

Abstract
Perturbation of the gut-associated microbial community may underlie many human illnesses, but the mechanisms that maintain homeostasis are poorly understood. We found depletion of butyrate-producing microbes by antibiotic treatment reduced epithelial signaling through the intracellular butyrate sensor PPAR-γ. Nitrate levels increased in the colonic lumen because epithelial expression of Nos2, the gene encoding inducible nitric oxide synthase (iNOS) was elevated in the absence of PPAR-γ-signaling. Microbiota-induced PPAR-γ-signaling also limits the luminal bioavailability of oxygen by driving the energy metabolism of colonic epithelial cells (colonocytes) towards β-oxidation. Therefore, microbiota-activated PPAR-γ-signaling is a homeostatic pathway that prevents a dysbiotic expansion of potentially pathogenic Escherichia and Salmonella by reducing the bioavailability of respiratory electron acceptors to Enterobacteriaceae in the lumen of the colon.
A balanced gut microbiota is characterized by the dominance of obligate anaerobic members of the phyla Firmicutes and Bacteroidetes, while an expansion of facultative anaerobic Enterobacteriaceae (phylum Proteobacteria) is a common marker of gut dysbiosis (1) (Fig. S1). Obligate anaerobic bacteria prevent dysbiotic expansion of facultative anaerobic Enterobacteriaceae, in part, by limiting the generation of host-derived nitrate and oxygen (2, 3). It is not known which host-signaling pathways are triggered by the gut microbiota to limit the availability of these respiratory electron acceptors. We found disruption of the gut microbiota by streptomycin treatment increased the bioavailability of host-derived nitrate in the lumen of the large intestine. Increased recovery of a wild-type E. coli strain by comparison with an isogenic derivative deficient for nitrate respiration (napA narG narZ mutant) was observed in mice (C57BL/6 from Jackson) infected with a 1:1 mixture of both strains (Fig. 1A). Supplementing streptomycin-treated mice with the iNOS inhibitor aminoguanidine hydrochloride (AG) abrogated the growth advantage conferred upon E. coli by nitrate respiration (Fig. 1A), supporting the notion that luminal nitrate was host-derived (2, 4).
Figure 1. The PPAR-γ agonist butyrate limits the availability of nitrate by repressing Nos2 expression.
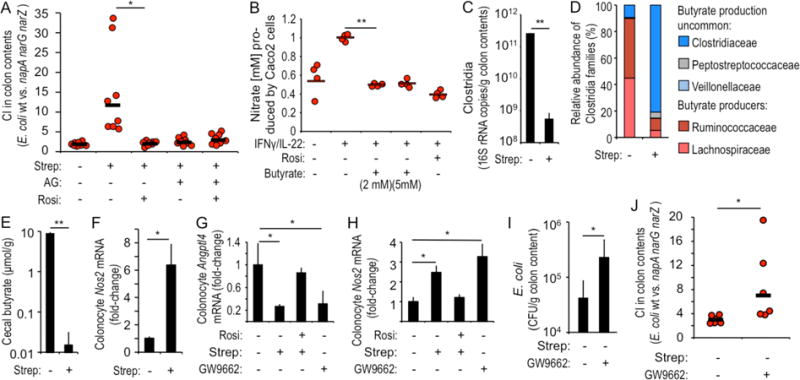
(A) Streptomycin (Strep)-treated mice (N = 8) were inoculated with a 1:1 mixture of E. coli wild type (wt) and napA narG narZ mutant and received rosiglitazone (Rosi) or aminoguanidine (AG) supplementation. The competitive index (CI; the ratio of wt and napA narG narZ mutant recovered from colon contents) was determined 3 days after inoculation. (B) Polarized Caco-2 cells (N = 4) grown in tissue culture medium received IFNγ/IL-22, Rosi or AG treatment. Nitrate produced in the apical compartment was determined by a modified Griess assay. (C–F) Mice (N = 8) were mock-treated (inoculation with vehicle control) or treated with Strep and organs collected 3 days later. (C) The abundance of Clostridia in colon contents was determined by quantitative real-time PCR using class-specific primers for Clostridia 16S rRNA genes. (D) Relative abundance of families belonging to the class Clostridia determined by 16S profiling of DNA isolated from colon contents. (E) The butyrate concentration was determined in cecal contents using gas chromatography and (F) transcript level of Nos2 in colonocyte preparations was determined by real-time PCR. (G and H) Colonocytes were isolated 3 days after treatment with streptomycin from mice (N = 8) receiving the indicated supplementation and transcript levels of Angptl4 (G) and Nos2 (H) determined by quantitative real-time PCR. (I and J) Mice (N = 6) were mock-treated or received the PPAR-γ antagonist GW9662 and were inoculated with E. coli indicator strains. Numbers of E. coli (I) and the CI of indicator strains (J) were determined 3 days after inoculation. (C and E–I) Bars represent geometric means ± standard error. (A, B and J) Dots represent measurements from individual animals (A and J) or wells (B) and bars represent geometric means. *, P < 0.05; **, P < 0.01.
To model nitrate production by the colonic epithelium, we induced NOS2 expression in human colonic epithelial cancer (Caco2) cells by stimulation with gamma interferon (IFNγ) and interleukin (IL)-22 (model epithelia). We exposed the model epithelia to butyrate, a fermentation product of the gut microbiota that serves as the main carbon source of colonic epithelial cells (colonocytes) (5). Butyrate significantly reduced NOS2 expression (P < 0.05) (Fig. S2A), lowered iNOS synthesis (P < 0.05)(6) (Fig. S2B) and diminished epithelial generation of nitrate (P < 0.05) (Fig. 1B), a product of nitric oxide decomposition in the intestinal lumen (4). The host can sense butyrate using the nuclear receptor PPAR-γ, which is synthesized at high levels in colonocytes (7) and does not respond to other short-chain fatty acids, such as acetate or propionate (8). To determine whether PPAR-γ repressed iNOS synthesis, we stimulated model epithelia with the PPAR-γ agonist rosiglitazone. Rosiglitazone-treatment significantly blunted NOS2 expression (P < 0.05) (Fig. S2A), reduced iNOS synthesis (P < 0.05) (Fig. S2B), lowered nitrate production (P < 0.01) (Fig. 1B) and induced synthesis and nuclear localization of PPAR-γ in model epithelia (Fig. S2C). Based on these data we hypothesized that microbiota-derived butyrate suppresses iNOS synthesis in the gut by stimulating PPAR-γ-signaling in colonocytes (Fig. S1).
Streptomycin-mediated depletion of a PPAR-γ agonist drives growth by nitrate respiration
To test our hypothesis, we used mice to investigate whether streptomycin treatment would deplete butyrate-producing bacteria, thereby increasing Nos2 expression in colonocytes. Streptomycin treatment reduced bacterial numbers in colon contents (Fig. S3A) and significantly (P < 0.01) reduced the abundance of Clostridia (phylum Firmicutes) (Fig. 1C and S3B), which are obligate anaerobes that include abundant butyrate-producers (9) (Fig. S3C), specifically Lachnospiraceae and Ruminococcaceae (Fig. 1D and S3D). The changes in the microbiota composition correlated with a significant (P < 0.01) drop in the cecal butyrate concentration (Fig. 1E) and significantly (P < 0.05) elevated Nos2 expression in murine colonocyte preparations (Fig. 1F).
