkler JR, Gray HB. J Am Chem Soc. 2001;123:3181–3182. doi: 10.1021/ja0043183. [DOI] [PubMed] [Google Scholar]
- 12.Shafaat HS, Griese JJ, Pantazis DA, Roos K, Andersson CS, Po-pović-Bijelić A, Gräslund A, Siegbahn PEM, Neese F, Lubitz W, Högbom M, Cox N. J Am Chem Soc. 2014;136:13399–13409. doi: 10.1021/ja507435t. [DOI] [PubMed] [Google Scholar]
- 13.Pizano AA, Lutterman DA, Holder PG, Teets TS, Stubbe J, Nocera DG. Proc Natl Acad Sci USA. 2012;109:39–43. doi: 10.1073/pnas.1115778108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berry BW, Martínez-Rivera MC, Tommos C. Proc Natl Acad Sci USA. 2012;109:9739–9743. doi: 10.1073/pnas.1112057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glover SD, Jorge C, Liang L, Valentine KG, Hammarström L, Tommos C. J Am Chem Soc. 2014;136:14039–14051. doi: 10.1021/ja503348d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore GF, Hambourger M, Gervaldo M, Poluektov OG, Rajh T, Gust D, Moore TA, Moore AL. J Am Chem Soc. 2008;130:10466–10467. doi: 10.1021/ja803015m. [DOI] [PubMed] [Google Scholar]
- 17.Megiatto JD, Jr, Méndez-Hernández DD, Tejeda-Ferrari ME, Teillout A-L, Llansola-Portolés MJ, Kodis G, Poluektov OG, Rajh T, Mujica V, Groy TL, Gust D, Moore TA, Moore AL. Nat Chem. 2014;6:423–428. doi: 10.1038/nchem.1862. [DOI] [PubMed] [Google Scholar]
- 18.Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Angew Chem Int Ed. 2011;50:2049–2053. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2011;123:2097–2101. [Google Scholar]
- 19.Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL. Proc Natl Acad Sci USA. 2012;109:21234–21239. doi: 10.1073/pnas.1212893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu F, Penner-Hahn JE, Pecoraro VL. J Am Chem Soc. 2013;135:18096–18107. doi: 10.1021/ja406648n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cangelosi VM, Deb A, Penner-Hahn JE, Pecoraro VL. Angew Chem Int Ed. 2014;53:7900–7903. doi: 10.1002/anie.201404925. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:8034–8037. [Google Scholar]
- 22.Plegaria JS, Dzul SP, Zuiderweg ERP, Stemmler TL, Pecoraro VL. Biochemistry. 2015;54:2858–2873. doi: 10.1021/acs.biochem.5b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plegaria JS, Herrero C, Quaranta A, Pecoraro VL. BBA—Bioenergetics. 2016;1857:522–530. doi: 10.1016/j.bbabio.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tebo AG, Hemmingsen L, Pecoraro VL. Metallomics. 2015;7:1555–1561. doi: 10.1039/c5mt00228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campagna S, Puntoriero F, Nastasi F, Bergamini G, Balzani V. Top Curr Chem. 2007;280:117–214. [Google Scholar]
- 26.Kalyanasundaram K. Coord Chem Rev. 1982;46:159–244. [Google Scholar]
- 27.Baron A, Herrero C, Quaranta A, Charlot MF, Leibl W, Vauzeilles B, Aukauloo A. Inorg Chem. 2012;51:5985–5987. doi: 10.1021/ic300227j. [DOI] [PubMed] [Google Scholar]
- 28.Baron A, Herrero C, Quaranta A, Charlot MF, Leibl W, Vauzeilles B, Aukauloo A. Chem Commun. 2011;47:11011–11013. doi: 10.1039/c1cc13683f. [DOI] [PubMed] [Google Scholar]
- 29.Harrowfield JM, Sobolev AN. Aust J Chem. 1994;47:763–767. [Google Scholar]
- 30.Candeias LP, Turconi S, Nugent J. Biochim Biophys Acta Bioenerg. 1998;1363:1–5. doi: 10.1016/s0005-2728(97)00077-7. [DOI] [PubMed] [Google Scholar]
- 31.Moser CC, Dutton PL. Biochim Biophys Acta Bioenerg. 1992;1101:171–176. [PubMed] [Google Scholar]
- 32.Miner KD, Pfister TD, Hosseinzadeh P, Karaduman N, Donald LJ, Loewen PC, Lu Y, Ivancich A. Biochemistry. 2014;53:3781–3789. doi: 10.1021/bi500353p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen JP, Kalman L, LoBrutto R, Williams JC. Nature. 1999;402:696–699. [Google Scholar]
- 34.Ma C, Barry BA. Biophys J. 1996;71:1961–1972. doi: 10.1016/S0006-3495(96)79394-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.