

Abstract
The growth of a metazoan body relies on a series of highly coordinated cell-fate decisions by stem cells, which can undergo self-renewal, reversibly enter a quiescent state, or terminally commit to a cell specification program. To guide their decisions, stem cells make frequent use of ubiquitylation, a posttranslational modification that can affect the activity, interaction landscape, or stability of stem cell proteins. In this review, we are discussing novel findings that have provided insight into ubiquitin-dependent mechanisms of stem cell control and revealed how an essential and highly conserved protein modification can shape metazoan development.
Keywords: ubiquitin, development, E3 ligase, stem cell, differentiation
The ubiquitin system in stem cells
To establish the staggering complexity of an adult human body with its more than 200 cell types, a developing embryo must commit to a series of coordinated and tightly regulated cell-fate decisions. At the center of this intricate developmental program are stem cells that either divide and multiply or exit the cell cycle and adopt specific functions [1, 2]. Stem cells continue to play important roles in adult organisms, where they help maintain or repair tissues that are subject to attrition or damage [3]. Preserving the correct number and functionality of stem cells is an essential task of human development, and problems in the underlying regulatory circuits are responsible for many diseases, ranging from birth defects to cancer. It is now known that posttranslational modification with ubiquitin is at the heart of many essential signaling networks that inform the critical cell-fate decisions made by stem cells throughout development.
Through its covalent attachment to intracellular targets, ubiquitin is able to regulate the activity or stability of thousands of metazoan proteins [4]. In human cells, the specificity of ubiquitin transfer is conferred by ~600 E3 ligases, which recruit select target proteins as well as an activated form of ubiquitin. Most E3 ligases share a characteristic RING-domain (RING: really interesting new gene) that depends on a ubiquitin-loaded E2 conjugating enzyme for catalysis [5–7] (Figure 1A). An additional set of ~50 E3 ligases possess either a HECT- or RBR-domain (HECT: homologous to E6-AP; RBR: Ring-in between-Ring) with an active site cysteine that is charged with ubiquitin prior to its transfer to substrates [8, 9]. E3 ligases are often expressed at specific developmental stages or in particular cell types [10–14], and as described below, mutations in genes encoding these enzymes are found with increasing frequency in diseases caused by aberrant stem cell function. These observations mirror genetic screens that have revealed important contributions of E3 ligases and their effectors to stem cell regulation [12, 15–17].
Figure 1. Hallmarks of ubiquitin-dependent signaling.

A. E3 ligases determine the specificity of ubiquitin transfer by recruiting target proteins as well as an activated form of ubiquitin (~ indicates a thioester bond between ubiquitin and an active site cysteine in an E2 or E3; - denotes an isopeptide bond). E3 ligases either possess a RING-domain (RING: really interesting new gene) to recruit an E2; a HECT-domain (HECT: homologous to E6-AP C-terminus) charged with ubiquitin prior to transfer; or an RBR arrangement (RING-in between RING-RING) that also contains an active site cysteine residue. B. Ubiquitin modifications differ in their topology and function. Examples for distinct ubiquitin chain types are shown, including their major function.
E3 ligases are able to decorate their targets with several types of ubiquitin modifications that adopt distinct topologies and encode different information [4, 18] (Figure 1B). In this manner, ubiquitylation can modulate the activity, binding partners, localization, or stability of stem cell proteins. The reversible attachment of a single ubiquitin, a reaction referred to as monoubiquitylation, was discovered as a modification of histone proteins [19], where it controls chromatin architecture to determine cell fate. E3 ligases that act upon histones, such as RING1, BMI1, RNF20 or Dzip3, and their counteracting deubiquitylases have accordingly emerged as crucial regulators of pluripotency and early development [20–26]. By contrast, polymeric ubiquitin chains were initially recognized for their ability to trigger protein degradation by the 26S proteasome [27], and K11-, K48-, and K11/K48-linked chains are well understood signals to mediate such proteolytic events [4]. Stem cells frequently use ubiquitin-dependent proteolysis to set the levels of transcription factors that establish pluripotency or specify a particular differentiation route. In addition, stem cells can assemble M1-linked, K63-linked, or M1/K63-hybrid chains that control the assembly of large signaling complexes and thereby safeguard the integrity of the stem cell genome or determine the responsiveness of stem cells to their environment [4]. The pervasive contribution of ubiquitylation to cellular regulation makes it impossible to discuss all roles of this modification in stem cell biology. Rather than listing each of ubiquitin’s activities in pluripotent cells, we will focus our discussion onto recent findings that highlight recurrent themes in the ubiquitin-dependent control of stem cell biology.
Control of stem cell self-renewal by ubiquitin
The ability of stem cells to divide while preserving their undifferentiated state is referred to as self-renewal (Figure 2). Self-renewal is essential for increasing the number of stem cells during development or in response to tissue damage, and it helps maintain a pool of stem cells that can repopulate adult tissues that are subject to frequent attrition, such as the skin or intestine [28]. Whether stem cells commit to self-renewal, remain quiescent, or initiate differentiation is dependent on environmental signals or mechanical cues that often originate in their close proximity, a compartment described as the stem cell niche [29–31]. Ubiquitylation plays essential roles in ensuring faithful communication between stem cells and their microenvironment, as illustrated by the self-renewal programs that depend on the secreted glycoproteins of the Wnt family (Figure 3). In fact, Wnt signaling provides a striking example for the many and mechanistically diverse functions in ubiquitylation in regulating stem cell biology.
