

Abstract
As sequencing technologies have evolved, the tools to analyze these sequences have made similar advances. However, for multi-species samples, we observed important and adverse differences in alignment specificity and computation time for bwa- mem (Burrows–Wheeler aligner-maximum exact matches) relative to bwa-aln. Therefore, we sought to optimize bwa-mem for alignment of data from multi-species samples in order to reduce alignment time and increase the specificity of alignments. In the multi-species cases examined, there was one majority member (i.e. Plasmodium falciparum or Brugia malayi) and one minority member (i.e. human or the Wolbachia endosymbiont wBm) of the sequence data. Increasing bwa-mem seed length from the default value reduced the number of read pairs from the majority sequence member that incorrectly aligned to the reference genome of the minority sequence member. Combining both source genomes into a single reference genome increased the specificity of mapping, while also reducing the central processing unit (CPU) time. In Plasmodium, at a seed length of 18 nt, 24.1 % of reads mapped to the human genome using 1.7±0.1 CPU hours, while 83.6 % of reads mapped to the Plasmodium genome using 0.2±0.0 CPU hours (total: 107.7 % reads mapping; in 1.9±0.1 CPU hours). In contrast, 97.1 % of the reads mapped to a combined Plasmodium–human reference in only 0.7±0.0 CPU hours. Overall, the results suggest that combining all references into a single reference database and using a 23 nt seed length reduces the computational time, while maximizing specificity. Similar results were found for simulated sequence reads from a mock metagenomic data set. We found similar improvements to computation time in a publicly available human-only data set.
Keywords: genome sequence alignment, BWA, dual-species alignment, Plasmodium, Brugia, Wolbachia
Abbreviations
BWA, Burrows–Wheeler aligner; CPU, central processing unit; LGT, lateral gene transfer; MEM, maximum exact matches; SRA, Sequence Read Archive; SW, Smith–Waterman Alignment.
Data Summary
The human dataset analyzed during the current study is available from the 1000 Genomes Project in the Sequence Read Archive (SRA) (http://www.internationalgenome.org, accession number ERR022446), the three Plasmodium–human datasets are available from the Wellcome Trust Sanger Institute in the SRA (accession numbers ERR015379 https://www.ncbi.nlm.nih.gov/sra/?term=ERR015379, ERR012739 https://www.ncbi.nlm.nih.gov/sra/?term=ERR012739, and ERR0153 60 https://www.ncbi.nlm.nih.gov/sra/?term=ERR015360), the Brugia–Wolbachia dataset is available from the University of Maryland Institute for Genome Sciences in the SRA (accession number SRR5188379 https://www.ncbi.nlm.nih.gov/sra/?term=SRR5188379) and the simulated data from the mock metagenome dataset is available from the University of Maryland Institute for Genome Sciences in Dryad doi:10.5061/dryad.m1m0p. Tables S1, S2, and S3 are available in the online Supplementary Material.
Impact Statement
As most organisms do not live in isolation, it can be informative to study combinations of organisms in co-culture. This has been greatly facilitated by ever improving sequencing data and tools. However, many of these tools were not developed for multi-species datasets, and it is not immediately apparent that optimization is needed or what optimization is needed. How multi-species datasets are handled varies even between generations of the same aligner, like the Burrows–Wheeler aligner (bwa)- aln and bwa- mem (maximum exact matches). In this case, optimization could be obtained with bwa- mem by using a combined reference, containing all of the genomes of the species present, and changing the seed stringency. There are many uses for aligners for multi-species datasets that will benefit from this optimization including: (a) use in dual-species RNASeq datasets where mis-mapping leads to inappropriate gene expression calculations, (b) use in identifying discordant read pairs that suggest horizontal/lateral gene transfer, and (c) use to cull datasets, for instance removing human reads from clinical pathogen data sets prior to data deposition. All of these cases will benefit from the increased specificity of the optimization here with little loss to sensitivity.
