

Abstract
Patients with BRAF V600E mutant melanoma are typically treated with targeted BRAF kinase inhibitors, such as vemurafenib and dabrafenib. Although these drugs are initially effective, they are not curative. Most of the focus to date has been upon genetic mechanisms of acquired resistance; therefore, we must better understand the global signaling adaptations that mediate escape from BRAF inhibition. In the current study, we have used activity-based protein profiling (ABPP) with ATP-analogue probes to enrich kinases and other enzyme classes that contribute to BRAF inhibitor (BRAFi) resistance in four paired isogenic BRAFi-naïve/resistant cell line models. Our analysis showed these cell line models, which differ in their PTEN status, have considerable heterogeneity in their kinase ATP probe uptake in comparing naïve cells and adaptations to chronic drug exposure. A number of kinases including FAK1, SLK and TAOK2 had increased ATP probe uptake in BRAFi resistant cells, while KHS1 (M4K5) and BRAF had decreased ATP probe uptake in the BRAFi-resistant cells. Gene ontology (GO) enrichment analysis revealed BRAFi resistance is associated with a significant enhancement in ATP probe uptake in proteins implicated in cytoskeletal organization and adhesion, and decreases in ATP probe uptake in proteins associated with cell metabolic processes. The ABPP approach was able to identify key phenotypic mediators critical for each BRAFi resistant cell line. Together, these data show that common phenotypic adaptations to BRAF inhibition can be mediated through very different signaling networks, suggesting considerable redundancy within the signaling of BRAF mutant melanoma cells.
Keywords: Melanoma, BRAF, Drug Resistance, Proteomics, Activity-Based Protein Profiling
Introduction
The number of new patients diagnosed with melanoma, the most lethal form of skin cancer, continues to rise.(1-4) If detected early, melanoma is curable with surgery; more than 90% of Stage I patients survive longer than 5 years. However, the prognosis is poor for metastatic patients; only 5-10% survived 5 years after diagnosis.(5) Prior to 2011, patients with advanced stage melanoma were mainly treated by conventional cytotoxic chemotherapy, which achieved a response rate of 10-30%.(6, 7) However, key driver mutations have been identified in melanoma, which enable the development and application of targeted therapy. Specifically, the V600E mutation in the serine-threonine protein kinase, BRAF, is typically observed in ∼50% of cutaneous melanomas.(8) US FDA-approved targeted therapies against mutant BRAF include vemurafenib and dabrafenib.(9, 10) In large-scale randomized clinical trials both drugs achieve clinical responses in more than 50% of patients by RECIST (Response Evaluation Criteria in Solid Tumors), and these responses are frequently associated with a remarkable reduction in tumor burden and related symptoms. Despite impressive initial responses, most responses are short lived (i.e. the median responses are on the order of 6-7 months before progression). Early studies showed reactivation of MAPK signaling to be a common occurrence following BRAFi treatment and strategies were developed to combine BRAF and MEK inhibitors. Despite the combination improving overall survival (currently ∼25 months), acquired resistance is common in the majority of individuals.(9, 11) Most resistance mechanisms identified to date are genetic and include: mutations in NRAS or MEK1, dimerization of aberrantly spliced BRAF, MEK1 mutations, amplification of BRAF in conjunction with mutations in MEK2, and the activation of PI3K-AKT pathway through PTEN loss or acquired mutations in AKT1.(12-18) Similar mechanisms of resistance have been observed between patients failing BRAFi monotherapy and the BRAFi-MEKi combinations.(19, 20) To date, attempts to understand the mechanisms underlying BRAFi resistance have centered upon genomic assessment, which can be complemented with additional information about the diversity of signaling adaptation seen following chronic BRAF inhibition.
Previous work from our groups has shown that the underlying genetics of BRAF mutant melanoma cells contribute to the mechanism of therapeutic adaptation, with PTEN status being a potential predictor of drug resistance.(21) In the current study, we have characterized four pairs of BRAF mutant melanoma cell lines with either intrinsic sensitivity to BRAFi (PTEN expressing) or relative resistance (PTEN null) and have determined how chronic BRAFi therapy leads to the rewiring of their signaling networks using activity-based protein profiling (ABPP);(22, 23) three pairs of cell lines were further examined with expression proteomics. This current work builds upon prior expression proteomics and phosphoproteomics of these and other melanoma cell line models(24-28) and complements recent publications examining phosphoproteomic responses to BRAFi and MEKi,(29) targeting ROCK in BRAF mutant melanoma,(24) and proteomics of human metastatic melanoma tumors.(30) The ABPP approach to examine the kinome and its rewiring in BRAFi resistance relies upon interaction of desthiobiotinylating ATP probes with conserved lysine residues in or near the ATP-binding pocket. Once the probe is in the ATP binding site, the ε-amino group of the conserved lysine in the binding pocket forms a covalent bond with the biotin moiety of the probe releasing ATP.(31) After trypsin digestion, streptavidin enrichment of desthiobiotinylated (DBT) peptides is combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS) to quantify probe uptake of each protein using these labeled peptides as surrogates, which can be informative of expression level as well as kinase activity.(32),(33) ABPP was used to identify enzymes with steady state differences in the ATP probe uptake between each of the four paired BRAFi sensitive and resistant cell lines. Our analysis revealed that BRAFi resistance is associated with marked changes in probe uptake of proteins associated with cell adhesion and the actin cytoskeleton. Although the network adaptations to BRAFi therapy were diverse and cell type specific, we identified cytoskeletal remodeling to be common to all of the cell line models. Together, our studies show that even though diverse patterns of signaling adaptation are seen in response to BRAFi treatment, these signaling changes are frequently associated with similar phenotypic behaviors.
Experimental
Cell Lines and Reagents
Unless otherwise noted, chemicals were ordered from Sigma-Aldrich (St. Louis, MO) in the highest available purity. Solvents (HPLC H2O and acetonitrile) were purchased from Burdick and Jackson (Honeywell, Muskegon, MI). The FAK inhibitor, PF573228, and JNK inhibitor, SB600125, were obtained from Selleck Chemicals (Houston, TX). The 1205Lu, WM793, and WM164 melanoma cell lines were a generous gift from Dr. Meenhard Herlyn (The Wistar Institute, Philadelphia, PA), while the A375 cell line was obtained from ATCC. All cell lines were verified by genotyping to confirm BRAF V600E mutation. Cell lines were grown in RPMI-1640 media supplemented with 5% FBS. BRAFi resistant cell lines were generated in the presence of chronic vemurafenib exposure (2 μM for WM164 and WM793, 2.5 μM for A375 and 3 μM for 1205Lu).(34, 35) For ABPP experiments, cells were plated on 150 mm Petri dishes and left overnight. Cells were harvested at 70% confluence. First, the media was aspirated and the plate washed with cold PBS. Cells were scraped, resuspended with additional cold PBS, and transferred to 15 ml collection tubes. Cell pellets were obtained by centrifugation for 5 minutes at 500 × g at 4°C.
Activity-based Protein Profiling (ABPP)
ABPP experiments were carried out using Pierce® kinase enrichment kits and ActivX® ATP Probes (#88310 and 88311, Thermo, San Jose, CA), according to the manufacturers' instructions and as previously published.(36) Briefly, cells were solubilized in 400 μl of lysis buffer and 4 μl of Halt™ phosphatase and protease inhibitor cocktail (Catalog #78440, Thermo). Samples were sonicated thrice at 1-minute intervals using a pulse of 30% duty cycle for 30 seconds (Cell Disruptor 200, Branson, Danbury, CT). Cell lysates were cleared by centrifugation at 14,000 × g for 20 minutes and the supernatant was buffer exchanged by Zeba spin desalting columns (Thermo). Protein concentrations were estimated using Bradford assays, and a total of 1 mg of protein from each sample was prepared for labeling, enrichment, and mass analysis. Equal amounts of cell lysate were first incubated with 20 mM MgCl2 followed by incubation with 10 μM of desthiobiotinylating ATP probe for 10 minutes.
Following labeling, proteins were denatured in 10 M urea and reduced with 5 mM DTT. Samples were alkylated by incubation with 40 mM iodoacetamide in the dark for 30 minutes at room temperature. Following a second round of buffer exchange (Zeba, Thermo), proteins were digested using trypsin (1:50 enzyme-to-substrate ratio) overnight at 37 °C. DBT-labeled peptides were captured by incubating the digests with 50 μl of high capacity streptavidin beads for 1 hour. The beads were sequentially washed with lysis buffer, PBS (Gibco), and HPLC water; bound peptides were eluted by addition of aqueous 50% acetonitrile with 0.1% trifluoroacetic acid (TFA). The eluted peptides were concentrated by vacuum centrifugation and resuspended in 20 μl of aqueous 2% acetonitrile with 0.1% formic acid for LC-MS/MS analysis.