Figure 2. Ubiquitylation controls stem cell quiescence, self-renewal, and differentiation.

Examples of E3 ligases discussed in this review are shown on the right.
Figure 3. Ubiquitin-dependent control of Wnt signaling.

A. Constitutive degradation of the β-catenin transcription factor following its phosphorylation by the destruction complex and ubiquitylation by the SCFβTrCP E3 ligase (SCF: Skp1-CUL1-Fbox; upper case denotes specific substrate adaptor βTrCP). The destruction complex is composed of the scaffolding proteins axin and APC, the tumor suppressor Wtx1, and the kinases CK1 and GSK3β. Wnt signals prevent the destruction complex from phosphorylating β-catenin, thereby allowing β-catenin to accumulate and drive a transcriptional program supporting stem cell self-renewal. B. Ubiquitin-dependent control of Wnt receptor abundance. The E3 ligases ZNRF3 and RNF43 use the Dishevelled protein (Dvl) as an adaptor to bind and ubiquitylate the Wnt receptor Frizzled, leading to its internalization by endocytosis and to its lysosomal degradation. Secreted R-spondin proteins and their membrane receptors Lgr4/5 sequester Frizzled proteins away from ZNFR3 and RNF43, thus amplifying the Wnt signals that ensure stem cell self-renewal.
The role of Wnt proteins in controlling self-renewal is best understood in crypt base columnar cells, the stem cells of the intestine that can replenish every cell type in this rapidly cycling tissue [28]. Secretion of Wnt by the intestinal stem cell niche results in a short-range Wnt gradient that neighboring stem cells sense through a co-receptor composed of the Frizzled and LRP5/6 proteins [32–34]. The ligand-engaged Frizzled-LRP5/6 complex inhibits the GSK3β kinase and recruits the axin protein to the membrane compartment [35, 36], which shuts off the “destruction complex” composed of the scaffolds axin and adenomatous polyposis coli, the tumor suppressor WTX, as well as the kinases CK1 and GSK3β [37]. In the absence of Wnt, this destruction complex phosphorylates the transcription factor β-catenin at residues in its amino-terminal domain [38], which mediates the recognition of β-catenin by the E3 SCFβTrCP for ubiquitylation and proteasomal degradation [39–41] (Figure 3A). By inhibiting the destruction complex, Wnt signals allow β-catenin to accumulate and enable it to team up with its partner Tcf/Lef to drive a transcriptional program that promotes stem cell self-renewal and survival [42]. Thus, stem cells depend on continuous input from their niche to prevent the degradation of a self-renewal factor, a setup that allows them to respond rapidly and initiate different pathways shortly after the cessation of Wnt signaling [43]. This regulatory flexibility, however, comes at a cost, and mutations in the Wnt-pathway that uncouple β-catenin stabilization from signals sent by the intestinal niche are sufficient to expand the stem cell compartment and account for the vast majority of inherited forms of colon cancer [37, 44, 45].
The sensitivity of intestinal stem cells towards Wnt is, at least in part, determined by the abundance of Frizzled and LRP5/6 receptors at the plasma membrane. In the absence of Wnt, the transmembrane E3 ligases ZNRF3 and RNF43 use the Disheveled protein as an adaptor to bind Frizzled and LRP6 [46–48] (Figure 3B). This results in ubiquitylation, internalization by endocytosis, and lysosomal degradation of Frizzled and LRP6. Underscoring the important role of restricting the membrane presentation of Wnt receptors, the function of ZNRF3 and RNF43 is conserved in C. elegans [49], and mutations in the RNF43 and ZNRF3 genes have been observed in cancer [50]. When needed, organisms can exploit secreted R-spondin proteins and their membrane receptors LGR4 and LGR5 to sequester ZNRF3 and RNF43 away from Frizzled [46, 51, 52], or they use the proto-oncogenic deubiquitylase USP6 to cleave ubiquitin off of Frizzled [53]. By protecting Frizzled from degradation, the R-spondin proteins release a ubiquitin-dependent break to Wnt signal transmission that is essential for stem cell function in multiple tissues [51, 54]; it is also a critical step in protocols for intestinal organoid formation in vitro [55]. Thus, ubiquitylation also controls the balance between receptor degradation and stabilization, thereby ensuring that stem cells remain responsive to signals emerging from their niche.
The ability of ubiquitylation to limit the pool of critical signaling molecules is not restricted to Wnt receptors. As mentioned above, β-catenin is degraded in a reaction that depends on prior phosphorylation by the destruction complex. The limiting component of the destruction complex, axin [56], is also tightly controlled by ubiquitin-dependent turnover: following its modification with a poly-ADP-ribosylation (PARsylation) tag by the poly-ADP-ribosylase Tankyrase [57], axin is recognized by the E3 ligase RNF146 [58, 59]. RNF146 converts binding to the PARsylation signal into allosteric activation of its E3 activity and subsequently decorates axin with a proteolytic ubiquitin mark [60]. In line with these observations, compounds that inhibit tankyrase stabilize axin and thereby dampen constitutive β-catenin signaling in cancer cells [57].