Introduction
One of the most popular tools for aligning genome sequencing reads to a reference genome is the Burrows–Wheeler aligner (bwa). The bwa software package allows users to align highly similar sequences to a large reference genome based on searches for exact matches with the Burrows–Wheeler transform [1]. There are three different bwa algorithms: bwa-aln (or bwa-backtrack), bwa-sw (Smith–Waterman Alignment) and bwa-mem (maximum exact matches). bwa-aln was the original bwa algorithm and was developed for 36–100 bp reads [1]. bwa-aln allows for gaps and mismatches, facilitating the identification of insertions and deletions [1]. When originally implemented, bwa-aln was 10–20× faster than the leading alignment tool, without sacrificing accuracy [1].
bwa-sw was originally used for mapping longer reads, like those originating from the 454 platform, but was updated for use with 200–1 000 000 bp sequencing reads [1]. As the length of sequencing reads increases, so does the error rate and the potential for structural variation within the reads. Therefore, alignment algorithms for longer reads must switch from global matches to local matches to accommodate this error rate and potential structural variation [1]. bwa also implemented a Smith–Waterman-like dynamic program to quickly align the query sequence to all suffix trees of the reference [1]. In addition to this seed-and-extend approach, heuristics were added to further increase the speed of bwa-sw [1]. However, as the amount of data generated by large-scale genomics projects increased and read length decreased, bwa-sw lacked the ability to analyze the data [1].
The most recent addition to the bwa suite is bwa-mem [2]. bwa-mem was implemented to increase the speed of aligning 100–1000 bp sequences to a large reference genome, while maintaining accuracy [2]. bwa-mem has the ability to perform chimeric alignments, can handle sequencing errors, and can choose between local or global/end-to-end alignments [2]. It is ideal for sequencing reads >70 bp and can handle reads up to 1 Mbp [2]. bwa-mem uses a seed-and-extend approach and extends the seed with an affine-gap Smith–Waterman algorithm, but also implements re-seeding to reduce incorrect mappings from a missing seed [2]. bwa-mem also employs heuristics that prevent it from extending an alignment through a poorly mapping area [2]. Another significant improvement of bwa-mem is that larger reference databases can be used. With bwa-aln, the complete reference index was limited to around the length of the human genome [1], but bwa-mem can now handle longer references [2]. bwa-mem is reported to be much faster than other aligners, and provides accurate alignments for both short and long reads [2].
Due to the speed and accuracy of bwa, it has been the aligner of choice for many different types of analyses from various organisms. bwa has been used for the analysis of all phases of the 1000 Genomes Project, studying multiple types of genetic variation in thousands of individuals across the globe [3–5]. In addition to studying this genetic variation, bwa has also been used to align cancer genome sequences to the human genome, including ovarian cancer [6], stomach cancer [7], prostate cancer [8], renal-cell cancers [9–11], cutaneous melanoma [12], head and neck squamous cell carcinoma [13], lung cancers [14, 15], papillary thyroid cancer [16], urothelial bladder carcinoma [17], acute myeloid leukemia [18], colon and rectal cancer [19], breast cancer [20], endometrial cancer [21], and glioblastoma [22].
While bwa was initially designed for single nucleotide polymorphism (SNP)-based analysis [2], it has also been used to identify structural variation. The Mobile Element Locator Tool (melt) [5, 23], which identifies mobile elements in eukaryotic sequencing samples, relies on bwa-generated alignments. melt was used in the 1000 Genomes Project to identify structural variants and LINE element insertion and deletion events in these samples [5, 23]. LGTSeek relies on bwa-aln to align read pairs to identify lateral gene transfers (LGTs) from a donor organism to a recipient organism [24]. LGTSeek can align sequencing reads to the recipient reference, then align the unmapped reads to the potential donor references, identifying pairs where one read maps to the host and the other read maps to the donor [24]. This approach relies on the speed and accuracy of bwa-aln to accurately identify these categories of reads.
While bwa-mem aligns the sequencing reads to the reference more quickly than bwa-aln [1], we have observed instances of low stringency/specificity of these alignments when using LGTSeek. This decreased specificity can result in the double mapping of sequencing reads to multiple references. For example, this was observed while examining dual-species transcriptome data [25] from Wolbachia endosymbionts and Drosophila ananassae, and was solved by combining the reference genomes. In this case, in preliminary work, bwa-aln and bwa-mem were both used for the alignments, but the counts were wildly different. More specifically, bwa-mem mapped bona fide Wolbachia sequencing reads to the D. ananassae reference genome, when only a very small portion of the read matched the Drosophila reference genome. This likely results from the fact that bwa was designed with the intent of mapping sequence data from a single organism to a single reference genome. However, as more tools use bwa for multi-species comparisons and more researchers use it in host–pathogen samples, these misalignments can significantly confound analyses.