Liquid Chromatography-Tandem Mass Spectrometry
LC-MS/MS data were acquired on a hybrid quadrupole-orbital ion trap instrument (QExactive Plus, Thermo, San Jose, CA) interfaced with a nanoflow ultra high performance liquid chromatograph (RSLC, Dionex, Sunnyvale, CA). The sample was first loaded on a trapping column (5mm × 300 μm ID packed with C18 PepMap100 reversed phase resin, 5 μm particle size, 100Å pore size) and washed for 8 minutes with aqueous 2% acetonitrile containing 0.04% TFA. The trapped peptides were then separated by using reversed phase chromatography (C18 PepMap100, 50 cm × 75 μm ID, PepMap 100, Dionex, Sunnyvale-CA) at flow rate of 300 nl/min using the following 120 minute gradient program: 95% solvent A (aqueous 2% acetonitrile with 0.1% formic acid) for 8 minutes, solvent B (aqueous 90% acetonitrile with 0.1% formic acid) ramped from 5% to 50% in 90 minutes, then increased from 50% to 90% in 7 minutes, holding 90% for 5 minutes, followed by decreasing solvent B from 90% to 5% over 1 minute and re-equilibration for 10 minutes. Data dependent acquisition was performed using the following parameters: 2.4 kV spray voltage, MS survey scans with resolution of 70,000 with an AGC target of 3E6, followed by high energy collision-induced dissociation of 16 peptide precursor ions at following each survey scan using a normalized collision energy (NCE) of 30, and an isolation window width of 2.0 Th with a 0.5 Th offset. MS/MS data was acquired at 17,500 resolution with an AGC target of 1E5. Dynamic exclusion was set to 40 seconds to prevent resampling.
Protein Identification and Quantification
Protein identification and label free relative quantification were performed by Andromeda and MaxQuant (v. 1.2.2.5) by searching raw MS and MS/MS data against human entries in the UniProt database (release: June 2014 for individual cell line searches and July 2014 for the combined searches).(37, 38) Trypsin was selected as the protease and up to 2 missed cleavages were allowed per peptide. Cysteine carbamidomethylation was set as a fixed modification, while desthiobiotinylation of lysine residues, methionine oxidation and N-terminal acetylation were set as dynamic modifications. Searches were performed with the “match between runs” feature accepting elution within a time window of 4 min. MaxQuant output was filtered to <2% false discovery rate (FDR) by retaining only those entries in the “Desthiobiotin-ATPSites.txt” file matching following criteria: PEP < 0.05, non-zero intensity, localization probability > 0.5, and Andromeda SCORE > 40 or 50. Searches and data analysis were performed two ways: all naïve vs. all resistant cell lines as one combined MaxQuant search and downstream analysis as well as individual MaxQuant searches for cell line pairs. In both data analysis schemes, DBT-peptides not identified reproducibly in at least one cell line were discarded. Data from the combined searches were normalized using Iterative Rank Order Normalization (IRON),(39) and log2 transformed prior to imputation of missing values (using half of the minimum value within that cell line). For the DBT-peptides compared in each isogenic naïve and resistant cell line pair, intensity values were log2 transformed and missing values were imputed by global minima in the data. Data were normalized by Z-Score to minimize sample variation, and fold change was calculated using the log2 ratio between BRAFi resistant and naïve cell lines. Initial candidate selection was determined by t-tests with unequal variance and further filtering of the hits was performed by restricting peptides with a fold change value greater or less than 2 standard deviations away from the mean. Hierarchical clustering and Z-score normalization were performed in Perseus,(40) companion software for the processing of MaxQuant data. Venn diagrams were created using Venny.(41) Pathway and interactome analysis was carried out using GeneGO (MetaCore, Thomson Reuters, New York, NY) and GO enrichment analysis was performed using Webgestalt.(42) Kinome tree mapping was done using Kinome Render.(43)
Tandem Mass Tag Chemical Labeling
The 6-plex tandem mass tag (TMT) experiment was performed with the BRAFi naïve and resistant A375, WM164 and 1205Lu cell lines. Each of the cell lines (300 μg total protein) was lysed for protein extraction (as described above) with 8M urea lysis buffer containing sodium orthovanadate, β-glycerophosphate and sodium pyrophosphate. The supernatant from each sample was subjected to reduction by triscarboxyethylphosphine (10 mM final concentration) and incubated at 55 °C for 1 hour. Proteins were alkylated by incubation with iodoacetamide (40 mM final concentration) in the dark for 30 minutes at room temperature. After a threefold dilution with 100 mM TEAB buffer, samples were digested overnight with trypsin (1:50 enzyme-to-substrate) at 37 °C. On the next day, samples were desalted (Sep-Pak, Thermo) and lyophilized. The lyophilized samples were re-suspended in 100 μl of 100 mM triethylammonium bicarbonate buffer and subjected to labeling with TMT 6-plex reagents using following design: 126 – WM164 naïve; 127 – WM164 BRAFi resistant; 128 – A375 naïve, 129 – A375 resistant, 130 – 1205Lu naïve, 131 – 1205Lu resistant. The labeling was quenched by addition of 8 μl of 50% hydroxylamine, and the samples were pooled. The pooled sample was subjected to high pH reversed phase chromatography yielding 96 fractions that were combined into 12 fractions using a variation of the method as described by Mertins et al.(44) Each fraction was run in duplicate by LC-MS/MS, as described above with the following adjustments: MS resolution of 70,000, AGC of 3E6 and maximum IT of 100 ms. Each survey scan was followed by data-dependent acquisition of top 16 precursor ions. MS2 settings included a resolution of 17,500, AGC target of 1E5 and maximum IT of 60 ms and NCE 35. RAW MS data was searched by MaxQuant v. 1.5.1.2 against human entries in the UniProt database (released August 2014) with N-terminal and K modification for TMT as well as methionine oxidation and N-terminal acetylation. Intensity values from “peptides.txt” were normalized by IRON, and peptides mapping to β-tubulin, actin and fibronectin were extracted for calculation of log2 ratio between the BRAFi naïve and resistant cell line pairs.
Immunofluorescence
Cells were plated on glass coverslips and allowed to attach overnight. On the next day, cells were fixed, permeabilized and stained using Tubulin-AlexaFluor 488 (Cell Signaling Technologies, Danvers, MA) and Phalloidin-AlexaFluor 647 (Life Technologies, Carlsbad, CA) in accordance with a previously described protocol.(45) Coverslips were mounted with ProLong Antifade with DAPI (Life Technologies) and imaged on a confocal microscope. Images were analyzed using Definiens® Developer v2.0 (Definiens AG, Munich, Germany) software suite. The total fluorescence intensity was calculated per nuclei (stained by DAPI).
siRNA Transfection
Cells were plated and allowed to attach overnight under normal culture conditions. Cells were transfected using TAOK siRNA or non-targeting Control A siRNA (Santa Cruz Biotechnologies, Dallas, TX) using Lipofectamine 2000 (Thermo Fisher, Waltham, MA) in serum-free conditions as described previously.(45) Media was changed to RPMI-1640 containing 5% serum following a 6-hour or 24-hour transfection. Lysates were analyzed 72 hours after transfection, and scratch assays were started 24 hours after transfection.
Western Blotting
Cells were plated and allowed to attach overnight. Cells were harvested, lysed and analyzed by western blot as described previously.(45) Membranes were probed with antibodies for fibronectin (Becton Dickinson, San Jose, CA), pFAK (Y397), pFAK (Y576/577), FAK (Cell Signaling Technologies, Danvers, MA), TAOK2 (Santa Cruz Biotechnologies, Dallas, TX), and GAPDH as a loading control (Sigma, St. Louis, MO).
Wound Healing Assay
Cells were plated and grown to confluence in a 6-well plate, then a “wound” was scratched down the middle of each treatment well using a 200 μl pipette tip. Wound healing cell migration was monitored and imaged every 24 hours. Wound closure was quantitated using Ibidi's (Madison, Wisconsin) online wound healing image analysis tool, “WimScratch” and expressed as percentage of surface area in an image not covered by the cells (scratch).
3D Spheroid Assay
Melanoma cells were cultured as a 3D spheroid following a protocol described previously.(46) Spheroids were treated with DMSO control or increasing concentrations of PF573228 for 72 hours, then stained with Calcein AM for viability at 30 minutes to 1 hour (Thermo Fisher, Waltham, MA), and imaged immediately.
Matrigel Invasion Assay
To measure Matrigel invasion, first a 1:1 mixture of Matrigel to PBS was used to coat the inside of a transwell membrane, then transwells were placed up-side down and 100,000 cells were plated on top of the transwell membrane and allowed to attach for 3 hours. Once the cells attached, the transwells were placed right-side up with serum-free medium containing PF573228 in the bottom well and 5% FBS/RPMI-1640 in the top chamber. Cells were allowed to migrate for 72 hours, then fixed and stained with Phalloidin-AlexaFluor 647. The invasion was quantified using Image-Pro Premier version 9.1 (Media Cybernetics, Rockville, MD). Cells were imaged in slices throughout the distance of the invasion using confocal microscopy. Average invasion distance was measured in micrometers from analysis of approximately 500 width measurements along the z plane for each sample.
Results and Discussion
Using four naïve cell lines and their isogenic counterparts resistant to the BRAFi, vemurafenib, we identified a total of 17,556 peptides and 2,852 proteins (Supplementary Tables 1A and 1C, Supplementary Figure S1A) spanning multiple enzyme classes including kinases. From this larger dataset, a total of 625 peptides mapped to 165 protein kinases, which represent ∼30% of the known human kinome (Supplementary Tables 1B and 1D, Supplementary Figure S1B). While overall protein identifications reveal ∼34% are observed across all cell types, only a subset of these proteins show differential ATP probe uptake between the naïve and resistant cell lines. A Venn diagram of the proteins (and kinases) that show differential probe uptake between naïve and BRAFi resistant cells illustrates the heterogeneity of mechanisms for therapeutic escape as only ∼100 proteins are observed in the center of the Venn diagram as commonly modulated in all four cell line pairs (Figure 1A). The 4 cell line pairs show a wide range of sensitivity and resistance to BRAF inhibition (Supplementary Figure S2).