As Axin, ZNRF3, and RNF43 are all β-catenin target genes [46, 47, 61], Wnt activation sets in motion a reaction cascade that allows this signaling system to return to its basal state. Similar negative feedback regulation is encountered in almost every development pathway [62]. Ubiquitylation also plays a central role in other network motifs that enable stem cells to compute environmental signals and integrate them into their self-renewal programs. An interesting example is provided by Disheveled: this developmental regulator acts both as an inhibitor of Wnt signaling that supports the turnover of Wnt receptors, as well as a positive factor that is required for Wnt signal transmission [48, 63]. Such apparently paradoxical functions are able to constitute incoherent feedforward loops [64], which can endow stem cells with the ability to detect fold-changes, rather than absolute differences, in receptor-bound Wnt [65, 66]. Stem cells also use ubiquitin-dependent degradation to implement positive feedback control [51, 67], a motif to amplify signaling or establish switch-like transitions between distinct states. Through its ability to rapidly turn off signal transducers, ubiquitylation is therefore often at the heart of network motifs that allow stem cells to accurately interpret signals emerging from their niche.
While we have discussed the role of ubiquitylation in controlling the self-renewal of intestinal stem cells, progenitor cells of other tissues rely on similar regulatory principles. For example, long-term hematopoietic stem cells employ the E3 ligase SCFFBW7 to efficiently ubiquitylate the transcription factor c-Myc [68, 69], one of the four original transcription factors to reprogram a differentiated fibroblast into an induced pluripotent stem cell [70]. Deletion of FBXW7 strongly impairs the proteasomal degradation of c-Myc and impedes the ability of LT-HSCs to self-renew, which was rescued by simultaneous loss of a single allele of the Myc gene [71]. In a similar manner, the E3 ligase CUL4-DDB1 supports the self-renewal of hematopoietic precursors [72], while the E3 mLin41/TRIM71 performs this task in neural precursors [13]. Extending these concepts to energy metabolism, SCFFBXO15, a stem cell-specific E3 that was initially used as a marker for induced pluripotent stem cells [70], ubiquitylates a regulator of mitochondrial biogenesis, which likely reduces the exposure of ESCs to reactive oxygen species [73]. By limiting the abundance of crucial receptors, transcription factors, and metabolic regulators, ubiquitylation allows stem cells of multiple tissues to translate signals emerging from their niche into efficient self-renewal.
Ubiquitin-dependent control of stem cell quiescence
When cultured in vitro, stem cells typically divide rapidly with short gap phases [74]. The situation is very different in living organisms, where many stem cells reversibly exit the cell cycle to enter a quiescent state. Quiescent stem cells can be readily reactivated to enter self-renewal and differentiation programs and thus help replenish a damaged tissue or repopulate differentiated cells that were lost by attrition. For example, while long-term hematopoietic stem cells spend most of their time in quiescence, chemotherapeutic ablation of the hematopoietic system triggers re-entry of these stem cells into the cell division program, followed by differentiation into the multiple cell types that constitute our blood system [71]. The ability to remain quiescent, a property ensured by the stem cell niche, likely protects stem cells from damage that is encountered during cell division, including replication or protein folding mistakes. Quiescence thus ensures the existence of a pristine stem cell pool throughout the life time of an organism, and the inability to regulate the interplay between cell cycle exit and re-entry contributes to stem cell depletion phenotypes observed during aging [75].
Consistent with a dominant role of ubiquitylation in cell division control [76], ubiquitin-dependent networks safeguard the transition of stem cells to and from quiescence. This often requires E3 ligases that target regulators of the core cell cycle machinery, but also ubiquitylate proteins that control the switch between continuous proliferation and reversible cell cycle exit. Underscoring this notion, the E3 ligase anaphase-promoting complex (APC/C) stabilizes the quiescent state by degrading kinases that drive entry into S phase, and it regulates stem cell differentiation by targeting transcription factors, regulators of mRNA translation, or even membrane bound receptors [77–82]. The E3 SCFFBW7, an enzyme very frequently mutated in human cancer [83], ensures stem cell quiescence by targeting the c-Myc transcription factor and cell cycle regulator cyclin E for degradation [68, 71, 84]. In a similar manner, the E3 HUWE1, which has been linked to intellectual disability and schizophrenia, targets regulators of cell division, cell survival, and transcription, allowing it to promote the entry of neural precursor cells into the quiescent state [85–89]. Dependent on the tissue of origin of diverse stem cells, the multiple activities of the APC/C, SCFFBW7, and HUWE1 likely endows these E3 ligases with the capacity to fine-tune the relationship between stem cell proliferation and quiescence programs, a function that for HUWE1 appears to be conserved all the way to planarians [90].
Stem cells require active protein management to maintain the quiescent state, a feature that is illustrated by progenitor cells that reside in the low-oxygen environment of the bone marrow. The relative absence of oxygen in their niche protects long-term hematopoetic stem cells from damage by reactive oxygen species and thus prevents exhaustion of this important cell population. The HIF1α transcription factor plays an important role in stabilizing stem cell quiescence in the presence of low oxygen, and degradation of HIF1α by the E3 ligase CUL2 and its substrate adaptor VHL helps these cells to reenter a division program when required [91]. As during self-renewal, multiple ubiquitylation pathways therefore appear to cooperate to ensure robust stem cell regulation during quiescence.