We next investigated the role of PPAR-γ in altering epithelial gene expression. Streptomycin treatment reduced epithelial expression of Angptl4, a gene positively regulated by PPAR-γ (8), and expression was restored in streptomycin-treated mice that received the PPAR-γ agonist rosiglitazone (Fig. 1G). Treatment of mice with the PPAR-γ antagonist 2-chloro-5-nitrobenzanilide (GW9662) mimicked the reduction (P < 0.05) in Angptl4 transcript levels observed after streptomycin treatment (Fig. 1G). The effects of each treatment on epithelial Nos2 expression (Fig. 1H) were opposite to those observed for expression of Angptl4 (Fig. 1G), which supported the idea that PPAR-γ negatively regulates Nos2 (Fig. S1).
Next, we used E. coli indicator strains to investigate whether silencing PPAR-γ signaling would increase the bioavailability of nitrate in the colon. To this end, mice were inoculated with a 1:1 mixture of a nitrate respiration-proficient indicator strain (E. coli wild type) and an isogenic nitrate respiration-deficient indicator strain (napA narG narZ mutant). Treatment with the PPAR-γ agonist rosiglitazone abrogated the fitness advantage conferred to wild-type E. coli by nitrate respiration in streptomycin-treated mice (Fig. 1A). To investigate whether inhibition of PPAR-γ signaling would support a nitrate respiration-dependent expansion of E. coli without antibiotic treatment, mice were mock-treated (inoculation with sterile PBS) or treated with the PPAR-γ antagonist GW9662 and then infected with E. coli indicator strains. Treatment with GW9662 significantly increased the overall number of E. coli recovered from the colon of mice (P < 0.05) (Fig. 1I) by driving a nitrate respiration-dependent E. coli expansion, as shown by increased recovery of the wild type over a nitrate respiration-deficient mutant (P < 0.05) (Fig. 1J). Next, we wanted to determine whether treatment with a PPAR-γ antagonist would increase the abundance of endogenous Enterobacteriaceae. While endogenous Enterobacteriaceae were not detected in C57BL/6 mice from Jackson, C57BL/6 mice from Charles River carried endogenous E. coli strains producing nitrate reductase activity (Fig. S4A). Treatment of Charles River mice with GW9662 significantly (P < 0.05) increased the abundance of endogenous E. coli, which could be abrogated by supplementation with the iNOS inhibitor AG (Fig. S4B).
Epithelial PPAR-γ-signaling limits luminal nitrate availability
To exclude the possibility that our results were due to off-target effects of chemical agonist or antagonists, we generated mice lacking PPAR-γ in the intestinal epithelium (Ppargfl/flVillincre/− mice) along wild-type littermate control animals (Ppargfl/flVillin−/− mice). Mice lacking epithelial PPAR-γ signaling exhibited significantly elevated transcript levels of Nos2 in the colonic epithelium (P < 0.01) (Fig. 2A), which resulted neither from a reduced abundance of butyrate-producing bacteria in their gut microbiota (Fig. 2B and Fig. S5) nor from lower butyrate levels in their cecal contents (Fig. 2C). Inoculation with E. coli indicator strains revealed that epithelial PPAR-γ-deficiency increased the bioavailability of nitrate through a mechanism that required iNOS activity, because treatment with the iNOS inhibitor AG abrogated the nitrate respiration-dependent growth advantage (P < 0.05) (Fig. 2D). Similar results were obtained when mice were infected with the murine E. coli isolate JB2 (Fig. S4C), which produced nitrate reductase activity (Fig. S4A). To test directly whether genetic ablation of epithelial PPAR-γ-signaling increased the concentration of nitrate in the intestinal lumen, we measured the concentration of this electron acceptor in colonic mucus scrapings, which revealed a significant increase (P < 0.01) in mice lacking epithelial PPAR-γ signaling compared to littermate controls (Fig. 2E).
Figure 2. Microbiota-induced epithelial PPAR-γ-signaling limits nitrate availability in the colon.
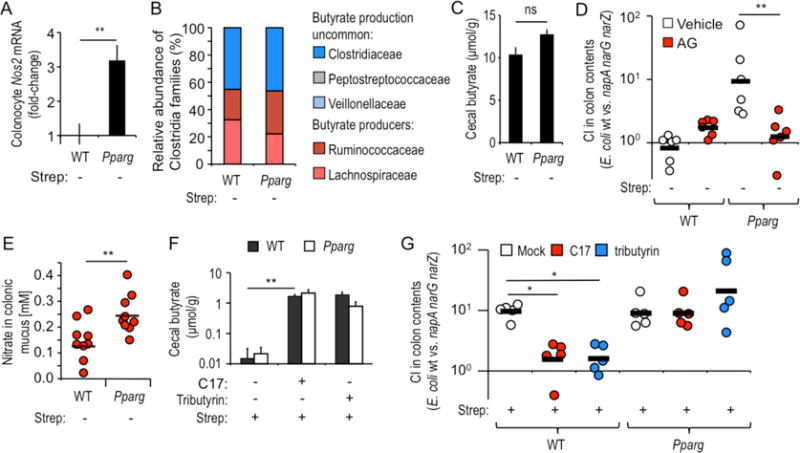
(A) Nos2 expression in the colonic epithelium of mice (N = 6) was determined by real-time PCR in Ppargfl/flVillincre/− mice (Pparg), which lack PPAR-γ in epithelial cells, and in littermate control Ppargfl/flVillin−/− mice (WT). (B) Relative abundance of families belonging to the class Clostridia in colon contents of mice (N = 6) was determined by 16S profiling. (C) The butyrate concentration was determined in cecal contents of mice (N = 6) using gas chromatography. (D) Mice (N = 6) were inoculated with a 1:1 mixture of E. coli wild type (wt) and napA narG narZ mutant and received aminoguanidine (AG) supplementation or vehicle control. The competitive index (CI; the ratio of wt and napA narG narZ mutant recovered from colon contents) was determined 3 days after inoculation. (E) The concentration of nitrate in the colonic mucus layer was determined in groups of animals (N = 9) by a modified Griess assay. (F and G) Streptomycin-treated mice (N = 6) were inoculated with E. coli indicator strains and received supplementation with tributyrin or a community of 17 human Clostridia isolates (C17). The butyrate concentration in cecal contents (F) and the CI in colon contents (G) were determined 3 days after inoculation. (A, C and F) Bars represent geometric means ± standard error. (D, E and G) Each dot represents data from an individual animal and black bars represent geometric means. *, P < 0.05; **, P < 0.01; ns, not statistically significantly different.
Mice lacking epithelial PPAR-γ-signaling were treated with streptomycin, infected the next day with E. coli indicator strains and inoculated one day later with a community of 17 human Clostridia isolates (10). Inoculation with the Clostridia isolates restored cecal butyrate concentrations (P < 0.01) (Fig. 2F) and suppressed nitrate respiration-dependent growth of E. coli in streptomycin-treated littermate control mice. However, nitrate respiration-dependent growth of E. coli was not suppressed in streptomycin-treated mice lacking epithelial PPAR-γ-signaling (P < 0.05) (Fig. 2G). To directly test whether butyrate was responsible for inhibiting nitrate respiration of E. coli in littermate control animals, mice were treated with streptomycin, infected the next day with E. coli indicator strains and inoculated one day later with 1,2,3-tributyrylglycerol (tributyrin). Tributyrin, a natural ingredient of butter, exhibits delayed absorption in the small intestine compared to butyrate and its degradation in the large intestine increases luminal butyrate concentrations (11). Tributyrin supplementation restored cecal butyrate concentrations (P < 0.01) (Fig. 2F), which abrogated nitrate respiration-dependent growth of E. coli in streptomycin-treated littermate control mice, but not in streptomycin-treated mice lacking epithelial PPAR-γ-signaling (P < 0.05) (Fig. 2G). Collectively, these data support the idea that microbiota-derived butyrate maintains gut homeostasis by inducing epithelial PPAR-γ-signaling, which in turn limits nitrate respiration-dependent dysbiotic E. coli expansion (Fig. S1).