Figure 1. Common Changes in ATP Probe Uptake across 4 Model Systems of BRAFi Resistance.
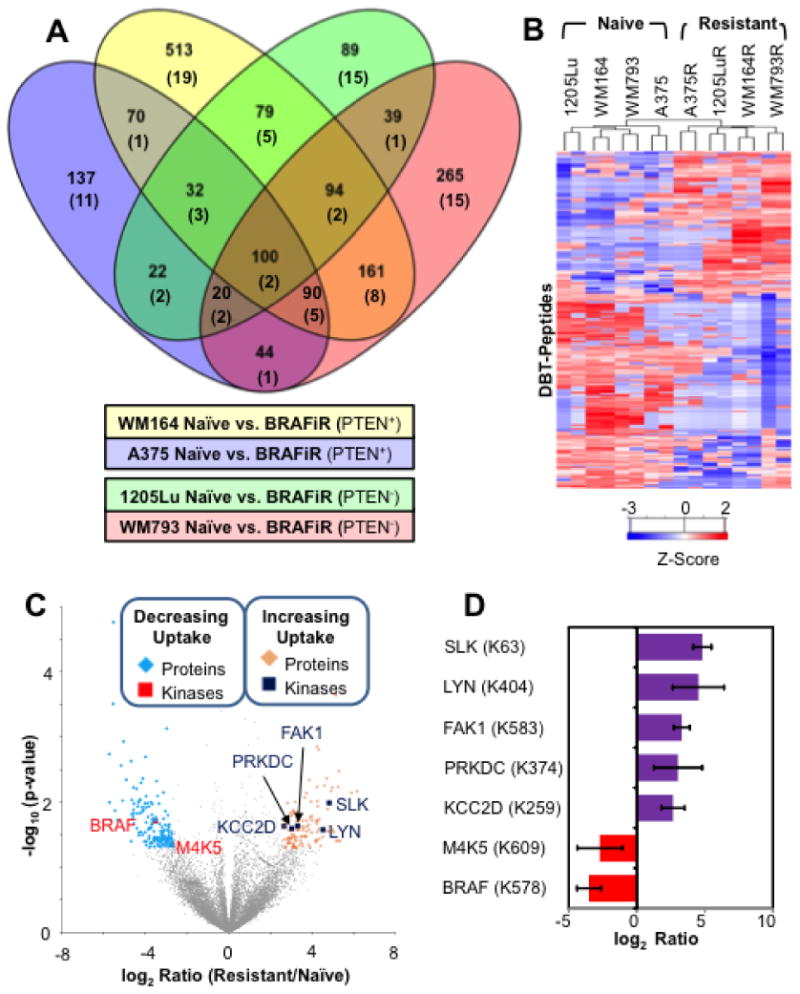
(A) Venn diagram showing overlap between the four isogenic naïve/BRAFi resistant cell line pairs in proteins with differential ATP probe uptake based on detection of DBT-peptides selected using p < 0.05; numbers of protein kinases with differential ion signals for DBT-peptides are given in parentheses. See Supplementary Figure S1 for additional details. (B) Heat map of desthiobiotinylated (DBT) peptides with consistently observed significant differences in ion signal (log2 ratio exceed 2 standard deviation away from the mean and p-value < 0.05) between naïve and BRAFi resistant cells produced by unsupervised clustering. (C) Volcano plot of ratios and p values for DBT-peptides indicating proteins (blue diamonds) and kinases (red squares) with decreasing ATP probe binding as well as proteins (gold diamonds) and kinases (blue squares) with increasing ATP probe binding in BRAFi resistant cell line. (D) Bar graph showing log ratio of the kinases (annotated with DBT-site) marked in the volcano-plot. Error bars represent standard deviation in BRAFi resistant to naïve ratio calculated for each set of replicate measurements.
Comparative analysis of all naïve vs. all BRAFi resistant cell lines
Two approaches were used to identify unique and common proteins contributing to drug resistance in these cell lines. First, we carried out an analysis by grouping all four naïve cell lines and comparing them to all four resistant lines. MaxQuant identified 10,974 peptides at <1% FDR after filtering based on PEP < 0.05, SCORE > 50, and localization probability > 0.5 (Supplementary Table 2A). A Welch test for significance identified a total of 469 DBT-peptides including 14 kinase specific peptides with differential ion signal intensities (based on p-value < 0.05) between the naïve and the resistant groups (Supplementary Table 2B). We next performed unsupervised clustering of ABPP peptides to determine relationships between the cell lines grouped as well as between groups of proteins with differential probe uptake.
A complete heat map of all DBT-peptides (Supplementary Figure S3) separates the cell lines based on PTEN status followed by BRAF inhibitor sensitivity, suggesting that many of the signaling events involving ATP binding proteins are PTEN dependent. To illustrate global fold change differences in relation to their significance, an unsupervised hierarchical analysis of DBT-peptides (selected based on log2 ratio differing from the average value for each experiment by more than 2 standard deviations, and p-value < 0.05; n=265, Supplementary Table 2C) was associated with a clear separation of naïve and resistant cell lines (Figure 1B). However, within the naïve and resistant groups, the individual cell lines did not cluster by their PTEN status. As can be seen in the heat map, the WM164 cell line groups with WM793 and 1205Lu rather than with A375 in a BRAFi resistant group (Figure 1B). DBT-peptides were graphed on a volcano plot of p-values from the Welch test against the log2 ratio between the resistant and naïve cell line groups. From the volcano plots, DBT-peptides with a fold change two standard deviations away from the mean and a p-value of < 0.05 were selected. We identified 113 DBT-peptides with higher ion signal in resistant cells and 153 DBT-peptides with higher ion signal in naïve cells. Within these selected hits, we identified 5 kinases (FAK1, SLK, LYN, PRKDC and KCC2D) with increased ATP probe uptake in the resistant cell line group and two kinases (BRAF and M4K5 or KHS1) with higher ATP probe uptake in the naïve cell line group (Figure 1C and 1D). The bar chart shows the magnitude of difference for each DBT-peptide and its modification site (Figure 1D). The decrease in BRAF ATP probe uptake was expected as the resistant cells are grown in presence of the BRAFi, and acquired BRAF inhibitor resistance is typically associated with the routing of MAPK signals through CRAF. (47, 48) The second candidate protein, KHS1, is a member of the STE family that has been shown to mediate signaling in the TNF-induced Stress Activated Protein Kinase (SAPK)/Jun amino-terminal kinase (JNK) pathway.(11) Our ABPP measurements suggest that this particular mechanism of activation of the SAPK pathway may be reduced in BRAFi resistant cell lines, as we see a decrease in the ATP probe uptake for KHS1 in BRAFi resistant cell lines. Other kinases, such as TAOK2, may compensate in order to maintain SAPK/JNK signaling (a possible mediator of BRAFi sensitivity) in resistant cell lines.(49) The decrease in probe uptake of KHS1 in resistant cell lines could also be due to off-target inhibitory effects of vemurafenib.(50)
GO term enrichment analysis of other enzyme classes was used to determine the biological processes that are associated with adaptive responses to BRAF inhibition. For this analysis, we only selected enzymes that showed differential ATP probe uptake based on both the fold change and the p-value cutoff (Figure 1C) as well as had a unique DBT-peptide identification. After applying these filters, 66 and 122 enzymes were detected with DBT-peptides that had significantly higher ion signal in the BRAFi resistant and naïve groups, respectively (Supplementary Table 3). GO term analysis for biological processes showed an enrichment of energy production pathways like tricarboxylic acid cycle (5 proteins), cellular respiration (9 proteins) and acetyl-CoA catabolic process (5 proteins) in the naïve cell lines (Table 1). These observations agree with multiple previous reports demonstrating that BRAF inhibitor resistance leads to metabolic reprogramming associated with increased oxidative respiration, the recovery of glycolysis and a greater dependence upon glutamine as an energy source.(51, 52), (53, 54) On the other hand, the resistant cell line group was mainly enriched for processes involved in cytoskeleton organization (18 proteins), actin filament bundle assembly (6 proteins) and cytoskeletal anchoring (3 proteins) (Table 1). In line with these observations, the resistant cells were found to exhibit a significant reorganization of cellular actin and tubulin (Figure 2, Supplementary Table 4).
Table 1. Biological Processes Related to BRAFi Resistance.
Top 10 GO categories (sorted by decreasing enrichment factor) representing enzymes excluding kinases with significantly different ATP probe uptake between the BRAFi sensitive and resistant cell line groups (log2 ratio differs by more than 2 standard deviations from the mean and p-value < 0.05).
| GO Biological Process (Naïve) | # | GO Biological Process (Resistant) | # |
|---|---|---|---|
| tricarboxylic acid cycle | 5 | cytoskeletal anchoring at plasma membrane | 3 |
| acetyl-CoA catabolic process | 5 | actin filament bundle assembly | 6 |
| monosaccharide biosynthetic process | 8 | establishment or maintenance of cell polarity | 6 |
| cellular respiration | 9 | cell junction assembly | 8 |
| energy derivation by oxidation of organic compounds | 13 | cell junction organization | 8 |
| generation of precursor metabolites and energy | 15 | actin cytoskeleton organization | 13 |
| carboxylic acid metabolic process | 22 | actin filament-based process | 14 |
| oxoacid metabolic process | 22 | cytoskeleton organization | 18 |
| organic acid metabolic process | 22 | cellular component assembly at cellular level | 18 |
| small molecule metabolic process | 42 | cellular component organization at cellular level | 29 |
Figure 2. Cytoskeletal remodeling in BRAFi resistant cell lines.