Flipping the switch from self-renewal to differentiation
Once stem cells have received an appropriate input, for example by encountering a long-range morphogen or by entering an embryonic territory that is destined to develop into a particular tissue, they can differentiate into the many specialized cell types that make up a metazoan body. Differentiation demands that stem cells exit their self-renewal program and commit to a series of terminal specialization events. This change in fate frequently relies on the degradation of pluripotency factors and differentiation inhibitors, and it is accompanied by the simultaneous synthesis of proteins that allow a cell type to perform its vital functions. Most transcription factors that are important for the pluripotent state, such as OCT4, NANOG, or SOX2, are short-lived proteins, poised for removal in case the right signal is detected and their synthesis comes to a halt [69, 92–94]. Conversely, inhibitors of differentiation, such as the REST or ID family of transcriptional regulators, are turned over in response to differentiation cues [81, 95], as are components of the general transcription factor TFIID that are being replaced by variants specific for a differentiated cell type [96]. In addition to effects on the transcriptional landscape, ubiquitin-dependent changes to the proteome can alter cell morphology and thus affect how stem cells communicate with their niche. Providing an interesting case in point, ubiquitin-dependent endocytosis of E-cadherin by the E3 ligase CBLL1 limits the abundance of adherens junctions and changes the ability of hESCs to engage in a mesenchymal differentiation program [29].
A major regulator of metazoan cell-fate decisions are cullin-RING ligases (CRLs), a large class of E3 ligases that are organized by cullin scaffold proteins and that recruit their substrates by virtue of ~350 specific adaptors [97–99]. The importance of these E3 for cell-fate decisions is underscored by the finding that regulators of CRLs, such as the CAND1 exchange factor or the COP9 signalosome, are essential for differentiation [100, 101]. Moreover, mutations in cullin proteins or their substrate adaptors have been tightly associated with diseases that can often be traced back to aberrant cell-fate decisions made by stem cells [102–105]. These diseases include autism, schizophrenia, myopathies, hypertension and cancer, and thus, affect a large patient population.
CRLs illustrate the versatility of ubiquitin-dependent control of stem cell behavior, as several members of the CRL family are able to inform differentiation decisions without affecting protein stability. This capacity was recently described for CUL3 and its substrate adaptor KBTBD8 (CUL3KBTBD8), a complex that is required for the specification of human embryonic stem cells into neural crest cells [11] (Figure 4A). Neural crest cells are migratory cells that originate at the border between the neural plate and non-neural ectoderm and are responsible for generating a wide range of specialized cell types, including chondrocytes, melanocytes, or glial cells [106, 107]. CUL3KBTBD8 drives neural crest formation by monoubiquitylating TCOF1 and NOLC1, which allows these paralogs to interact with each other and recruit multiple enzymes required for the production of new, and likely modified, ribosomes [11] (Figure 4A). Reflecting the growing appreciation for the role of regulated protein synthesis in cell-fate decisions [108–110], ribosomes produced upon CUL3KBTBD8-dependent ubiquitylation translate specific mRNAs with different efficiency than ribosomes present in stem cells, suggesting that ubiquitin-dependent changes in the mRNA translation landscape underlie neural crest specification. This regulatory circuit is essential for human development, and genetic lesions that reduce the levels of the essential CUL3KBTBD8-substrate TCOF1 result in the neurocristopathy Treacher Collins Syndrome [111].
Figure 4. Regulation of stem cell fate and function by CUL3-dependent monoubiquitylation.
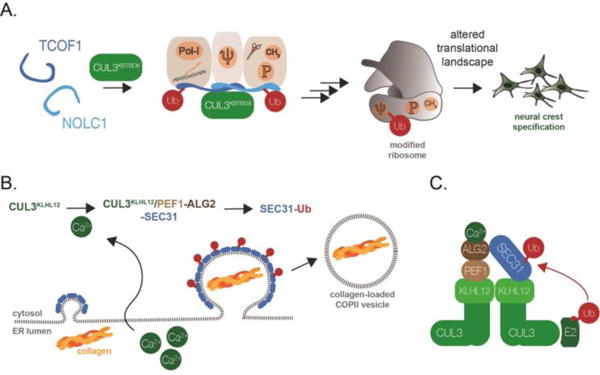
A. CUL3KBTBD8 controls neural crest specification by catalyzing the monoubiquityation of TCOF1 and NOLC1. This modification allows TCOF1 and NOLC1 to organize a ribosome biogenesis platform that includes RNA polymerase I (Pol-I), the H/ACA complex catalyzing pseudouridilyation (depicted as Ψ), and the SSU processome (CH3; P for its methylation and phosphorylation activities). This ubiquitin-dependent platform leads to the production of modified ribosomes that translate specific mRNAs to drive neural crest specification. B. CUL3KLHL12 controls COPII vesicle size and collagen secretion, a reaction critical for formation of a stem cell niche and bone. CUL3KLHL12 catalyzes the monoubiquitylation of the COPII coat protein SEC31, thereby allowing this protein to support the formation of large COPII vesicles that accelerate the process of collagen secretion. C. To perform its cellular functions, CUL3KLHL12 depends on a calcium-dependent co-adaptor specific for the substrate SEC31. The ALG2 subunit is only able to engage SEC31 after calcium has been released from the endoplasmic reticulum.
Although stem cell differentiation has long been assumed to produce a terminal state, it is now known that re-expression of certain transcription factors is able to change the fate of specialized cell populations [70]. Reminiscent of the situation in quiescent stem cells, organisms actively maintain the differentiated state, and this also requires ubiquitylation. As described above, the E3 ligase SCFFBW7 typically inhibits stem cell proliferation, a function that is important for its role as a tumor suppressor. In the pancreas, however, the tissue-specific loss of SCFFBW7 resulted in transdifferentiation of ductal cells into insulin-producing β-cells [112]. SCFFBW7 fulfills its role in stabilizing the ductual cell fate by protecting ductal cells from the accumulation of neurogenin-3, a transcription factor that drives an alternative β-cell neogenesis program [112]. Through its tight grip onto transcription, mRNA translation, and protein stability, ubiquitylation thus provides a powerful mechanism to trigger decisive switches between different cell fates, as well as to ensure the long-term maintenance of a specified cellular state.