Lack of epithelial PPAR-γ-signaling increases colonocyte oxygenation during colitis
Colonocytes obtain energy through β-oxidation of microbiota-derived butyrate, which consumes a considerable amount of oxygen, thereby rendering the epithelium hypoxic (12). However, after a streptomycin-mediated depletion of butyrate (Fig. 1E), colonocytes switch their energy metabolism to converting glucose into lactate (anaerobic glycolysis) (11). Consistent with this metabolic reprogramming, streptomycin treatment increased the concentration of lactate (Fig. 3A) and reduced ATP levels (Fig. 3B) in primary murine colonocyte preparations. Anaerobic glycolysis does not consume oxygen, which then permeates through the epithelium into the gut lumen (3, 11). This scenario was supported by increased recovery of aerobic respiration-proficient (Nissle 1917 wild type) E. coli over an E. coli strain that is impaired for aerobic respiration under microaerophilic conditions (cydAB mutant) from the colon of streptomycin-treated mice (Fig. 3C). Similar results were obtained when the result was repeated with a different E. coli strain (MG1655) (Fig. S6A). Increasing the concentration of the PPAR-γ antagonist butyrate in streptomycin-treated mice either by inoculation with a community of 17 human Clostridia isolates or by supplementation with tributyrin (Fig. S6B) appeared to reduce the bioavailability of oxygen, as E. coli indicator strains that were proficient (wild type) or deficient (cydAB mutant) for aerobic respiration under microaerophilic conditions were recovered equally in the gut (Fig. 3C). Furthermore, the aerobic growth benefit observed in streptomycin-treated mice inoculated with E. coli indicator strains was abrogated by treatment with the PPAR-γ agonist rosiglitazone (Fig. 3C). Surprisingly, E. coli indicator strains were recovered at the same ratio from mice lacking epithelial PPAR-γ signaling and from their littermate controls (Fig. 3D), suggesting that reducing PPAR-γ signaling alone was not sufficient for increasing the bioavailability of oxygen.
Figure 3. Lack of epithelial PPAR-γ-signaling increases colonocyte oxygenation during colitis.
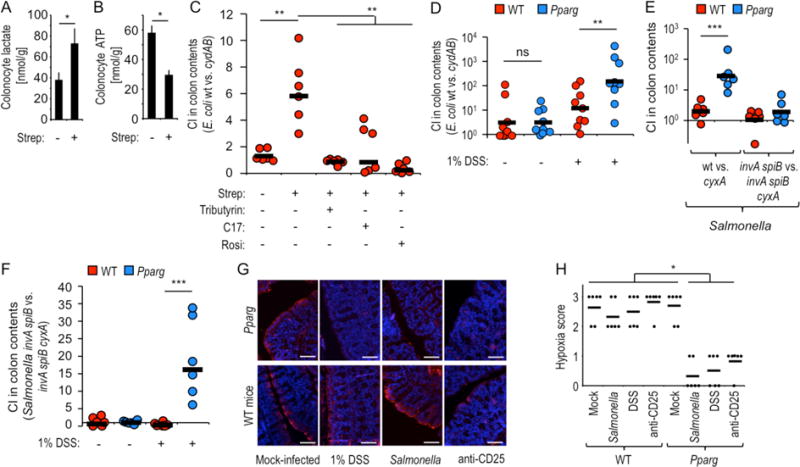
(A and B) Groups of mice (N = 5) were mock treated or treated with streptomycin (Strep) and colonocytes isolated one day later to measure intracellular concentrations of lactate (A) or ATP (B). (C) Groups (N = 6) of streptomycin-treated or mock-treated mice were inoculated with a 1:1 mixture of E. coli wild type (wt) and cydAB mutant and received supplementation with rosiglitazone (Rosi), tributyrin or a community of 17 human Clostridia isolates (C17). (D) Groups of mice (N = 6) receiving no supplementation or water supplemented with 1% dextran sulfate sodium (DSS) were inoculated with a 1:1 mixture of E. coli wild type (wt) and cydAB mutant. (C and D) The competitive index (CI) was determined 3 days after inoculation. (E and F) Groups of mice (N = 6) were inoculated with the indicated Salmonella strain mixtures. (F) Mice received no supplementation or water supplemented with 1% DSS. (G and H) Mice (N = 6) were treated as indicated and were injected intraperitoneally with pimonidazole one hour before euthanasia. Binding of pimonidazole was detected using hypoxyprobe-1 primary antibody and a Cy-3 conjugated goat anti-mouse secondary antibody (red fluorescence) in sections of the colon that were counter stained with DAPI nuclear stain (blue fluorescence). (G) Representative images are shown. Scale bars represent 50 μm. (H) A veterinary pathologist scored blinded sections for hypoxia staining. Each dot represents data from one animal. (A-B) Bars represent geometric means ± standard error. (C–F) Each dot represents data from an individual animal and black bars represent geometric means. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically significantly different.
PPAR-γ-signaling activates mitochondrial β-oxidation in alternatively activated (M2) macrophages (13). Since IFNγ-signaling drives the energy metabolism of macrophages towards anaerobic glycolysis (13), we hypothesized that in addition to silencing PPAR-γ-signaling, metabolic reprogramming of colonocytes might also require an inflammatory signal (Fig. S1). To test this idea, we turned to S. enterica serovar Typhimurium (S. Typhimurium), a pathogen that employs two type III secretion systems to trigger intestinal inflammation and uses the cyxAB genes, encoding cytochrome bd-II oxidase, for its subsequent aerobic expansion in the intestinal lumen (3). When mice were infected with S. Typhimurium strains that were proficient (wild type) or deficient (cyxA mutant) for aerobic respiration under microaerophilic conditions, a benefit provided by aerobic respiration was observed in mice lacking epithelial PPAR-γ-signaling, but not in littermate control animals (Fig. 3E). To investigate whether inflammatory responses elicited by Salmonella virulence factors were required to increase the bioavailability of oxygen, we inactivated the two type III secretion systems essential for Salmonella enteropathogenicity through mutations in invA and spiB (3). Consistent with our hypothesis, aerobic respiration no longer provided a benefit to the pathogen in mice lacking epithelial PPAR-γ-signaling when mice were infected with avirulent S. Typhimurium strains that were either proficient (invA spiB mutant) or deficient (invA spiB cyxA mutant) for aerobic respiration under microaerophilic conditions (Fig. 3E).