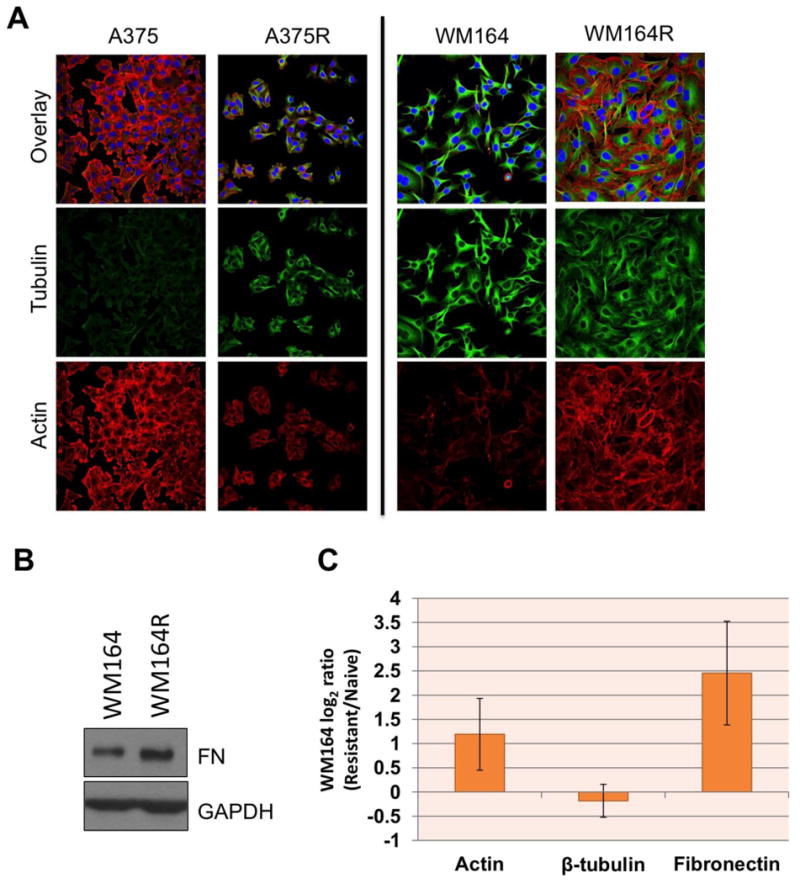
(A) Immunofluorescent staining showing different modes of cytoskeletal remodeling in different BRAFi resistant lines. The WM164 BRAFi resistant cell line shows cytoskeletal remodeling is driven by actin (Phalloidin- AlexaFluor 647, red) while in A375 resistant it is driven by tubulin (Tubulin-AlexaFluor 488, green). (B) Western blot analysis shows an increase in fibronectin expression in the WM164R cell line compared to treatment naïve WM164. (C) Protein expression data for actin, tubulin and fibronectin extracted from TMT expression proteomics highlights a similar trends as observed by cell staining experiments. Error bars denote standard deviation between log2 ratios of all the peptides mapping these proteins. For more details, see Supplementary Table 4.
Cellular localization of these hits also reflected their biological processes. The GO cellular compartment enrichment identified the cytoskeleton (25 proteins) as the most enriched cellular component for the proteins with increased ATP probe uptake in the resistant cell lines, while the proteins with decreased ATP probe uptake in the resistant lines or increased ATP probe uptake in inhibitor naïve cell lines were enriched for the mitochondrial compartment (34 proteins), as shown in Table 2.
Table 2. Cellular Compartments of Proteins Involved in BRAFi Resistance.
Top 10 GO categories (sorted by decreasing enrichment factor) representing enzymes, excluding kinases, with significantly different ATP probe uptake between naïve and resistant cell line groups (log2 ratio differs by more than 2 standard deviations from the mean and p-value < 0.05).
| GO Biological Process (Naïve) | # | GO Biological Process (Resistant) | # |
|---|---|---|---|
| mitochondrial part | 22 | actin filament bundle | 3 |
| mitochondrion | 35 | stress fiber | 6 |
| cytoplasmic part | 79 | actomyosin | 6 |
| intracellular organelle part | 75 | adherens junction | 8 |
| organelle part | 75 | anchoring junction | 8 |
| cytoplasm | 95 | actin cytoskeleton | 13 |
| intracellular organelle | 98 | cytosol | 14 |
| organelle | 98 | cytoplasmic part | 18 |
| intracellular part | 111 | cytoplasm | 18 |
| intracellular | 113 | intracellular part | 29 |
Pairwise comparison of ATP probe uptake in each cell line model
Since we expected some heterogeneity in the ATP probe uptake in each cell line due to differences in the biological background and varying levels of intrinsic resistance to BRAFi, MaxQuant searches were used to identify and evaluate the differences in the ATP probe uptake between paired BRAFi-naïve and resistant cell lines. Table 3 summarizes the DBT-peptides identified from each cell line pair and the final kinase specific DBT-peptides used for kinome analysis (Supplementary Table 5-8).
Table 3. Summary of DBT-Peptides identified from cell line specific MaxQuant searches.
| Category | A375 N/R | WM164 N/R | 1205Lu N/R | WM793 N/R |
|---|---|---|---|---|
| Total DBT-Peptides | 6,286 | 7,614 | 5,509 | 7,761 |
| Selected DBT-Peptides (p-value < 0.05) | 737 | 2,605 | 721 | 1,459 |
| Selected Kinase DBT-Peptides (p-value < 0.05) | 35 | 67 | 40 | 52 |
The union of filtered kinase DBT-peptides from individual cell lines (Table 3) resulted in a list of 92 kinases with differential ATP probe uptake in the four isogenic BRAFi naïve and resistant cell line pairs. We mapped each kinase (n = 69) that was identified by a unique DBT-peptide (132 peptides) on the human kinome tree (Figure 3).(55) To remove ambiguity in linking DBT-peptides and kinases, we provide additional tabular data for 40 DBT-peptides that were selected based on p-value, but mapped to multiple kinases as well as multiple peptides mapping to DNA-PK (Supplementary Table 9). The detected kinases represent all major kinase families, as well as the atypical kinases; cell lines show different patterns of kinase use under continuous BRAFi treatment. Overall, the patterns of ATP probe uptake across the resistant cell lines indicate that each exhibits a unique signaling pattern (Figure 3). This wide divergence in adaptive signaling is expected given that melanomas harbor one of the highest mutational loads of all cancers and display great genetic and phenotypic heterogeneity.(56)
Figure 3. Mapping ABPP Data to the Kinome Tree for Four Cell Line Models of BRAFi Resistance.