Regulating the regulators
To make informed cell-fate decisions, stem cells rely on signals that are sent and received at specific times and locations within a growing organism. Given the role of ubiquitylation in development, it is important to understand how stem cells are able to translate such organismal signals into proper activation or inhibition of critical ubiquitylation enzymes. In many cases, it is the abundance of E3 ligases or deubiquitylases that is tightly controlled, with several enzymes being preferentially expressed in the pluripotent state [10, 11, 24, 73, 113]. The activity of these enzymes can be further fine-tuned by binding partners, such as the R-spondin proteins that titrate the ZNRF3 and RNF43 E3 ligases away from their membrane localized substrates [46, 47]. In the case of CUL3 E3 ligases, recent work pointed to target-specific co-adaptors as a novel class of regulatory factors that allow stem cells to turn on crucial E3 ligases at the right time and place during development [114].
The role of the CUL3 E3 ligase in stem cell biology was initially discovered by a genetic screen that linked a CUL3KLHL12 complex to secretion of collagen, a crucial component of stem cell niches [10]. CUL3KLHL12 monoubiquitylates the COPII vesicle protein SEC31, which in turn results in formation of COPII vesicles that are large enough to package collagen molecules for their secretion into the extracellular space [10] (Figure 4B). To efficiently recognize and ubiquitylate SEC31 in cells, CUL3KLHL12 depends on two additional factors, PEF1 and ALG2, which form a heterodimeric complex that bridges KLHL12 and SEC31 and provides an additional binding site for the substrate on the CUL3KLHL12 E3 ligase [114] (Figure 4C). Importantly, the PEF1-ALG2 co-adaptor depends on calcium release from the endoplasmic reticulum to engage its cognate substrate SEC31[114]. Calcium also plays important roles in the transcriptional circuits that specify neural crest fate and drive collagen synthesis [115, 116]. Moreover, the highest calcium levels in metazoan organisms are found in bones [117], and thus, at the location where the developing organism is expected to activate CUL3KLHL12 and deposit collagen. These findings document how regulation of E3 ligases by organismal signals, including calcium, can help coordinate a series of developmental events that in this case start with a stem cell and end with a mineralized bone.
As our understanding of ubiquitin-dependent control of stem cell biology is increasing, so are calls to modulate the activity of ubiquitylation enzymes for therapeutic benefit. Proof of concept has been provided by thalidomide and related compounds that bind to a CUL4-dependent E3 ligase build around the substrate adaptor cereblon [118]. By interacting with both the CUL4cereblon complex and cellular proteins referred to as neo-substrates, thalidomide induces degradation events that are beneficial for the treatment of leukemia [119–121]. Thalidomide has become infamous for its teratogenic role in interfering with limb development, suggesting that the CUL4cereblon complex plays an important, yet still poorly understood role in cell-fate decisions. Similar to thalidomide, the anti-tumorigenic sulfonamide indisulam induces the CUL4-dependent ubiquitylation and proteasomal degradation of a protein, the spliceosomal regulator RMB39, that is essential for cancer cell survival [122]. These observations raise the exciting possibility that clarifying the role of ubiquitylation in stem cells and early human development will provide an avenue towards developing novel, and safe, therapeutic approaches to correct the inaccurate cell-fate decisions that are at the origin of many inherited human diseases.
Conclusions
While the roles of epigenetic and transcriptional control in stem cells have been investigated in much detail, less is known about the functions of posttranslational modifications for early cell fate decisions. Based on a flurry of recent studies, this review highlights how modification with ubiquitin regulates almost every level of stem cell behavior, including the decision of stem cells to undergo self-renewal, temporarily pause with division, or adopt a specialized and non-dividing fate (Figure 2). By inducing the degradation of membrane receptors that receive input from the niche or transcription factors that determine cell fate, the ubiquitin system is key to successful communication between stem cells and their environment. Ubiquitin can also act non-proteolytically, as documented by its role in establishing the chromatin architecture or translational landscape that supports cell fate decisions. The versatility of ubiquitylation, encoded in the many different ubiquitin signals, is a major reason for its recurrent use as a regulatory system in such an important cell population as stem cells.
It was eloquent biochemistry that had revealed how the distinct ubiquitin signals are able to encode diverse biological information. Yet, in recent years, we have also learned how these ubiquitylation marks are implemented in the control of living cells. As an untransformed population that has to make important decisions about fate and function, stem cells have been an ideal model to investigate the ubiquitin system in vivo and at the same time revealed key reactions that shape human development. New technologies, such as CRISPR/Cas9-dependent genome editing, high-throughput shRNA screens, ribosome profiling, or more sensitive and quantitative proteomic approaches provide an exciting foundation from which to continue our exploration of the ubiquitin system. In fact, these new experimental platforms should make it possible to combine biochemistry and developmental biology to provide mechanistic insight into the ubiquitin-dependent control of stem cell behavior. Translating the insights that will surely emerge from such studies into novel therapeutic approaches will be a most exciting frontier of future work.
Trends Box.
Ubiquitylation controls stem cell self-renewal by restricting the presentation of receptor complexes that can detect niche signals and by controlling the stability of transcription factors that determine cell fate.