To test whether, in addition to genetic ablation of PPAR-γ-signaling, an inflammatory signal was needed to increase luminal oxygen bioavailability, mice received low-dose (1% in drinking water) dextran sodium sulfate (DSS)-treatment, which elicited inflammatory changes as indicated by a reduction in colon length (Fig. S6C). Inoculation with E. coli indicator strains revealed that aerobic respiration provided a larger growth benefit in DSS-treated mice lacking epithelial PPAR-γ-signaling compared to their DSS-treated littermate controls (Fig. 3D). Similarly, inoculation with avirulent S.. Typhimurium strains that were either proficient (invA spiB mutant) or deficient (invA spiB cyxA mutant) for aerobic respiration under microaerophilic conditions provided evidence for increased oxygen bioavailability only in DSS-treated mice that lacked epithelial PPAR-γ-signaling (Fig. 3F).
Next, we investigated whether either DSS treatment or infection with wild-type S. Typhimurium would increase epithelial oxygenation in mice lacking epithelial PPAR-γ-signaling. To this end, we visualized the hypoxia of surface colonocytes using the exogenous hypoxic marker pimonidazole, which is reduced under hypoxic conditions to hydroxylamine intermediates that irreversibly bind to nucleophilic groups in proteins or DNA (14, 15). Genetic ablation of PPAR-γ-signaling was not sufficient to reduce epithelial hypoxia. However, DSS-treatment or infection with wild-type S. Typhimurium increased epithelial oxygenation in mice lacking epithelial PPAR-γ-signaling, while hypoxia staining remained unchanged in littermate control animals (Fig. 3G and 3H).
PPAR-γ-signaling and Tregs cooperate to maintain colonocyte hypoxia
While streptomycin treatment reduced PPAR-γ-signaling (Fig. 1G) by depleting microbiota-derived butyrate (Fig. 1E and S6B), the findings shown above suggested that reducing PPAR-γ-signaling was necessary, but not sufficient for increasing oxygen bioavailability in the colon, because DSS-induced inflammation or Salmonella virulence factors were required for increasing colonocyte oxygenation (Fig. 3D–3H). Although streptomycin treatment does not lead to overt inflammation, disruption of the gut microbiota reduced concentrations of other microbiota-derived short-chain fatty acids (Fig. S6D) besides butyrate (Fig. S6B), that signal through G-protein-coupled receptor (GPR)43, GPR109A and histone deacetylases expressed by T cells, dendritic cells and macrophages, respectively, to reduce intestinal inflammation (16–19). Engagement of these host cell receptors by short-chain fatty acids induces maturation and expansion of regulatory T-cells (Tregs) in the colon, a cell type that limits pro-inflammatory responses (20–24). Consistent with previous reports showing that an antibiotic-mediated depletion of short-chain fatty acids leads to a contraction of the Treg population in the colonic mucosa (20, 23), we observed that streptomycin treatment shrank the pool of colonic Tregs (CD3+-enriched CD4+ FOXP3+ cells) to one third of its normal size (Fig. 4A, S6E and S7). Thus, during antibiotic treatment, the second input that increases oxygen bioavailability in the colon might be provided by contraction of the colonic Treg population, which increases the inflammatory tone of the mucosa (Fig. S1).
Figure 4. Microbiota-induced PPAR-γ-signaling and Tregs cooperate to limit the bioavailability of oxygen in the colon.
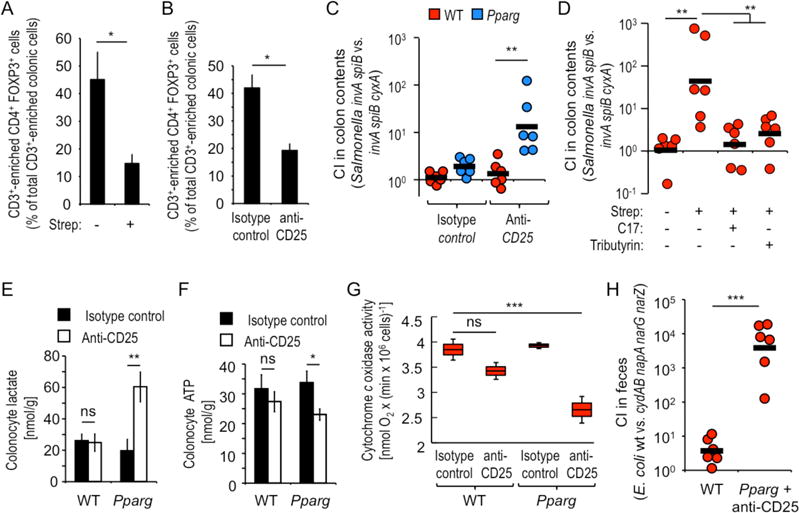
(A and B) Groups of mice (N = 4) were treated with streptomycin (A) or with anti-CD25 antibody (B) and CD3+-enriched live colonic cells analyzed for expression of CD4 and FOXP3 by flow cytometry. (C) Groups of mice (N = 6) were treated with anti-CD25 antibody or isotype control and 10 days later inoculated with a 1:1 mixture of an avirulent Salmonella strain (invA spiB mutant) and an avirulent Salmonella strain lacking cytochrome bdII oxidase (invA spiB cyxA mutant). (D) Groups (N = 6) of streptomycin-treated or mock-treated mice were inoculated with Salmonella indicator strains and received supplementation with tributyrin or a community of 17 human Clostridia isolates (C17). (C and D). The CI was determined 4 days after inoculation. (E–G) Groups of mice (N = 6) were treated with anti-CD25 antibody or isotype control antibody and colonocytes isolated to measure intracellular concentrations of lactate (E), ATP (F) or mitochondrial cytochrome c oxidase activity. (G) Boxes in Whisker plots represent the standard error, while lines indicate the standard deviation. (A–B and E–F) Bars represent geometric means ± standard error. (C, D and H) Each dot represents data from an individual animal and black bars represent geometric means. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically significantly different.
Treatment with anti-CD25 antibody reduced the pool of colonic Tregs (Fig. 4B and S6F) by a magnitude similar to that observed after streptomycin treatment (Fig. 4A) and elicited inflammatory changes in mice lacking epithelial PPAR-γ-signaling, as indicated by a reduction in colon length (Fig. S6G). When anti-CD25-treated mice were infected with avirulent S. Typhimurium strains that were either proficient (invA spiB mutant) or deficient (invA spiB cyxA mutant) for aerobic respiration under microaerophilic conditions, there was no benefit provided by aerobic respiration to S. Typhimurium in wild-type mice. Hence, depletion of Tregs was not sufficient for increasing oxygen bioavailability. In contrast, depletion of Tregs increased oxygen bioavailability in mice lacking epithelial PPAR-γ-signaling, but not in wild-type littermate control mice (Fig. 4C and S6H). Genetic ablation of PPAR-γ-signaling combined with Treg-depletion phenocopied the effects of streptomycin treatment on the recovery of avirulent S. Typhimurium strains proficient or deficient for aerobic respiration under microaerophilic conditions (Fig. 4D). Depletion of Tregs increased epithelial oxygenation in mice lacking epithelial PPAR-γ-signaling, but not in littermate control mice (Fig. 3G and 3H). Consistent with metabolic reprogramming towards anaerobic glycolysis, Treg-depletion increased intracellular lactate levels and lowered ATP concentrations in colonocyte preparations from mice lacking epithelial PPAR-γ-signaling, but not from littermate controls (Fig. 4E and 4F). Measurement of mitochondrial cytochrome c oxidase activity revealed that Treg-depletion caused a significant (P < 0.01) reduction in oxygen consumption in colonocyte preparations of mice lacking epithelial PPAR-γ-signaling, but not in littermate control animals (Fig. 4G).