Here, we sought to optimize bwa-mem in eukaryote/eukaryote and eukaryote/prokaryote dual-species datasets by altering various options to prevent incorrect mappings. We identified appreciable effects upon altering the seed length of bwa-mem alignments, which determines the minimum base pair length of an exact match, and by default is 19 nt. Since bwa-mem can handle larger reference databases, we also separately analyzed the effect of combining host–pathogen references into one larger database. Both methods decrease computational time, while increasing or maintaining specificity of the alignments. During this research, we also observed that the computational time spent on human genome sequence alignments, in general, could be decreased by increasing the seed length, which we attribute to the time spent attempting to align poor quality, off-target, non-human contaminant sequences to the reference genome.
Methods
Sample description
Human dataset
The 100 bp paired-end Illumina human dataset is available from the 1000 Genomes Project in the Sequence Read Archive (SRA) (http://www.internationalgenome.org, ERR0 22446). The data is from a Colombian male and the sample (HG01133) of B-lymphocytes was collected from the peripheral vein [5].
Plasmodium-human dataset
The 76 bp paired-end Illumina Plasmodium–human datasets are available from the Wellcome Trust Sanger Institute in the SRA (ERR015379, ERR012739 and ERR015360) from a study that extracted DNA from leukocyte-depleted blood samples or after short-term in vitro culture [26]. These samples (PK0055-C, PK0076-C and PK0044-C) all originated in Burkina Faso in 2008 [26].
Brugia– Wolbachia dataset
The 101 bp paired-end Brugia–Wolbachia dataset is available from the University of Maryland Institute for Genome Sciences in the SRA (SRR5188379) from a currently unpublished study designed to test DNA amplification methods on low input DNA samples. DNA was extracted from a population of Brugia malayi worms. Qiagen Repli-G (Qiagen) was used to amplify 2 ng genomic DNA with tetramethylammonium chloride (TMAC)[27]. A genomic DNA library was constructed for sequencing on the Illumina platform using the KAPA Hyper Prep kit (Kapa Biosystems) according to the manufacturer’s protocol. DNA was fragmented with the Covaris E210. The DNA was purified between enzymatic reactions and the size selection of the library was performed with SPRI-select beads (Beckman Coulter Genomics). The PCR amplification step was performed with primers containing a 7 nt index sequence. The indexed libraries were pooled and sequenced on a on a Hiseq2500 sequencer (Illumina).
Simulated metagenomic dataset
A dataset of 101 bp paired-end sequencing reads from a mock metagenomic community was simulated with art, version 2.3.7 28]. An even 8× sequencing depth with a HiSeq2500 error profile, a minimum fragment length of 300 bp and sd of 105 bp was generated across the reference genomes: Acinetobacter baumanii ATCC17978; Actinomyces odontolyticus ATCC17982; Bacillus cereus ATCC10987; Bacteroides vulgatus ATCC8482; Clostridium beijerinckii ATCC51743; Deinoccous radiodurans DSM20539; Enterococcus faecalis ATCC47077; Escherichia coli ATCC700926; Helicobacter pylori ATCC700392; Lactobacillus gasseri DSM 20243; Listeria monocytogenes ATCCBAA-679; Methanobrevibacter smithii ATCC35061; Neisseria meningitidis ATCCBAA-335; Propionibacterium acnes DSM16379; Pseudomonas aeruginosa ATCC47085; Rhodobacter sphaeroides ATCC17023; Staphylococcus aureus ATCCBAA-1718; Staphylocococcus epidermidis ATCC12228; Streptococcus agalactiae ATCCBAA-611; Streptococcus mutans ATCC7 00610; and Streptococcus pneumoniae ATCCBAA-334.