Ubiquitin-dependent signaling also governs the reversible exit from the cell cycle into a quiescent state, often by targeting cell cycle regulators and transcription factors for degradation.
Ubiquitylation plays an essential role in differentiation, in part by non-proteolytic regulation of chromatin architecture or mRNA translation.
Essential ubiquitylation enzymes are tightly regulated in stem cells and during early development.
Outstanding questions.
What are the targets of disease-linked ubiquitylation enzymes? Recent genome-wide association studies tightly linked aberrant ubiquitylation to many developmental diseases, yet the responsible targets remain mostly unknown.
What is the extent of crosstalk between ubiquitylation and other posttranslational modifications? E3 ligases have been reported to require phosphorylation, oxidation, or PARsylation to recognize their substrates, indicative of substantial crosstalk between multiple signaling pathways.
Do ubiquitylation enzymes cooperate with each other to establish robust stem cell regulation? Recent studies suggested that E3 ligases cooperate to modify proteins with complex ubiquitylation signals, including branched ubiquitin chains. This observation suggests that the interplay between distinct ubiquitylation enzymes can fine-tune the signaling function of this essential posttranslational modification.
Can we target ubiquitylation with small molecules to alter cell fate? Thalidomide and related compounds suggest that small molecules could alter the activity or substrate-binding properties of E3 ligases as a potential strategy to change cell-fate decisions in stem cells.
Acknowledgments
We apologize to all scientists whose work could not be discussed within the space confines of this review. We thank members of our lab for continuous help and discussions, and we are grateful to Julia Schaletzky for comments on this manuscript. AGM was supported by a Postdoctoral Fellowship, PF-15-215-01 – DCC from the American Cancer Society; AW is a recipient of a NIDCR K99 pathway to independence award (K99DE025314); MR is the Dr. K. Peter Hirth chair of cancer biology at the University of California at Berkeley and an Investigator with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Becker AJ, et al. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature. 1963;197:452–4. doi: 10.1038/197452a0. [DOI] [PubMed] [Google Scholar]
- 3.Leblond CP, Walker BE. Renewal of cell populations. Physiol Rev. 1956;36(2):255–76. doi: 10.1152/physrev.1956.36.2.255. [DOI] [PubMed] [Google Scholar]
- 4.Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18(6):579–86. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- 5.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 6.Saha A, et al. Essential role for ubiquitin-ubiquitin-conjugating enzyme interaction in ubiquitin discharge from Cdc34 to substrate. Mol Cell. 2011;42(1):75–83. doi: 10.1016/j.molcel.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plechanovova A, et al. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489(7414):115–20. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10(6):398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 9.Wenzel DM, et al. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474(7349):105–8. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin L, et al. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482(7386):495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werner A, et al. Cell-fate determination by ubiquitin-dependent regulation of translation. Nature. 2015;525(7570):523–7. doi: 10.1038/nature14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buckley SM, et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell. 2012;11(6):783–98. doi: 10.1016/j.stem.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, et al. The ubiquitin ligase mLin41 temporally promotes neural progenitor cell maintenance through FGF signaling. Genes Dev. 2012;26(8):803–15. doi: 10.1101/gad.187641.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okita K, et al. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 15.Vilchez D, et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature. 2012;489(7415):304–8. doi: 10.1038/nature11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin H, et al. Systematic identification of barriers to human iPSC generation. Cell. 2014;158(2):449–61. doi: 10.1016/j.cell.2014.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chia NY, et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature. 2010;468(7321):316–20. doi: 10.1038/nature09531. [DOI] [PubMed] [Google Scholar]
- 18.Komander D, Rape M. The ubiquitin code. Annual review of biochemistry. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 19.Goldknopf IL, et al. Presence of protein A24 in rat liver nucleosomes. Proc Natl Acad Sci U S A. 1977;74(12):5492–5. doi: 10.1073/pnas.74.12.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Endoh M, et al. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet. 2012;8(7):e1002774. doi: 10.1371/journal.pgen.1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pengelly AR, et al. Transcriptional repression by PRC1 in the absence of H2A monoubiquitylation. Genes Dev. 2015;29(14):1487–92. doi: 10.1101/gad.265439.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu Y, et al. The histone H2A deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem cell function. Proc Natl Acad Sci U S A. 2016;113(1):E51–60. doi: 10.1073/pnas.1517041113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoue D, et al. Dzip3 regulates developmental genes in mouse embryonic stem cells by reorganizing 3D chromatin conformation. Sci Rep. 2015;5:16567. doi: 10.1038/srep16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs G, et al. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Molecular cell. 2012;46(5):662–73. doi: 10.1016/j.molcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheuermann JC, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–7. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W, et al. The histone H2A deubiquitinase Usp16 regulates embryonic stem cell gene expression and lineage commitment. Nat Commun. 2014;5:3818. doi: 10.1038/ncomms4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chau V, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243(4898):1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 28.Clevers H, et al. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346(6205):1248012. doi: 10.1126/science.1248012. [DOI] [PubMed] [Google Scholar]
- 29.Przybyla L, et al. Tissue Mechanics Orchestrate Wnt-Dependent Human Embryonic Stem Cell Differentiation. Cell Stem Cell. 2016 doi: 10.1016/j.stem.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McBeath R, et al. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6(4):483–95. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 31.Losick VP, et al. Drosophila stem cell niches: a decade of discovery suggests a unified view of stem cell regulation. Dev Cell. 2011;21(1):159–71. doi: 10.1016/j.devcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farin HF, et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature. 2016;530(7590):340–3. doi: 10.1038/nature16937. [DOI] [PubMed] [Google Scholar]