To further study how colonic Tregs and PPAR-γ-signaling cooperate to limit respiratory growth of facultative anaerobic bacteria, mice lacking epithelial PPAR-γ-signaling were treated with anti-CD25 antibody and infected with a 1:1 mixture of wild-type E. coli and a mutant lacking cytochrome bd oxidase and nitrate reductases (cydAB napA narG narZ mutant). The competitive index was approximately 1,000-fold greater (P < 0.01) in anti-CD25-treated mice lacking epithelial PPAR-γ compared to wild-type littermate control animals (Fig. 4H). Similar results were obtained when mice were infected with individual bacterial strains (Fig. S6I).
The emerging picture is that epithelial hypoxia maintains anaerobiosis in the colon to drive the microbial community towards a dominance of obligate anaerobes, which produce short-chain fatty acids. In turn, short-chain fatty acids sustain Tregs and epithelial PPAR-γ-signaling, which cooperatively drives the energy metabolism of colonocytes towards β-oxidation of microbiota-derived butyrate to preserve epithelial hypoxia, thereby closing a virtuous cycle maintaining homeostasis of a healthy gut. PPAR-γ-signaling also activates expression of beta-defensins, which might contribute to shaping the intestinal environment (25). An antibiotic-induced lack of epithelial PPAR-γ-signaling and a contraction of the Treg pool cooperatively drive a metabolic reorientation of colonocytes towards anaerobic glycolysis, thereby increasing epithelial oxygenation and consequently elevating oxygen bioavailability to promote an expansion of Enterobacteriaceae (Fig. S1), a common marker of dysbiosis (1). Thus, an expansion of Enterobacteriaceae in the gut-associated microbial community is a microbial signature of epithelial dysfunction, which has important ramifications for targeting PPAR-γ-signaling as a potential intervention strategy.
MATERIALS AND METHODS
Bacterial culture conditions
The 17 human Clostridia isolates were kindly provided by K. Honda (20, 26) and were cultured individually as described previously (10). E. coli strains (wild-type E. coli Nissle 1917, E. coli Nissle 1917 napA narZ narG mutant (2), E. coli Nissle 1917 cydAB mutant, E. coli Nissle 1917 cydAB napA narZ narG mutant, E. coli JB2 (27), E. coli MG1655, E. coli MG1655 cydA mutant) and S. Typhmurium strains (wild-type ATCC14028 resistant to nalidixic acid [IR715], IR715 invA spiB mutant, IR715 cyxA mutant or IR715 invA spiB cyxA mutant) (3) were routinely grown aerobically at 37°C in LB broth (BD Biosciences) or on LB plates. When necessary, antibiotics were added to the media at the following concentrations: 0.1 mg/ml carbenicillin, and 0.05 mg/ml kanamycin. Single colonies of endogenous coliforms isolated on McConkey agar from the feces of Charles River mice were subjected to species identification using EnteroPluri Test (Liofilchem, Italy) per the manufacturer`s recommendations.
Construction of E. coli mutants
To construct an E. coli Nissle 1917 cydA mutant, upstream and downstream regions of approximately 0.5kb in length flanking the cydA gene were amplified by PCR and purified using the MiniElute kit (Qiagen). The pRDH10 suicide vector was digested with SalI, run on an agarose gel, purified using the MiniElute kit (Qiagen) and assembled with the fragments using the Gibson Assembly Master Mix (NEB) to form plasmid pYL9. Plasmid pYL9 was then transformed into E. coli S17-1 λpir and conjugation performed with E. coli Nissle 1917 carrying the temperature-sensitive plasmid pSW172 for counter selection (4). Conjugation was performed at 30°C and exconjugants in which the suicide plasmid had integrated into the chromosome were recovered on LB plates containing carbenicilin and chloramphenicol. Subsequent sucrose selection was performed on sucrose plates (5 % sucrose, 8 g/l nutrient broth base, 15 g/l agar) to select for a second crossover events. PCR was performed to detect events that lead to the unmarked deletion of the cydA gene. Plasmid pSW172 was cured by cultivating the bacteria at 37°C. Plasmid pCAL61 (2) was transformed into the E. coli Nissle 1917 cydA mutant strain to introduce a selective marker.
To generate an E. coli Nissle 1917 cydA napA narG narZ mutant, E. coil Nissle 1917 cydA (pSW172) served as recipient for sequential conjugations and sucrose selections using conjugational donor strains carrying plasmids pSW224, pSW225 and pSW237 (4), respectively. Finally, plasmid pSW172 was cured and plasmid pCAL61 introduced by transformation into the E. coil Nissle 1917 cydA napA narG narZ mutant strain to introduce a selective marker.
To generate a cydA mutant of E. coli strain MG1655, an internal fragment of the cydA gene was amplified by PCR using the primers MGcydA_F: GAGGTACCGCATGCGATATCGAGCTATGAATCCGGCTACGAAATG and MGcydA_R: TAGCGATCGAATTCCCGGGAGAGCTCCTGGTCGGTAGAACCAGAA. The cydA PCR product was then cloned into suicide vector pGP704 using Gibson Assembly to create pFRC12. Plasmid pFRC12 was propagated in E. coli DH5α λpir and introduced into a streptomycin resistant E. coli MG1655 strain by conjugation using E. coli S17-1 λpir as a donor strain. Exconjugants were selected on LB agar plates containing streptomycin and carbenicillin to select for integration of the suicide plasmid. Chromosomal integration of the plasmid into the cydA gene was verified by PCR and the resulting strain was designated FRC91.
Animal Experiments
The Institutional Animal Care and Use Committee at the University of California at Davis approved all experiments in this study. Female and male C57BL/6J wild-type mice, aged 8–10 weeks, were obtained from The Jackson Laboratory. Female and male C57BL/6 Ppargfl/flVillincre/− and littermate Ppargfl/flVillin−/− (control) mice were generated at UC Davis by mating Ppargfl/fl mice with Villincre/− mice (The Jackson Laboratory). C57BL/6, Ppargfl/flVillincre/− and Ppargfl/flVillin−/− mice were treated with 20 mg/animal streptomycin via oral gavage or mock treated and orally inoculated 24 hours later with 1 × 109 CFU of 1:1 mixture of the indicated E. coli or S. Typhimurium strains. For tributyrin supplementation, mice were mock treated or received tributyrin (5 g/kg) by oral gavage at 1 and 2 days after infection. In some experiments, animals were inoculated with a mixture of 17 human Clostridia strains cultures individually in an anaerobic chamber by oral gavage at 1 day after infection. When necessary, mice were treated intragastrically at 0, 1, and 2 days after streptomycin with 8 mg/kg/day of the PPARγ agonist Rosiglitazone (Cayman) diluted in 0.1 ml of sterile PBS solution. For the treatment with PPARγ antagonist GW9662 (Cayman, Ann Arbor, MI), mice were treated intragastrically for 3 days with 5 mg/kg/day of PPARγ antagonist GW9662 (Cayman, Ann Arbor, MI) diluted in 0.2 ml of sterile PBS solution.
DSS treatment: Mice were given 1% DSS salt (MP Biomedicals) in their drinking water continuously for 8 days as described previously (2). DSS was replaced with fresh DSS solution every 2 to 3 days during this time. At day 4 of DSS treatment, mice were inoculated with 1 × 109 CFU of a 1:1 mixture of E. coli Nissle 1917 and E. coli Nissle 1917 cydA mutant, or 1:1 mixture of a S. Typhimurium invA spiB mutant and a S. Typhimurium invA spiB cyxA mutant. Samples were collected at 4 days after infection.