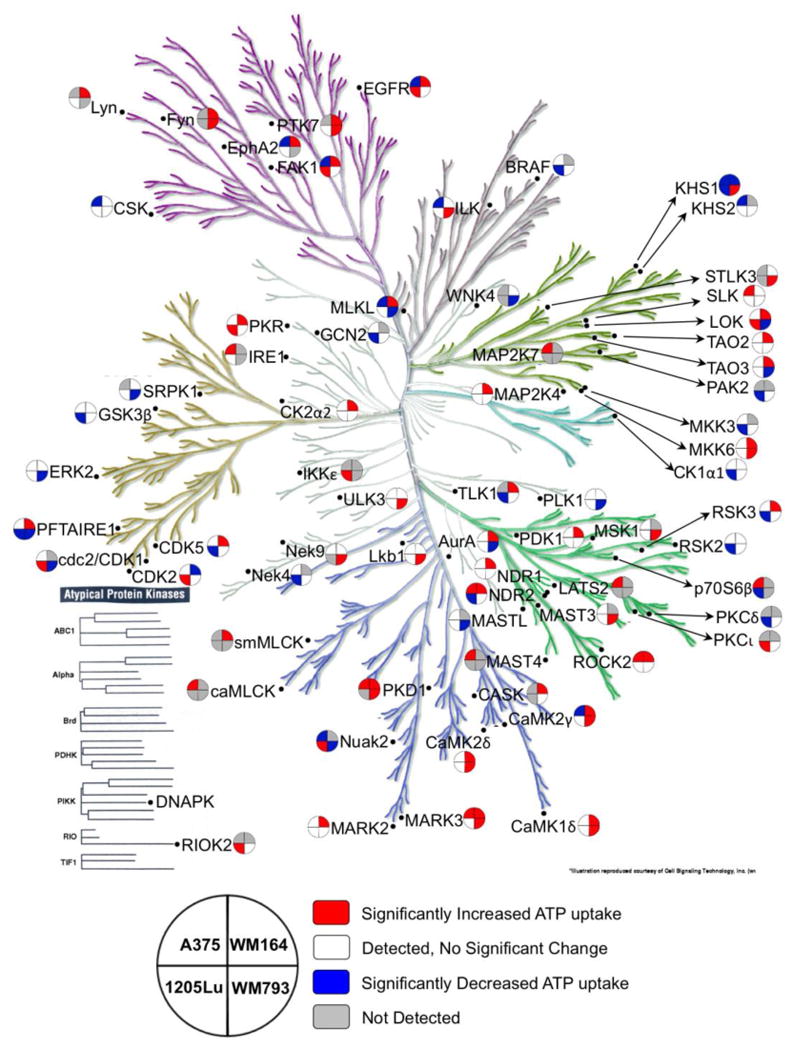
To provide an overview of the diverse signaling events associated with BRAFi resistance, selected kinase peptide-level data (DBT peptide with p < 0.05 in at least one naïve/BRAFi resistant cell line pair) is mapped onto the kinome tree. The key describes the layout of cell lines; the color coding includes DBT peptide not detected (gray), DBT peptide detected but not significantly changed (white), significantly increased (red), and significantly decreased in the drug resistance (blue). The kinome tree is used with permission from Cell Signaling Technology (Manning G et. al. Science 2002).55
The two PTEN WT cell lines, A375 and WM164 shared 7 kinases (EphA2, EGFR, FAK1, STK38L, KHS1, CAMK2G and MLKL) that showed differential ATP probe uptake in their isogenic BRAFi naïve and resistant lines in which KHS1 and STK38L showed consistent probe uptake. The PTEN Null cell lines, 1205Lu and WM793, shared 5 kinases (LOK, CDK14, CDK1, NUAK2 and KHS1) that showed differential ATP probe uptake. Within these 5 kinases, only CDK14 showed consistent increase in the ATP probe uptake in the BRAFi naïve 1205Lu and WM793 cell lines (Supplementary Table 9). These observations further strengthen the fact that heterogeneity in BRAFi response and mechanisms of resistance in melanoma cell lines extends beyond receptor tyrosine kinases and EMT signature proteins as reported previously.(57, 58)
Within individual cell lines there were several kinases that showed differential ATP probe uptake between the isogenic naïve and resistant pairs. We identified 31, 49, 33 and 41 unique kinase-specific DBT-peptides with differential ATP probe uptake (p-value <0.05 filtered) in the naïve and resistant pair in the A375, WM164, 1205Lu and WM793 cell line models respectively (see Supplementary Figures S4 and S5).
We further explored BRAF mutant, PTEN WT WM164 cells, which provided the highest number of candidate proteins with differential ATP probe uptake. The 49 DBT-peptides mapped to 31 kinases, which were narrowed down to 10 unique kinases with fold change values greater than 2 standard deviations away from the mean (Figures 4A-C). Selected kinases passing the p-value filter formed a well-connected interactome starting with signal initiation from EphA2 and EGFR (Figure 4D). There is already evidence linking both EGFR and EphA2 signaling to BRAF inhibitor adaptation. Chronic treatment of melanoma cells with BRAF inhibitors was associated with the induction of a TGF-β-mediated increase in EGFR expression that led to the adoption of a slow-growing phenotype which allowed melanoma cells to persist in the presence of BRAF and BRAF-MEK inhibition.(59) Likewise, increased EphA2 expression was found to convey resistance to BRAF inhibition in both melanoma cell lines and post-treatment tissue samples, with its shRNA knockdown or small molecule EphA2 inhibitors found to restore BRAFi sensitivity.(60) Further work also showed the ability of ligand independent EphA2 signaling to mediate the switch to a metastatic phenotype in the presence of chronic BRAFi treatment.(34) Another kinase identified through our analysis was TAOK2, a relatively understudied protein that was first identified in a screen for RNAs with increased expression in human prostate cancer.(61)
Figure 4. Comparison of Peptides Representing ATP Probe Uptake by Kinases in Naïve and BRAFi Resistant WM164 Melanoma Cell Lines.
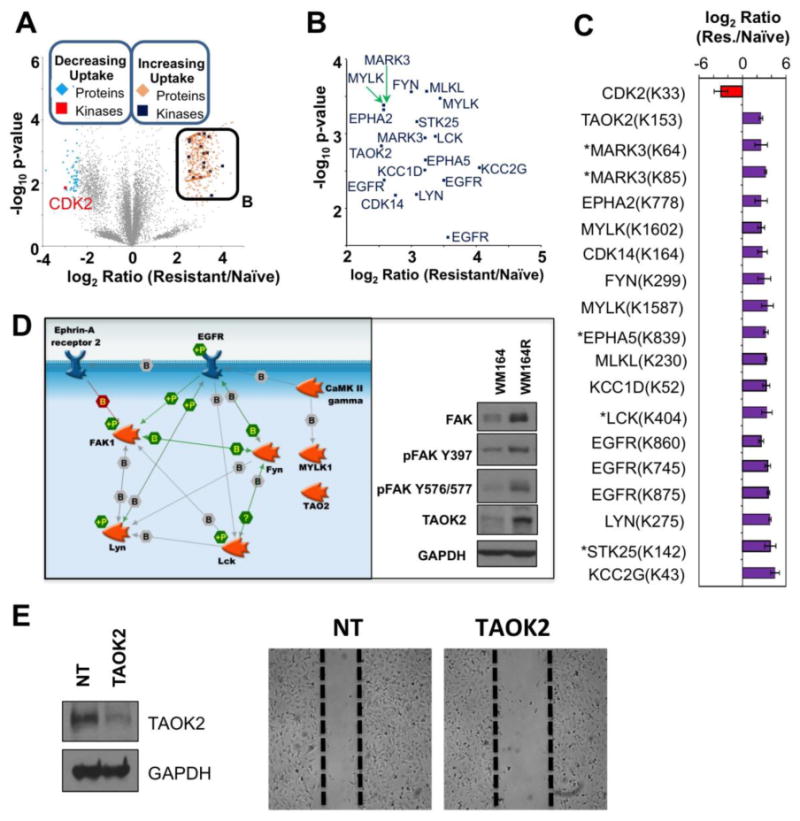
(A) Volcano plot of DBT-peptides observed WM164 naïve and BRAFi resistant cells using the following cutoffs: log2 ratio exceeds 2 standard deviations away from the mean and p-value < 0.05. (B) Inset shows kinases with significantly increased ATP probe uptake in the WM164 BRAFi resistant cell line from panel A. (C) Bar-graph displaying log2 ratio for the significantly different kinases in WM164 cell line comparison and the DBT labeled lysine site marked on the volcano plots. Error bars represent standard deviation in fold change per replicate measurement, * represents kinase not identified by a unique DBT-peptide. (D) Interactome of selected kinases with increased ATP probe uptake in the WM164 BRAFi resistant cell line, using p-value < 0.05. Additional unconnected nodes (e.g. MLKL, CAMK1D) not shown for clarity. (E) Western blot analysis (left) confirms siRNA-mediated knockdown of TAOK2, which leads to a decrease in migratory capacity of WM164R in a wound healing assay (right).
TAOK2 is a serine/threonine protein kinase that is involved in multiple processes including DNA damage response and MAPK14/p38 MAPK stress activated pathway signaling.(49) The DBT-peptide, DVK(dbt)AGNIIISEPGIVK, in TAOK2 is modified at K153 adjacent to the active site D151. Western blot validation confirmed an increased expression of TAOK2 and FAK in the WM164 BRAFi-resistant cells compared to the naïve parental line (Figure 4D). As TAOK2 is a known facilitator of JNK signaling, TAOK2 activation may act as a compensatory mechanism for sustained JNK signaling in the resistant cell line (49). The JNK signaling pathway is an important mediator of escape from BRAF inhibition. Through validation studies, we have confirmed that combined targeting of JNK (using the inhibitor SB600125) sensitized WM793 and 1205Lu to BRAF inhibition and prevented clonal outgrowth in long term colony formation assays (Supplementary Figure S6A). Furthermore, immunoblots confirm increased activation of MKK3/6/p38 signaling, a pathway with known crosstalk with JNK/Jun signal transduction (Supplementary Figure S6B).(62, 63) A phenotypic role for TAOK2 in drug resistant WM164R cells was suggested by the modest reduced cell migration following siRNA knockdown of TAOK2 (Figure 4E). No significant changes in cell cycle, cell growth, or sensitivity to vemurafenib were observed with knockdown of TAOK2 in the WM164R BRAFi-resistant cells (Supplementary Figure S7).
In the BRAF mutant and PTEN null 1205Lu cells, 33 DBT-peptides (p value < 0.05) represent 27 unique kinases. Focusing on fold changes of 2 standard deviations away from the mean reduced the number of significantly different kinases to 5 (CDK19, CDK14, NEK4, M4K5 and BRAF) that showed decreased ATP probe uptake in the resistant cell line and 3 kinases (DNA-PK, FAK1 and RIOK2) that showed increased ATP probe uptake in the resistant cell line (Figure 5A and 5B).
Figure 5. Comparison of Peptides Representing ATP Probe Uptake by Kinases in Naïve and BRAFi Resistant 1205Lu Melanoma Cell Line.