- 33.Janda CY, et al. Structural basis of Wnt recognition by Frizzled. Science. 2012;337(6090):59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhanot P, et al. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382(6588):225–30. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 35.Stamos JL, et al. Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6. Elife. 2014;3:e01998. doi: 10.7554/eLife.01998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang E, et al. Wnt pathway activation by ADP-ribosylation. Nat Commun. 2016;7:11430. doi: 10.1038/ncomms11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 38.Aberle H, et al. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16(13):3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winston JT, et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999;13(3):270–83. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latres E, et al. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999;18(4):849–54. doi: 10.1038/sj.onc.1202653. [DOI] [PubMed] [Google Scholar]
- 41.Hart M, et al. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999;9(4):207–10. doi: 10.1016/s0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 42.Hernandez AR, et al. Kinetic responses of beta-catenin specify the sites of Wnt control. Science. 2012;338(6112):1337–40. doi: 10.1126/science.1228734. [DOI] [PubMed] [Google Scholar]
- 43.Purvis JE, Lahav G. Encoding and decoding cellular information through signaling dynamics. Cell. 2013;152(5):945–56. doi: 10.1016/j.cell.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinzler KW, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253(5020):661–5. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 45.Nishisho I, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–9. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 46.Hao HX, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. 2012;485(7397):195–200. doi: 10.1038/nature11019. [DOI] [PubMed] [Google Scholar]
- 47.Koo BK, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488(7413):665–9. doi: 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- 48.Jiang X, et al. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol Cell. 2015;58(3):522–33. doi: 10.1016/j.molcel.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 49.Moffat LL, et al. The conserved transmembrane RING finger protein PLR-1 downregulates Wnt signaling by reducing Frizzled, Ror and Ryk cell-surface levels in C. elegans. Development. 2014;141(3):617–28. doi: 10.1242/dev.101600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koo BK, et al. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43;Znrf3-mutant neoplasia. Proc Natl Acad Sci U S A. 2015;112(24):7548–50. doi: 10.1073/pnas.1508113112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Lau W, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476(7360):293–7. doi: 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- 52.Zebisch M, et al. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat Commun. 2013;4:2787. doi: 10.1038/ncomms3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Madan B, et al. USP6 oncogene promotes Wnt signaling by deubiquitylating Frizzleds. Proc Natl Acad Sci U S A. 2016;113(21):E2945–54. doi: 10.1073/pnas.1605691113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Planas-Paz L, et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat Cell Biol. 2016;18(5):467–79. doi: 10.1038/ncb3337. [DOI] [PubMed] [Google Scholar]
- 55.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 56.Lee E, et al. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS biology. 2003;1(1):E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang SM, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 58.Callow MG, et al. Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PloS one. 2011;6(7):e22595. doi: 10.1371/journal.pone.0022595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nature cell biology. 2011;13(5):623–9. doi: 10.1038/ncb2222. [DOI] [PubMed] [Google Scholar]
- 60.DaRosa PA, et al. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature. 2015;517(7533):223–6. doi: 10.1038/nature13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Amerongen R, et al. Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell. 2012;11(3):387–400. doi: 10.1016/j.stem.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 62.Uriu K. Genetic oscillators in development. Dev Growth Differ. 2016;58(1):16–30. doi: 10.1111/dgd.12262. [DOI] [PubMed] [Google Scholar]
- 63.Wallingford JB, Habas R. The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development. 2005;132(20):4421–36. doi: 10.1242/dev.02068. [DOI] [PubMed] [Google Scholar]
- 64.Hart Y, Alon U. The utility of paradoxical components in biological circuits. Mol Cell. 2013;49(2):213–21. doi: 10.1016/j.molcel.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 65.Goentoro L, Kirschner MW. Evidence that fold-change, and not absolute level, of beta-catenin dictates Wnt signaling. Mol Cell. 2009;36(5):872–84. doi: 10.1016/j.molcel.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goentoro L, et al. The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol Cell. 2009;36(5):894–9. doi: 10.1016/j.molcel.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yue S, et al. Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. Elife. 2014;3 doi: 10.7554/eLife.02555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yada M, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23(10):2116–25. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Welcker M, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101(24):9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 71.Reavie L, et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat Immunol. 2010;11(3):207–15. doi: 10.1038/ni.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao J, et al. The CUL4-DDB1 ubiquitin ligase complex controls adult and embryonic stem cell differentiation and homeostasis. Elife. 2015;4 doi: 10.7554/eLife.07539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Donato V, et al. The TDH-GCN5L1-Fbxo15-KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat Cell Biol. 2017 doi: 10.1038/ncb3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li VC, et al. Gap 1 phase length and mouse embryonic stem cell self-renewal. Proc Natl Acad Sci U S A. 2012;109(31):12550–5. doi: 10.1073/pnas.1206740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adams PD, et al. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell. 2015;16(6):601–12. doi: 10.1016/j.stem.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mocciaro A, Rape M. Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. Journal of cell science. 2012;125(Pt 2):255–63. doi: 10.1242/jcs.091199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang Y, et al. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science. 2009;326(5952):575–8. doi: 10.1126/science.1177087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shao R, et al. Cdh1 regulates craniofacial development via APC-dependent ubiquitination and activation of Goosecoid. Cell Res. 2016;26(6):699–712. doi: 10.1038/cr.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang J, et al. A Cdh1-APC/FMRP Ubiquitin Signaling Link Drives mGluR-Dependent Synaptic Plasticity in the Mammalian Brain. Neuron. 2015;86(3):726–39. doi: 10.1016/j.neuron.2015.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Konishi Y, et al. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303(5660):1026–30. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- 81.Lasorella A, et al. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature. 2006;442(7101):471–4. doi: 10.1038/nature04895. [DOI] [PubMed] [Google Scholar]