For Treg depletion, mice were intraperitoneally injected with 250 μg/mouse of PC61 mAb or isotype control, as previously described (28). At day 10 after treatment, mice were inoculated with 1 × 109 CFU of a 1:1 mixture of the indicated E. coli or S. Typhimurium strains. Samples were collected at 4 days after infection.
For microbiota analysis, mice were euthanized at 3 days after streptomycin treatment, and DNA from the colon contents was extracted using the PowerSoil DNA Isolation kit (Mo-Bio) according to the manufacturer’s protocol. Generation and analysis of sequencing data is described below.
Colonocyte isolation
The proximal colon was flushed with ice-cold phosphate buffered saline (PBS) (Gibco) using a 10 mL syringe fitted with an 18-gauge blunt needle before opening lengthwise, cutting into 2–4 cm pieces and placing in a 15 mL conical centrifuge tube filled with 10 mL ice-cold PBS (Gibco) on ice. Tubes were gently inverted four times, PBS (Gibco) wash was removed and replaced with additional ice-cold PBS (Gibco) before transfer to sterile petri dishes where colon tissue was cut into <5 mm pieces using a razor blade. Tissue pieces were placed in crypt chelating buffer (2 mM EDTA, pH 8 in PBS) and incubated for 30 minutes with gentle rocking buried in ice. Chelating buffer was removed and replaced with cold dissociation buffer (54.9 mM D-sorbitol, 43.4 mM sucrose in PBS) before shaking vigorously by hand for 8 minutes. Cell suspension was transferred to a new 15 mL conical centrifuge tube, leaving any large remnant colon tissue, and cells were pelleted by centrifugation at 4000 rpm for 10 minutes at 4°C. Supernatant was removed and the cell pellet was resuspended in 1 mL Tri Reagent (Molecular Research Center) for subsequent RNA and protein extraction. Cytochrome c oxidase activity was evaluated as described before in detail (29).
Caco-2 cell culture
Caco-2 cells were cultured on 1× Minimal Essential Media (Gibco), with 10% Fetal Bovine Serum (Gibco) and 1× Glutamax (Gibco) at at 37°C and 5% CO2. The cells were seeded at 1 × 105 cells per six-well plates overnight prior to experiments. When necessary, cells were pre-treated with rhIL-22 (100ng/mL, R&D) and rhIFNγ (40 ng/mL, R&D) for 30 minutes and cytokines (rhIL-22 and rhIFNγ), 2mM sodium butyrate (Sigma-Aldrich), 5mM sodium butyrate (Sigma-Aldrich) or 5μM Rosiglitazone (Cayman) were added to the wells and kept throughout the experiments. At 8 or 20 hours after treatments, cells were collected for gene expression or protein analysis by Western Blot, as described below. For nitrate measurement experiments, a trans well plate model of polarized intestinal cells was used as previously described (30). Briefly, Caco-2 cells were maintained on minimum essential medium (MEM) containing 10% fetal bovine serum (FBS). To polarize Caco-2 cells, 0.5 ml of medium containing approximately 1 × 105 cells was seeded apically in 0.4-μm 12-mm Transwell plates (polycarbonate membrane, Corning-Costar), and 1.0 ml of medium was added to the basolateral compartment. The medium was changed every 2 days, and the transepithelial electrical resistance was measured after a week and the day before the experiment using the Millicell-ERS electrical resistance system. When necessary, cells were pre-treated with rhIL-22 (100ng/mL, R&D) and rhIFNγ (40 ng/mL, R&D) for 30 minutes and cytokines (rhIL-22 and rhIFNg), 2mM sodium butyrate (Sigma-Aldrich), 5mM sodium butyrate (Sigma-Aldrich) or 5μM Rosiglitazone (Cayman) were added to the wells and kept throughout the experiments. At 24 hours after treatments, supernatants from each well were collected and immediately used for nitrate measurements as described below. All experiments were repeated at least four times independently.
Real time PCR
RNA from caco-2 cells or colonocytes was isolated with Tri-reagent (Molecular Research Center) according to the instructions of the manufacturer. A reverse transcriptase reaction was performed to prepare complementary DNA (cDNA) using TaqMan reverse transcription reagents (Applied Biosystems). A volume of 4 μl of cDNA was used as template for each real-time PCR reaction in a total reaction volume of 25 μL. Real-time PCR was performed using SYBR Green (Applied Biosystems). Data were analyzed using the comparative Ct method (Applied Biosystems). Transcript levels of Nos2 and were normalized to mRNA levels of the housekeeping gene Act2b, encoding β-actin.
Short-chain fatty acid measurements
Cecum content samples were weighed, diluted with nanopure water to 1 mg/10 μL, and homogenized in the shaker overnight. The samples were then centrifuged at 21130×g for 20 minutes, and 20μL of supernatant was mixed with 20 μL 0.2 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride in 5% pyridine/water (v/v) and 40 μL 0.1 M 2-nitrophenylhydrazine in 80% ACN/water (v/v) with 0.05 M HCl. The mixture was allowed to react at 40 °C for 30 min and cool to room temperature. Then 400 μL of 10% ACN/water (v/v) was added. The solution was centrifuged at 4000×g for 30 minutes. The supernatant was injected to ultra high performance liquid chromatography triple quadrupole mass spectrometer (UHPLC-QqQ-MS) for short chain fatty acid analysis.
Microbiota analysis
Primers 515F (AATGATACGGCGACCACCGAGATCTACACNNNNNNNNTATGGTAATTGTGTGCCAGCMGC CGCGGTAA) and 806R (CAAGCAGAAGACGGCATACGAGATNNNNNNNNAGTCAGTCAGCCGGACTACHVGGGWTC TAAT) were used to amplify the V4 domain of the bacterial 16S rRNA. Both forward and reverse primers contained a unique 8 nt barcode (N), a primer pad, a linker sequence, and the Illumina adaptor sequences. Each sample was barcoded with a unique forward and reverse barcode combination. PCR contained 1 Unit Kapa2G Robust Hot Start Polymerase (Kapa Biosystems), 1.5 mM MgCl2, 10 pmol of each primer and 1ul of DNA. PCR conditions were: an initial incubation at 95°C for 2 min, followed by 30 cycles of 95°C for 10 s, 55°C for 15 s, 72°C for 15 s and a final extension of 72°C for 3 min. The final product was quantified on the Qubit instrument using the Qubit High Sensitivity DNA kit (Invitrogen) and individual amplicon libraries were pooled, cleaned by Ampure XP beads (Beckman Coulter), and sequenced using a 250 bp paired-end method on an Illumina MiSeq instrument in the Genome Center DNA Technologies Core, University of California, Davis. Qiime open-source software (http://qiime.org) (Caporaso et al., 2010) was used for initial identification of operational taxonomic units (OTU), clustering, and phylogenetic analysis. Principal Coordinate (PC) analysis taxa summaries using weighted UniFrac were created through QIIME. Subsequent data transformation and analysis was performed using R open-source software (www.r-project.org) and the packages Phyloseq and Gggplot (31). Profiling data were deposited at the BioSample database (https://www.ncbi.nlm.nih.gov/biosample/) under accession numbers SAMN07152105 through SAMN07152132.