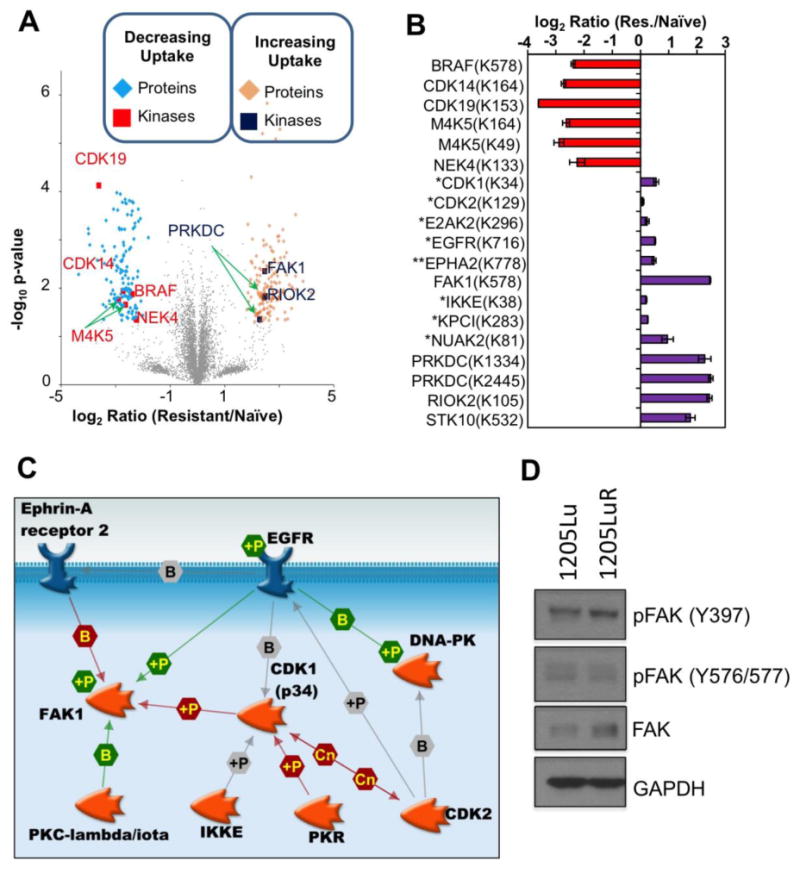
(A) Volcano plot of DBT-peptides observed 1205Lu naïve and BRAFi resistant cells using the following cutoffs: log2 ratio exceeds 2 standard deviations away from the mean and p-value < 0.05. (B) Bar-graph displaying log2 ratio for the significantly different kinases and the ATP labeled lysine site marked on the volcano plots in 1205Lu, * represents kinases with significantly increased ATP probe uptake (p-value < 0.05) in the BRAFi resistant cell line that do not pass fold change criteria, ** EphA2 selected based on prior literature evidence for its role in melanoma drug resistance. Error bars represent standard deviation in fold change per replicate measurement. (C) Interactome of kinases with increased ATP probe uptake in the 1205Lu BRAFi resistant cell line (p-value < 0.05) Proteins with unconnected nodes (RIOK2, NUAK2, STK10) not shown for clarity. (D) Western blot showing differences in levels of FAK expression and phosphorylation between 1205Lu and 1205LuR cell lines.
The DBT-peptide mapping to CDK19 was also shared by CDK8, a colorectal cancer oncogene whose suppression results in histone variant macroH2A-dependent inhibition of melanoma proliferation in cell line models.(64) The decreases in ATP probe uptake for BRAF and M4K5 in the BRAFi resistant 1205LuR cell line confirm the direct and off target inhibitory actions of vemurafenib, while CDK14 has been implicated in maintaining various hallmarks of cancer in several tumor types including ovarian, pancreatic, breast and gastric cancer as well as glioma.(65-69) Never in Mitosis Gene A (NIMA)-related protein kinase, NEK4, plays an important role in microtubule positioning and its deletion makes cancer cells either sensitive or resistant to microtubule poisons.(70) Apart from this, deletion of NEK4 was also found to delay replicative senescence and DNA damage repair in human fibroblast cells.(71) Both these observations suggest NEK4 could be a potential therapeutic target in melanoma as it shows decreased ATP probe uptake in the resistant cell lines. To create an interaction network for these kinases, we included all kinases that were selected based on p-value alone irrespective of their fold change and also included EphA2.(34, 60) Similar to the interaction network for WM164, signaling could be initiated from the receptor tyrosine kinases, EGFR and EphA2, with FAK1 and CDK1 acting as hub proteins (Figure 5C).
Increased FAK1 Signaling Mediates Increased Invasiveness in BRAFi Resistant Cells
Signal initiation from EphA2 and EGFR is crucial for downstream signaling to promote survival of BRAFi resistant cell lines.(34, 59, 60) EphA2 is a receptor tyrosine kinase known to bind to FAK1 leading to recruitment of Shp2, which dephosphorylates FAK1 and inhibits integrin signaling in a ligand dependent manner.(72) However, in our dataset, we observed increased ATP probe uptake of both EphA2 and FAK1 consistent with ligand-independent activity of EphA2 recently reported in melanoma progression.(34) Together, our results suggest increased activity of processes involved in cytoskeleton remodeling, actin filament bundle assembly and cytoskeletal anchoring in BRAFi resistant cell lines mediated through FAK1 (Figure 2). FAK1 is a non-receptor tyrosine kinase implicated in cell adhesion and migration as well as cytoskeleton remodeling. FAK is known to be overexpressed in several cancer types including colon, ovary, breast, pancreas, and prostate.(73) Small molecule FAK inhibitors are currently being evaluated in early-stage clinical trials for a variety of epithelial cancers. In the four melanoma cell line models, we observed multiple DBT-peptides from FAK1. In WM164 cells, the FAK1 DBT-peptide passed our p-value filter of 0.05; in 1205Lu cells, we observed more than 4-fold increase in ATP probe uptake in the BRAFi resistant cell line compared to its naïve counterpart. Western blots confirmed increases in the expression and phosphorylation of FAK at Y397 in the 1205LuR cell line (Figure 5D). The Y397 position in FAK1 is the autophosphorylation site, which is required for the binding with the Src-family kinases.(74) A role for FAK signaling in the therapy-adapted phenotype was suggested by the ability of the FAK inhibitor, PF573228, to inhibit melanoma cell migration into a scratch wound and invasion in both a 3D spheroid assay and Matrigel invasion assay (Figure 6A-D). The reliance upon FAK signaling in melanoma cells with acquired resistance to BRAFi is distinct from what has been observed when melanoma cells are subjected to acute MEK inhibition.(75) In this instance, treatment of the cells with MEK inhibitor for 24-48 hours was associated with an increased reliance upon Src, and enhanced protease mediated invasion, with little change in FAK signaling noted.(75) Other work implicated an EGFR/Src/STAT3 signaling loop in both BRAF inhibitor resistance and metastatic dissemination to the lungs.(76) However, no changes were observed between the naïve and resistant 1205Lu cell lines in either Src family kinase ATP probe uptake in the ABPP experiment or in SRC family tyrosine autophosphorylation by immunoblotting (data not shown). These data are consistent with a lack of increased FAK1 phosphorylation at Y576/577 (Figure 5D). Further analysis of the quantitative ABPP data also revealed little change in STAT3 ATP probe uptake between the sensitive and resistant cell lines (data not shown). Additional experiments showed that FAKi treatment did not induce significant BRAF inhibitor sensitivity in either 1205Lu cells or 1205LuR cells (Supplementary Figure S8). A FAK-inactive cell line A745R did not show sensitivity to FAK inhibition in 3D spheroid invasion and wound scratch assays (Supplementary Figure S9).
Figure 6. FAK signaling mediates invasion in BRAFi resistant cell lines.
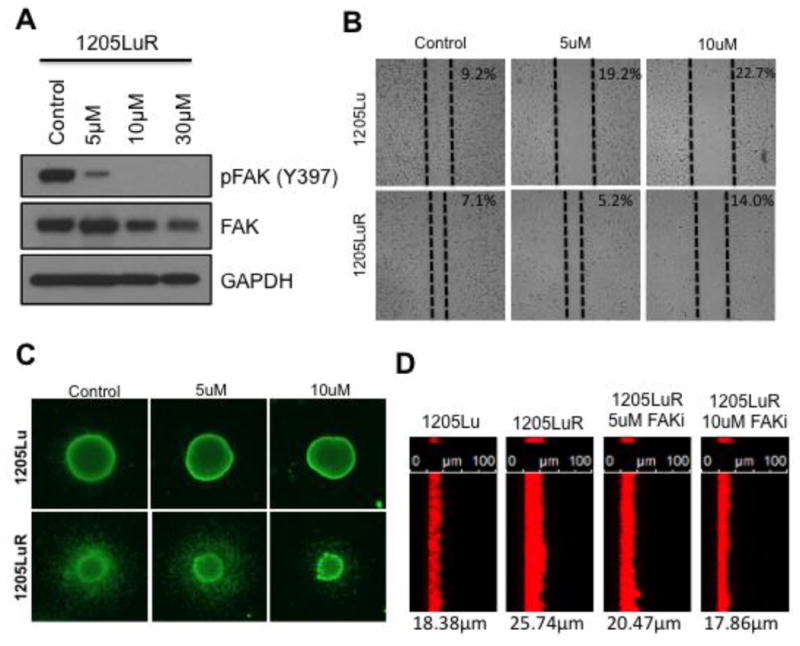
(A) Western blot analysis confirms efficacy in inhibiting FAK phosphorylation with increasing doses of the FAK inhibitor, PF573228, in 1205LuR cells (2 hours after treatment). (B) Wound healing assay shows increasing concentrations of the FAK inhibitor (24 hour treatment after wound scratch) can block the increased migratory capacity of 1205LuR cells. Values represent the percentage of scratch surface area (not covered by cells). (C) Immunofluorescent viability (Calcein-AM) staining of 1205Lu and 1205LuR in 3D spheroid invasion assay. Increasing doses of FAKi block the increased invasive capacity of 1205LuR cells. (D) Matrigel invasion assay shows FAKi blocks the increased invasive capacity of 1205LuR (48 hour treatment). Cells were plated on top of Matrigel and allowed to invade towards an increased FBS gradient with and without FAKi treatment. Cells were then fixed and stained for actin cytoskeleton using Phalloidin-AlexaFluor 647. Values represent average distance of invasion in microns.