- 82.Stegmuller J, et al. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50(3):389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 83.Cheng Y, Li G. Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev. 2012;31(1–2):75–87. doi: 10.1007/s10555-011-9330-z. [DOI] [PubMed] [Google Scholar]
- 84.Takeishi S, et al. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23(3):347–61. doi: 10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 85.Urban N, et al. Return to quiescence of mouse neural stem cells by degradation of a proactivation protein. Science. 2016;353(6296):292–5. doi: 10.1126/science.aaf4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Forget A, et al. Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev Cell. 2014;29(6):649–61. doi: 10.1016/j.devcel.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 87.Zhong Q, et al. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–95. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 88.Zhao X, et al. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol. 2008;10(6):643–53. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Adhikary S, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005;123(3):409–21. doi: 10.1016/j.cell.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 90.Henderson JM, et al. Identification of HECT E3 ubiquitin ligase family genes involved in stem cell regulation and regeneration in planarians. Dev Biol. 2015;404(2):21–34. doi: 10.1016/j.ydbio.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 91.Takubo K, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 92.Fang L, et al. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol Cell. 2014;55(4):537–51. doi: 10.1016/j.molcel.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 93.Bahnassawy L, et al. TRIM32 modulates pluripotency entry and exit by directly regulating Oct4 stability. Sci Rep. 2015;5:13456. doi: 10.1038/srep13456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liao B, Jin Y. Wwp2 mediates Oct4 ubiquitination and its own auto-ubiquitination in a dosage-dependent manner. Cell Res. 2010;20(3):332–44. doi: 10.1038/cr.2009.136. [DOI] [PubMed] [Google Scholar]
- 95.Guardavaccaro D, et al. Control of chromosome stability by the beta-TrCP-REST-Mad2 axis. Nature. 2008;452(7185):365–9. doi: 10.1038/nature06641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li L, et al. A specific E3 ligase/deubiquitinase pair modulates TBP protein levels during muscle differentiation. Elife. 2015;4:e08536. doi: 10.7554/eLife.08536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 98.Lydeard JR, et al. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14(12):1050–61. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Skaar JR, et al. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14(6):369–81. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pan L, et al. Protein competition switches the function of COP9 from self-renewal to differentiation. Nature. 2014;514(7521):233–6. doi: 10.1038/nature13562. [DOI] [PubMed] [Google Scholar]
- 101.Dubiel D, et al. CAND1-dependent control of cullin 1-RING Ub ligases is essential for adipogenesis. Biochim Biophys Acta. 2013;1833(5):1078–84. doi: 10.1016/j.bbamcr.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 102.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gupta VA, Beggs AH. Kelch proteins: emerging roles in skeletal muscle development and diseases. Skelet Muscle. 2014;4:11. doi: 10.1186/2044-5040-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Betancur P, et al. Assembling neural crest regulatory circuits into a gene regulatory network. Annu Rev Cell Dev Biol. 2010;26:581–603. doi: 10.1146/annurev.cellbio.042308.113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Simoes-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142(2):242–57. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kondrashov N, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145(3):383–97. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xue S, et al. RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature. 2015;517(7532):33–8. doi: 10.1038/nature14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Signer RA, et al. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509(7498):49–54. doi: 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dixon J, et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006;103(36):13403–8. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sancho R, et al. Loss of Fbw7 reprograms adult pancreatic ductal cells into alpha, delta, and beta cells. Cell Stem Cell. 2014;15(2):139–53. doi: 10.1016/j.stem.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jung M, et al. A data integration approach to mapping OCT4 gene regulatory networks operative in embryonic stem cells and embryonal carcinoma cells. PloS one. 2010;5(5):e10709. doi: 10.1371/journal.pone.0010709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McGourty CA, et al. Regulation of the CUL3 Ubiquitin Ligase by a Calcium-Dependent Co-adaptor. Cell. 2016;167(2):525–538 e14. doi: 10.1016/j.cell.2016.09.026. [DOI] [PubMed] [Google Scholar]
- 115.Lin SS, et al. Cav3.2 T-type calcium channel is required for the NFAT-dependent Sox9 expression in tracheal cartilage. Proc Natl Acad Sci U S A. 2014;111(19):E1990–8. doi: 10.1073/pnas.1323112111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tomita M, et al. Calcineurin and NFAT4 induce chondrogenesis. J Biol Chem. 2002;277(44):42214–8. doi: 10.1074/jbc.C200504200. [DOI] [PubMed] [Google Scholar]
- 117.Karsenty G, et al. Genetic control of bone formation. Annu Rev Cell Dev Biol. 2009;25:629–48. doi: 10.1146/annurev.cellbio.042308.113308. [DOI] [PubMed] [Google Scholar]
- 118.Ito T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–50. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 119.Fischer ES, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lu G, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–9. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kronke J, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–5. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Han T, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017 doi: 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]