Within the class Clostridia, butyrate production is most common for the Lachnospiraceae (cluster XIVa) and the Ruminococcaceae (cluster IV) and OTUs belonging to these families were classified as predicted butyrate producers (32). For OTUs assigned to other classes of bacteria, the presence in available bacterial genomes of genes generating butyrate through the acetyl-CoA pathway, the glutarate pathway, the 4-aminobutyrate pathway or the lysine pathway has been described previously (9) and was used to assign each OTUs to a family containing predicted butyrate-producers or a family predicted not to contain butyrate-producers.
Fluorescent microscopy
A single microfluidic chamber (8×3×0.075mm, length × width × height) was used for culturing the Caco2 cells. The microfluidic devices were fabricated using standard soft-lithography protocols by replica molding polydimethylsiloxane (PDMS) (33). Caco2 cell culture inside the microfluidic devices was modified from a previous cell culture protocol for neurons and cancer cells (34). Briefly, the microfluidic platforms were first sterilized under UV for 1h. Collagen solution (0.2mg/ml) was then coated on the glass coverslip for 4 h at 37°C inside a cell incubator. The devices were washed with 1× PBS once and then filled with culture media to provide a hospitable environment before cell seeding. At the same time of washing, the Caco2 cells grown in T25 flask (in MEM media supplemented with 10% FBS and 1% Glutamax) were harvested and re-suspended in fresh media at a density of 5,000 cells per microliter of media. After removing the excess media, a 15~20 μl of the cell suspension was loaded into the upstream reservoir. The microfluidic platforms were then placed in a cell culture incubator with 5% CO2 at 37°C to allow Caco2 cells to attach to the collagen-coated glass coverslips. After 2h, the seeding solution was removed and approximately 200μl of culture media was added to each reservoir, and further cultured overnight. The following day, 400μl treatment solution containing rhIL-22 (100ng/mL, R&D Systems) and rhIFNg (40 ng/mL, R&D Systems) was added for 30 minutes. Incubation of cytokines with treatments, either 2mM or 5mM sodium butyrate (Sigma-Aldrich) or 5μM Rosiglitazone (Cayman Chemical) was added to the upstream reservoir and a 100μl of fresh media was added to the downstream reservoir to create a continuous flow. After 3 days of treatment, the cells in the device were immunostained for PPAR-γ. Immunostaining began by washing the cells in the chambers with 1× PBS and filling the channel with 4% paraformaldehyde with 0.2% Triton X-100 for 15min at room temperature. The surface was blocked by 10% Goat serum with 0.3M Glycine for 1h at room temperature. Subsequently, the primary PPAR-γ antibody (1:100, Santa Cruz Biotechnology) was incubated overnight at 4°C. The secondary antibody (1:1000, BD Pharmigen) was then incubated for 1h at room temperature. Finally, the cells were incubated with Phlloidin for labeling actin filaments. Between each step, the devices were washed by 1× PBS for 10min. The stained cells were imaged using a Nikon Eclipse Ti-S fluorescent microscope (Melville, NY) equipped with the Nikon DAPI-FITC-TRITC triple excitation filter set. An LED lamp (Lumencor, Beaverton, OR) was used for illumination. 16-bit images were captured by the Zyla sCMOS camera (Concord, MA) and processed by the NIS-Elements imaging software.
Western Blot
Total protein was extracted from Caco-2 cells with lysis buffer (50mM Tris-HCl pH 7.6, 150mM NaCl, 0.1% v/v NP-40, 1mM EDTA) mixed with protease inhibitor (Calbiochem). Bradford assay was conducted for protein concentration, and 9 μg were loaded and separated by a 6% SDS-PAGE and transferred onto a Immobilon-P PVDF transfer membrane (Millipore Sigma). Membranes were further probed with antibodies against iNOS (2ug/ml, R&D) or alpha/beta tubulin (1:200, Cell Signaling). Proteins of interest were detected with HRP-conjugated goat anti-mouse IgG antibody (1:1000, Jackson) and donkey anti-rabbit IgG (1:1000, Jackson) and visualized with Supersignal West Pico Chemiluminescent Substrate (Termoscientific) according to manufacturers’ instructions. All experiments were repeated at least four times independently.
Nitrate measurements
Intestinal nitrate measurements were performed as described previously (30). Briefly, uninfected mice were euthanized, and the intestine was removed and divided along its sagittal plane. The mucus layer was gently scraped from the tissue and homogenized in 200 μl PBS and then placed on ice. Samples were centrifuged at 5,000 × g for 10 min at 4°C to remove the remaining solid particles. The supernatant was then filter sterilized (0.2-μm Acrodisc syringe filter, Pall Life Sciences). Measurement of intestinal nitrate followed an adaptation of the Griess assay. In this assay, nitrate was first reduced to nitrite by combining 50 μl of each sample with 50 μl of Griess reagent 1 containing vanadium(III) chloride (0.5 M HCl, 0.2 mM VCl3, 1% sulfanilamide), and then the mixture was incubated at room temperature for 10 min. Next, 50 μl of Griess reagent 2 [0.1% (1-naphthyl)ethylenediamine dichloride] was added to each sample. Absorbance at 540 nm was measured immediately after the addition of Griess reagent 2 to detect any nitrite present in the samples. The samples were then incubated for 8 h at room temperature (to allow for reduction of nitrate to nitrite), and the absorbance at 540 nm was measured again. The initial absorbance (prior to reducing nitrate to nitrite) was subtracted from the absorbance after 8 h to determine nitrate concentrations in the cecal mucus layer. Samples were tested in duplicate, and all measurements were standardized to the initial sample weight. To measure nitrate for in vitro assays, polarized Caco-2 cell medium was changed to MEM containing 10% fetal bovine serum (FBS) medium without phenol, the day before experiment. At 24 hours post treatments, cell supernatant was collected, and nitrate was measured using the Griess assay as described above.
Nitrate reductase activity assay
Triplicate aerobic overnight cultures grown in Luria-Bertani (LB) Broth with or without appropriate antibiotic depending on strain were diluted in fresh LB Broth with 40 mM NaNO3 and incubated for 3 hours at 37°C without shaking in closed tubes. After incubation, cultures were placed on ice, an aliquot was transferred to a microcentrifuge tube and pelleted. Supernatant was removed and the cells were resuspended in Phosphate Buffered Saline (PBS, Gibco) before measuring A600. An aliquot of the washed cells was permeabilized by vortexing in 0.1% SDS and chloroform. Nitrate reductase activity was then assayed by adding equal volumes of 2 mM methyl viologen and bicarbonate/dithionate/nitrate solution (8 mg/mL NaHCO3, 8 mg/mL Na2S2O4, 0.5 M NaNO3). The reduction of nitrate to nitrite was allowed to progress for 5 minutes before vortexing vigorously to terminate the assay. To visualize the amount of nitrite produced, equal volumes of sulfanilic/HCl (1% Sulfanilic Acid, 20% HCl) and Marshall’s Reagent (0.13% N-1-napthylethylenediamine 2 HCl) were added, vortexed and incubated for 10 minutes at room temperature. The A540 and A420 were measured and relative activity units were determined with the following equation: units = {[A540 – (0.72 × A420)]/(t × v × A600)} × 100.