bwa alignments
All alignments were written as sam output files using bwa v. 0.7.12. The qsub -m function, as implemented in GE 6.2u5 lx24-amd64 dated December 1, 2009, was used to generate a summary report from each experiment including the central processing unit (CPU) time for each script that was executed. To reduce variation in CPU times, all jobs were executed on a single multi-core node ensuring that <40 % of the total number of cores and <75 % of total memory were used. Each job was executed in triplicate, and no replicate jobs were running simultaneously. For bwa-aln/sampe alignments, the script contained aln commands for each fastq file executed sequentially followed by the sampe command, which were all executed with the default parameters. For bwa-mem alignments, the script contained a single command to align the sequencing read pairs to their respective references; default parameters were used except where seed length was altered. The Ensembl GRCh37 human reference containing the decoy sequence was used for alignments to the human genome. The B. malayi genome [29] version 4 was obtained from WormBase (www.wormbase.org) and was used as the combined reference for Brugia and Wolbachia. The Wolbachia wBm genome [30] reference was obtained by pulling the Bm_006 contig from the combined reference and the Brugia reference was made by using all contigs except Bm_006. The Plasmodium reference used was version 3.0 of high quality Plasmodium falciparum 3D7 draft genome released by PlasmoDB-9.3 [31]. The combined human–Plasmodium reference was created by adding the Plasmodium contigs to the GRCh37 fasta file. samtools flagstat v.1.1 was used to determine the number of total reads, secondary reads, and mapped reads in each sample. To correct for the number of secondary reads, the percentage of reads mapping was calculated as (mapped reads – secondary reads)/(paired in sequencing reads) ×100.
Results
Datasets
Four types of datasets were used for this study: eukaryote/prokaryote, eukaryote/eukaryote, eukaryote only and simulated metagenomic. The first dataset contained 101 bp paired-end Illumina sequencing reads from a population of B. malayi worms and their Wolbachia endosymbiont strain wBm from an unpublished study examining DNA amplification methods; it will be referred to as the Brugia– Wolbachia dataset (Table 1). Using the Perl 5.8.8 rand() and srand() functions, 250 000 first reads were randomly selected and the taxonomy of best hits for these reads was generated with a mega-blast (v. 2.2.17) search against the NCBI nucleotide database NT. We typically find that in mega-blast searches of this nature that some percentage of reads have no match, and that the best criterion to use is the percentage of reads with a best match relative to the number of reads that have any match, and this criterion was used in all other instances. Based on this criterion, this dataset was estimated to contain 79.7 % reads with matches having best matches to B. malayi and 14.5 % reads with best matches to Wolbachia wBm. However, the version of NT used for these searches does not have the B. malayi genome, so most reads did not have matches, making this measurement skewed in this case. If we instead focus on all reads, 14.1 % of reads had a best match to B. malayi and 2.6 % of reads had a best match to Wolbachia wBm, which is likely a better reflection of the composition of this data.
Table 1. Total number of reads for each dataset.
The datasets are listed that were used in this study, with the total number of reads.
| Dataset | Accession no. | Total no. of reads |
|---|---|---|
| Brugia–Wolbachia | SRR5188379 | 31 494 916 |
| Plasmodium–human | ERR015379 | 6 051 406 |
| Plasmodium–human | ERR012739 | 8 080 550 |
| Plasmodium–human | ERR015360 | 25 867 194 |
| 1000 Genomes | ERR022446 | 219 493 146 |
| Simulated metagenome | Dryad acc. (doi:10.5061/dryad.m1m0p) | 5 490 726 |
The second dataset that we initially focused on contained 76 bp paired-end Illumina sequence reads from P. falciparum sequenced from a human sample from the SRA (ERR015379) [26]; it will be referred to as the Plasmodium–human dataset (Table 1). Parsing the taxonomy of best hits of 250 000 randomly selected first reads generated with a mega-blast search against NT, this dataset was estimated to contain 87.5 % reads with best matches to P. falciparum reads and 7.4 % reads with best matches to human sequences.
The third dataset contained 101 bp paired-end Illumina sequence reads that have been simulated from a mock metagenomic community (Dryad doi:10.5061/dryad.m1m0p); it will be referred to as the mock metagenomic dataset (Table 1). The final dataset examined was 100 bp Illumina paired-end human sequencing reads from the 1000 Genomes Project (ERR022446) [5]; it will be referred to as the 1000 Genomes dataset (Table 1). Parsing the taxonomy of best hits of 250 000 randomly selected first reads generated with a mega-blast search against NT, this dataset was estimated to contain 94.7 % of reads with matches having best matches to human sequences.