Conclusions
The ATP-probe-based ABPP approach provides important new insights into how chronic BRAF inhibition rewires signaling networks across different BRAF mutant melanoma cell lines. We show for the first time that the adaptations to the BRAF inhibitor, vemurafenib, are heterogeneous, with very limited overlap between the patterns of kinase usage observed in isogenic sensitive and resistant cell lines. Despite this signaling diversity, acquired resistance is associated with common phenotypic behavior including altered metabolism and marked rearrangement of the cytoskeleton. Our data highlight the inherent redundancy in signaling networks and suggest many different signal transduction routes reach the same cellular behaviors. These findings have important implications for the development of therapy combinations and suggest that therapeutic adaptation will only be abrogated when key combinations of non-redundant signaling nodes are identified and targeted, a task complicated by the genetic complexity and heterogeneity of melanoma. The observation that different cell lines have high levels of heterogeneity in kinase utilization yet often demonstrate similar phenotypic adaptations certainly poses a very complicated challenge to addressing therapy resistance, and something our group has considered previously through the use of inhibitors of HSP90 as a tool for targeting multiple resistance mediators simultaneously (35, 77). The ultimate goal for continued research into the links between genotype and phenotypic adaptation is the development of improved, clinically relevant, targeted therapy combinations.
Supplementary Material
Acknowledgments
The Moffitt Proteomics Core is supported by the National Cancer Institute (P30-CA076292) and the Moffitt Foundation. This project has been supported by R01 CA161107, R21 CA198550, and Skin SPORE grant P50 CA168536 (Project 2: VKS and KSSM, DRP: JK) from the National Institutes of Health. IF is supported by the Melanoma Research Foundation. The authors would like to thank Proteome Sciences, particularly Ian Pike and Vikram Mitra, for discussions about TMT labeling and analysis.
The Koomen lab is funded in part by a sponsored research agreement from Proteome Sciences (Electrophoretics), but that support has not been used for the completion of this project.
Footnotes
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflict of Interest: Conflict is noted because this manuscript involves TMT chemical labeling.
Supporting Information: Supporting information is available online free of charge; the datasets have been provided for public access as partial submissions at PRIDE (http://www.ebi.ac.uk/pride/archive/) (78) using the following identifiers: Activity-Based Protein Profiling of BRAF Inhibitor Resistance in Melanoma Cell Lines: PXD004343 (Username: reviewer43181@ebi.ac.uk and Password: hoZ6v4Ps) and Expression Proteomics of BRAF Inhibitor Resistance in Melanoma Cell Lines: PXD004342 (Username: reviewer01718@ebi.ac.uk Password: aIIO6VjT). Supplementary Tables include selected DBT-peptide data from ABPP datasets as well as specific calculations referenced in the text; Supplementary Figures includes additional Venn diagrams, ABPP data for the cell lines not featured in the body of the manuscript, and additional biological follow up data.
References
- 1.U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999–2012 Incidence and Mortality Web-based Report. Atlanta: U.S Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2015. http://www.cdc.gov/uscs. [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 4.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 5.Ugurel S, Rohmel J, Ascierto PA, Flaherty KT, Grob JJ, Hauschild A, Larkin J, Long GV, Lorigan P, McArthur GA, Ribas A, Robert C, Schadendorf D, Garbe C. Survival of patients with advanced metastatic melanoma: The impact of novel therapies. Eur J Cancer. 2016;53:125–34. doi: 10.1016/j.ejca.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Serrone L, Zeuli M, Sega FM, Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19(1):21–34. [PubMed] [Google Scholar]
- 7.Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob JJ, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A, Roesch A, Hauschild A. Melanoma. Nat Rev Dis Primers. 2015;1:15003. doi: 10.1038/nrdp.2015.3. [DOI] [PubMed] [Google Scholar]
- 8.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, Group BS. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trinh VA, Davis JE, Anderson JE, Kim KB. Dabrafenib therapy for advanced melanoma. Ann Pharmacother. 2014;48(4):519–29. doi: 10.1177/1060028013513009. [DOI] [PubMed] [Google Scholar]
- 11.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Jr, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 12.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–67. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, Misteli T, Chapman PB, Sosman JA, Graeber TG, Ribas A, Lo RS, Rosen N, Solit DB. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480(7377):387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, Fukunaba-Kalabis M, Wubbenhorst B, Chen TY, Liu Q, Sproesser K, DeMarini DJ, Gilmer TM, Martin AM, Marmorstein R, Schultz DC, Speicher DW, Karakousis GC, Xu W, Amaravadi RK, Xu X, Schuchter LM, Herlyn M, Nathanson KL. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4(6):1090–9. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–9. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA, Golub TR. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, Howle J, Hayward NK, Johansson P, Scolyer RA, Kefford RF, Rizos H. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- 20.Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, Johannessen CM, McKenna A, Cibulskis K, Kryukov G, Hodis E, Lawrence DP, Fisher S, Getz G, Gabriel SB, Carter SL, Flaherty KT, Wargo JA, Garraway LA. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4(1):61–8. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, Ribas A, Palma MD, Nathanson KL, Koomen JM, Messina JL, Smalley KS. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–60. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao Y, Guo L, Wang Y. A targeted quantitative proteomics strategy for global kinome profiling of cancer cells and tissues. Mol Cell Proteomics. 2014;13(4):1065–75. doi: 10.1074/mcp.M113.036905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Worboys JD, Sinclair J, Yuan Y, Jorgensen C. Systematic evaluation of quantotypic peptides for targeted analysis of the human kinome. Nat Methods. 2014;11(10):1041–4. doi: 10.1038/nmeth.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smit MA, Maddalo G, Greig K, Raaijmakers LM, Possik PA, van Breukelen B, Cappadona S, Heck AJ, Altelaar AF, Peeper DS. ROCK1 is a potential combinatorial drug target for BRAF mutant melanoma. Mol Syst Biol. 2014;10:772. doi: 10.15252/msb.20145450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh CK, George J, Nihal M, Sabat G, Kumar R, Ahmad N. Novel downstream molecular targets of SIRT1 in melanoma: a quantitative proteomics approach. Oncotarget. 2014;5(7):1987–99. doi: 10.18632/oncotarget.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qendro V, Lundgren DH, Rezaul K, Mahony F, Ferrell N, Bi A, Latifi A, Chowdhury D, Gygi S, Haas W, Wilson L, Murphy M, Han DK. Large-scale proteomic characterization of melanoma expressed proteins reveals nestin and vimentin as biomarkers that can potentially distinguish melanoma subtypes. J Proteome Res. 2014;13(11):5031–40. doi: 10.1021/pr5006789. [DOI] [PubMed] [Google Scholar]
- 27.Old WM, Shabb JB, Houel S, Wang H, Couts KL, Yen CY, Litman ES, Croy CH, Meyer-Arendt K, Miranda JG, Brown RA, Witze ES, Schweppe RE, Resing KA, Ahn NG. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell. 2009;34(1):115–31. doi: 10.1016/j.molcel.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cholewa BD, Pellitteri-Hahn MC, Scarlett CO, Ahmad N. Large-scale label-free comparative proteomics analysis of polo-like kinase 1 inhibition via the small-molecule inhibitor BI 6727 (Volasertib) in BRAF(V600E) mutant melanoma cells. J Proteome Res. 2014;13(11):5041–50. doi: 10.1021/pr5002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuart SA, Houel S, Lee T, Wang N, Old WM, Ahn NG. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol Cell Proteomics. 2015;14(6):1599–615. doi: 10.1074/mcp.M114.047233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welinder C, Pawlowski K, Sugihara Y, Yakovleva M, Jonsson G, Ingvar C, Lundgren L, Baldetorp B, Olsson H, Rezeli M, Jansson B, Laurell T, Fehniger T, Dome B, Malm J, Wieslander E, Nishimura T, Marko-Varga G. A protein deep sequencing evaluation of metastatic melanoma tissues. PLoS One. 2015;10(4):e0123661. doi: 10.1371/journal.pone.0123661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry. 2007;46(2):350–8. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- 32.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, Kozarich JW. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol. 2011;18(6):699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McAllister FE, Niepel M, Haas W, Huttlin E, Sorger PK, Gygi SP. Mass spectrometry based method to increase throughput for kinome analyses using ATP probes. Anal Chem. 2013;85(9):4666–74. doi: 10.1021/ac303478g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paraiso KH, Das Thakur M, Fang B, Koomen JM, Fedorenko IV, John JK, Tsao H, Flaherty KT, Sondak VK, Messina JL, Pasquale EB, Villagra A, Rao UN, Kirkwood JM, Meier F, Sloot S, Gibney GT, Stuart D, Tawbi H, Smalley KS. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov. 2015;5(3):264–73. doi: 10.1158/2159-8290.CD-14-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smyth T, Paraiso KH, Hearn K, Rodriguez-Lopez AM, Munck JM, Haarberg HE, Sondak VK, Thompson NT, Azab M, Lyons JF, Smalley KS, Wallis NG. Inhibition of HSP90 by AT13387 delays the emergence of resistance to BRAF inhibitors and overcomes resistance to dual BRAF and MEK inhibition in melanoma models. Mol Cancer Ther. 2014;13(12):2793–804. doi: 10.1158/1535-7163.MCT-14-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang B, Hoffman MA, Mirza AS, Mishall KM, Li J, Peterman SM, Smalley KS, Shain KH, Weinberger PM, Wu J, Rix U, Haura EB, Koomen JM. Evaluating kinase ATP uptake and tyrosine phosphorylation using multiplexed quantification of chemically labeled and post-translationally modified peptides. Methods. 2015;81:41–9. doi: 10.1016/j.ymeth.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10(4):1794–805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 38.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 39.Welsh EA, Eschrich SA, Berglund AE, Fenstermacher DA. Iterative rank-order normalization of gene expression microarray data. BMC Bioinformatics. 2013;14:153. doi: 10.1186/1471-2105-14-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics. 2012;13(Suppl 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliveros JC. Venny. An interactive tool for comparing lists with Venn's diagrams. 2007-2015 http://bioinfogp.cnb.csic.es/tools/venny/index.html.