Hypoxia staining
For detection of hypoxia, mice were treated with 60mg/kg of pimonidazole HCl i.p. (Hypoxyprobe™-1 kit, Hypoxyprobe) one hour prior to euthanasia. Colon samples were fixed in 10% buffered formalin and paraffin-embedded tissue was probed with mouse anti-pimonidazole monoclonal IgG1 (MAb 4.3.11.3) and then stained with Cy-3 conjugated goat anti-mouse antibody (Jackon Immuno Research Laboratories). Samples were counterstained with DAPI using SlowFace Gold mountant. Samples were scored based on the degree of colonic epithelial hypoxia (0: no hypoxia; 1: mild focal hypoxia; 2: moderate multifocal hypoxia; 3: intense diffuse hypoxia) Representative images were obtained using a Zeiss Axiovert 200 M fluorescent microscope and brightness adjusted (Adobe Photoshop CS2).
Flow cytometry
Preparation of mouse intestinal lymphocytes has been described previously(35). After initial isolation, intestinal lymphocytes preparation was enriched for T cells using a CD3 enrichment kit (Easysep™ mouse T cell enrichment kit, STEMCELL technologies) according to manufacturer’s instructions. T (CD3+) cell-enriched intestinal cell population was resuspended in 2 ml Dulbecco’s phosphate-buffered saline (PBS) without calcium and magnesium and stained with aqua live/dead cell discriminator (Invitrogen) in accordance with the manufacturer’s protocol. Cells were then rinsed and stained for Treg population using a Treg specific kit (Mouse regulatory T cell staining kit #2, eBioscience) according to manufacturer’s instructions. Stained cells were analyzed with an LSR II flow cytometer (Becton Dickinson, San Jose, CA). The data were analyzed by using FlowJo software (TreeStar, Inc., Ashland, OR). Gates were set on singlets and then on live cells. Subsequent gates were based on fluorescence-minus-one and unstained controls Fig S6).
Lactate measurements
For determination of intracellular lactate measurements, primary colonocytes were isolated as described above. Lactate measurement in colonocyte lysates was performed by using a Lactate Colorimetry Assay Kit II (Biovision, Milpitas, CA) according to manufacturer’s instructions.
ATP measurements
For determination of intracellular ATP measurements, primary colonocytes were isolated as described above. ATP measurement in colonocyte lysates was performed by using a ATP Colorimetry Assay Kit (Biovision), according to manufacturer’s instructions.
Statistical Analysis
Fold changes of ratios (bacterial competitive index, mRNA levels and protein levels), percentages (flow cytometry) and bacterial numbers were transformed logarithmically prior to statistical analysis. An unpaired Student’s t test was used on the transformed data to determine whether differences between groups were statistically significant (p < 0.05). When more than two treatments were used, statistically significant differences between groups were determined by one-way ANOVA followed by Tukey’s HSD test (between > 2 groups). Significance of differences in hypoxia scores was determined by a one-tailed non-parametric test (Kruskal Wallis).
Supplementary Material
Acknowledgments
We acknowledge the Host-Microbe Systems Biology Core (HMSB Core) at the UC Davis School of Medicine for expert technical assistance with microbiota sequence analysis. The data reported in the manuscript are tabulated in the main paper and in the supplementary materials. M.X.B. and A.J.B. filed invention report number 0577501-16-0038 at iEdison.gov for a treatment to prevent post-antibiotic expansion of Enterobacteriaceae. This work was supported by Public Health Service Grants AI060555 (S.A.C.), TR001861 (E.E.O.), AI112241 (C.A.L.), DK087307 (C.G.), AI109799 (R.M.T), AI112258 (R.M.T), AI112949 (A.J.B. and R.M.T.), AI096528 (A.J.B.), AI112445 (A.J.B.), and AI114922 (A.J.B.).
Footnotes
References and Notes
- 1.Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33:496–503. doi: 10.1016/j.tibtech.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 2.Spees AM, et al. Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. MBio. 2013;4:e00430–00413. doi: 10.1128/mBio.00430-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rivera-Chavez F, et al. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe. 2016;19:443–454. doi: 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winter SE, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donohoe DR, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13:517–526. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marion-Letellier R, Butler M, Dechelotte P, Playford RJ, Ghosh S. Comparison of cytokine modulation by natural peroxisome proliferator-activated receptor gamma ligands with synthetic ligands in intestinal-like Caco-2 cells and human dendritic cells– potential for dietary modulation of peroxisome proliferator-activated receptor gamma in intestinal inflammation. Am J Clin Nutr. 2008;87:939–948. doi: 10.1093/ajcn/87.4.939. [DOI] [PubMed] [Google Scholar]
- 7.Lefebvre M, et al. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J Endocrinol. 1999;162:331–340. doi: 10.1677/joe.0.1620331. [DOI] [PubMed] [Google Scholar]
- 8.Alex S, et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2013;33:1303–1316. doi: 10.1128/MCB.00858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio. 2014;5:e00889. doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atarashi K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 11.Kelly CJ, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuta GT, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xavier MN, et al. PPARgamma-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe. 2013;14:159–170. doi: 10.1016/j.chom.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terada N, Ohno N, Saitoh S, Ohno S. Immunohistochemical detection of hypoxia in mouse liver tissues treated with pimonidazole using “in vivo cryotechnique”. Histochem Cell Biol. 2007;128:253–261. doi: 10.1007/s00418-007-0324-4. [DOI] [PubMed] [Google Scholar]
- 15.Kizaka-Kondoh S, Konse-Nagasawa H. Significance of nitroimidazole compounds and hypoxia-inducible factor-1 for imaging tumor hypoxia. Cancer science. 2009;100:1366–1373. doi: 10.1111/j.1349-7006.2009.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maslowski KM, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millard AL, et al. Butyrate affects differentiation, maturation and function of human monocyte-derived dendritic cells and macrophages. Clin Exp Immunol. 2002;130:245–255. doi: 10.1046/j.0009-9104.2002.01977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang B, Morinobu A, Horiuchi M, Liu J, Kumagai S. Butyrate inhibits functional differentiation of human monocyte-derived dendritic cells. Cell Immunol. 2008;253:54–58. doi: 10.1016/j.cellimm.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111:2247–2252. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atarashi K, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arpaia N, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furusawa Y, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 23.Smith PM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh N, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peyrin-Biroulet L, et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc Natl Acad Sci U S A. 2010;107:8772–8777. doi: 10.1073/pnas.0905745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narushima S, et al. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut microbes. 2014;5:333–339. doi: 10.4161/gmic.28572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Behnsen J, et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40:262–273. doi: 10.1016/j.immuni.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur J Immunol. 2010;40:780–786. doi: 10.1002/eji.200939613. [DOI] [PubMed] [Google Scholar]
- 29.Barrientos A. In vivo and in organello assessment of OXPHOS activities. Methods. 2002;26:307–316. doi: 10.1016/S1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 30.Rivera-Chavez F, et al. Energy Taxis toward Host-Derived Nitrate Supports a Salmonella Pathogenicity Island 1-Independent Mechanism of Invasion. MBio. 2016;7 doi: 10.1128/mBio.00960-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 33.Xia YN, Whitesides GM. Soft lithography. Annual Review of Materials Science. 1998;28:153–184. [Google Scholar]
- 34.Gao Y, et al. A versatile valve-enabled microfluidic cell co-culture platform and demonstration of its applications to neurobiology and cancer biology. Biomed Microdevices. 2011;13:539–548. doi: 10.1007/s10544-011-9523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keestra AM, et al. Early MyD88-dependent induction of interleukin-17A expression during Salmonella colitis. Infect Immun. 2011;79:3131–3140. doi: 10.1128/IAI.00018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.