- 42.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33(Web Server issue):W741–8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chartier M, Chenard T, Barker J, Najmanovich R. Kinome Render: a stand-alone and web-accessible tool to annotate the human protein kinome tree. PeerJ. 2013;1:e126. doi: 10.7717/peerj.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10(7):634–7. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fedorenko IV, Abel EV, Koomen JM, Fang B, Wood ER, Chen YA, Fisher KJ, Iyengar S, Dahlman KB, Wargo JA, Flaherty KT, Sosman JA, Sondak VK, Messina JL, Gibney GT, Smalley KS. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene. 2016;35(10):1225–35. doi: 10.1038/onc.2015.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fedorenko IV, Fang B, Koomen JM, Gibney GT, Smalley KS. Amuvatinib has cytotoxic effects against NRAS-mutant melanoma but not BRAF-mutant melanoma. Melanoma Res. 2014;24(5):448–53. doi: 10.1097/CMR.0000000000000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, Santiago-Walker AE, Letrero R, D'Andrea K, Pushparajan A, Hayden JE, Brown KD, Laquerre S, McArthur GA, Sosman JA, Nathanson KL, Herlyn M. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Z, Cobb MH. Regulation of stress-responsive mitogen-activated protein (MAP) kinase pathways by TAO2. J Biol Chem. 2001;276(19):16070–5. doi: 10.1074/jbc.M100681200. [DOI] [PubMed] [Google Scholar]
- 50.Vin H, Ojeda SS, Ching G, Leung ML, Chitsazzadeh V, Dwyer DW, Adelmann CH, Restrepo M, Richards KN, Stewart LR, Du L, Ferguson SB, Chakravarti D, Ehrenreiter K, Baccarini M, Ruggieri R, Curry JL, Kim KB, Ciurea AM, Duvic M, Prieto VG, Ullrich SE, Dalby KN, Flores ER, Tsai KY. BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. Elife. 2013;2:e00969. doi: 10.7554/eLife.00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H, Viros A, Martin M, Galbraith L, Girotti MR, Dhomen N, Gottlieb E, Marais R. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol. 2016;10(1):73–84. doi: 10.1016/j.molonc.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286(49):42626–34. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23(3):302–15. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, Scolyer RA, Rizos H, Lo RS, Cullinane C, Ayer DE, Ribas A, Johnstone RW, Hicks RJ, McArthur GA. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014;4(4):423–33. doi: 10.1158/2159-8290.CD-13-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 56.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gutman M, Singh RK, Radinsky R, Bar-Eli M. Intertumoral heterogeneity of receptor-tyrosine kinases expression in human melanoma cell lines with different metastatic capabilities. Anticancer Res. 1994;14(5A):1759–65. [PubMed] [Google Scholar]
- 58.Kim JE, Leung E, Baguley BC, Finlay GJ. Heterogeneity of expression of epithelial-mesenchymal transition markers in melanocytes and melanoma cell lines. Front Genet. 2013;4:97. doi: 10.3389/fgene.2013.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, Zecchin D, Hobor S, Bajpe PK, Lieftink C, Mateus C, Vagner S, Grernrum W, Hofland I, Schlicker A, Wessels LF, Beijersbergen RL, Bardelli A, Di Nicolantonio F, Eggermont AM, Bernards R. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–22. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 60.Miao B, Ji Z, Tan L, Taylor M, Zhang J, Choi HG, Frederick DT, Kumar R, Wargo JA, Flaherty KT, Gray NS, Tsao H. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015;5(3):274–87. doi: 10.1158/2159-8290.CD-14-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moore TM, Garg R, Johnson C, Coptcoat MJ, Ridley AJ, Morris JD. PSK, a novel STE20-like kinase derived from prostatic carcinoma that activates the c-Jun N-terminal kinase mitogen-activated protein kinase pathway and regulates actin cytoskeletal organization. J Biol Chem. 2000;275(6):4311–22. doi: 10.1074/jbc.275.6.4311. [DOI] [PubMed] [Google Scholar]
- 62.Fey D, Croucher DR, Kolch W, Kholodenko BN. Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front Physiol. 2012;3:355. doi: 10.3389/fphys.2012.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 64.Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, Polsky D, Osman I, Garcia BA, Hernando E, Bernstein E. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468(7327):1105–9. doi: 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan S, Zhao C, Zhang L, Dai S, Ren J, Zhang X, Ban N, He X, Yang L, Bao Z, Chen W, Sun J, Gao Y, Tao T. Knockdown of PFTK1 Inhibits the Migration of Glioma Cells. J Mol Neurosci. 2015;57(2):257–64. doi: 10.1007/s12031-015-0600-z. [DOI] [PubMed] [Google Scholar]
- 66.Gu X, Wang Y, Wang H, Ni Q, Zhang C, Zhu J, Huang W, Xu P, Mao G, Yang S. Upregulated PFTK1 promotes tumor cell proliferation, migration, and invasion in breast cancer. Med Oncol. 2015;32(7):195. doi: 10.1007/s12032-015-0641-8. [DOI] [PubMed] [Google Scholar]
- 67.Yang L, Zhu J, Huang H, Yang Q, Cai J, Wang Q, Zhu J, Shao M, Xiao J, Cao J, Gu X, Zhang S, Wang Y. PFTK1 Promotes Gastric Cancer Progression by Regulating Proliferation, Migration and Invasion. PLoS One. 2015;10(10):e0140451. doi: 10.1371/journal.pone.0140451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W, Liu R, Tang C, Xi Q, Lu S, Chen W, Zhu L, Cheng J, Chen Y, Wang W, Zhong J, Deng Y. PFTK1 regulates cell proliferation, migration and invasion in epithelial ovarian cancer. Int J Biol Macromol. 2016;85:405–16. doi: 10.1016/j.ijbiomac.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 69.Zheng L, Zhou Z, He Z. Knockdown of PFTK1 inhibits tumor cell proliferation, invasion and epithelial-to-mesenchymal transition in pancreatic cancer. Int J Clin Exp Pathol. 2015;8(11):14005–12. [PMC free article] [PubMed] [Google Scholar]
- 70.Doles J, Hemann MT. Nek4 status differentially alters sensitivity to distinct microtubule poisons. Cancer Res. 2010;70(3):1033–41. doi: 10.1158/0008-5472.CAN-09-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nguyen CL, Possemato R, Bauerlein EL, Xie A, Scully R, Hahn WC. Nek4 regulates entry into replicative senescence and the response to DNA damage in human fibroblasts. Mol Cell Biol. 2012;32(19):3963–77. doi: 10.1128/MCB.00436-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2(2):62–9. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 73.Golubovskaya VM, Kweh FA, Cance WG. Focal adhesion kinase and cancer. Histol Histopathol. 2009;24(4):503–10. doi: 10.14670/HH-24.503. [DOI] [PubMed] [Google Scholar]
- 74.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680–8. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ferguson J, Arozarena I, Ehrhardt M, Wellbrock C. Combination of MEK and SRC inhibition suppresses melanoma cell growth and invasion. Oncogene. 2013;32(1):86–96. doi: 10.1038/onc.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, Zambon A, Sinclair J, Hayes A, Gore M, Lorigan P, Springer C, Larkin J, Jorgensen C, Marais R. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3(2):158–67. doi: 10.1158/2159-8290.CD-12-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paraiso KH, Haarberg HE, Wood E, Rebecca VW, Chen YA, Xiang Y, Ribas A, Lo RS, Weber JS, Sondak VK, John JK, Sarnaik AA, Koomen JM, Smalley KS. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res. 2012;18(9):2502–14. doi: 10.1158/1078-0432.CCR-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41(Database issue):D1063–9. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.