Percentage of mapped read pairs using different seed lengths with bwa-mem
bwa-mem was used to align three different whole genome sequencing datasets to the respective reference databases with varying seed lengths from 18 to 30 nt. In the two dual-species datasets, the sequencing reads were aligned to the reference genome of each species separately, as well as a combined reference of both genomes. The 1000 Genomes dataset was only aligned to the human reference genome. The lowest variation in percentage of reads mapped with bwa-mem was observed in the 1000 Genomes dataset, where the total percentage of reads mapped only varied by 0.49 % across all seed lengths tested (Fig. 1, Table 2).
Fig. 1.

The percentage of mapped read pairs for all bwa-mem datasets. The percentage of mapped read pairs for all datasets against all references is shown for each seed length for (a) the human–Plasmodium dataset, (b) the Brugia–Wolbachia dataset and (c) the human-only dataset. The sequencing reads were aligned to the reference genome of each species separately, as well as a combined reference of both genomes, as indicated in the legend. Mappings to the reference genome of the minority member in the sample (human for the Plasmodium–human dataset and Wolbachia for the Brugia–Wolbachia dataset) were plotted on the secondary y-axis on the right, while all others were plotted on the primary y-axis on the left. The sum of the mappings to individual reference genomes is illustrated with a dashed line to enable comparisons.
Table 2. The percentage of read pairs that aligned to each reference.
The percentage is provided of read pairs from a subset of datasets that aligned to each reference for bwa-aln with default parameters and bwa-mem with seed lengths from 18 to 30 nt.
| Dataset | Reference | aln | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasmodium–human | Human | 10.2 | 24.1 | 22.4 | 21.2 | 20.4 | 19.9 | 19.6 | 19.4 | 19.2 | 19.1 | 19.0 | 18.8 | 18.7 | 18.5 |
| Plasmodium–human | Plasmodium | 78.1 | 83.6 | 83.3 | 83.1 | 82.9 | 82.8 | 82.6 | 82.4 | 82.3 | 83.6 | 83.3 | 83.1 | 82.9 | 82.8 |
| Plasmodium–human | Combined reference | 88.3 | 97.1 | 96.9 | 96.7 | 96.5 | 96.3 | 96.1 | 95.9 | 95.7 | 95.5 | 95.3 | 95.1 | 94.9 | 94.6 |
| Brugia–Wolbachia | Brugia | 91.9 | 94.4 | 94.0 | 93.8 | 93.8 | 93.7 | 93.7 | 93.6 | 93.6 | 93.6 | 93.6 | 93.5 | 93.5 | 93.5 |
| Brugia–Wolbachia | Wolbachia | 2.7 | 3.3 | 3.0 | 2.9 | 2.9 | 2.9 | 2.9 | 2.9 | 2.9 | 2.9 | 2.8 | 2.8 | 2.8 | 2.8 |
| Brugia–Wolbachia | Combined reference | 94.5 | 96.4 | 96.2 | 96.1 | 96.1 | 96.1 | 96.1 | 96.0 | 96.0 | 96.0 | 96.0 | 96.0 | 96.0 | 96.0 |
| 1000 Genomes | Human | 95.9 | 99.7 | 99.6 | 99.6 | 99.6 | 99.6 | 99.5 | 99.5 | 99.4 | 99.4 | 99.4 | 99.3 | 99.2 | 99.2 |
In the Brugia– Wolbachia dataset, reads mapping to the reference genome of the minority member (Wolbachia) decreased markedly from 3.3 to 2.9 % when increasing the seed length from 18 to 20 nt (Fig. 1, Table 2). While this is only a 0.4 % difference, it is important to consider it is >10 % of the reads that mapped to this genome. This problem of read pairs aligning to both references was resolved, meaning they were mapped to the best position, by combining the Brugia and Wolbachia references into a single reference sequence (Fig. 1, Table 2). The percentage of reads that mapped to Brugia only varied by 0.87 % across all seed lengths (Fig. 1, Table 2). However, the extensive amount of recent LGT to B. malayi from its Wolbachia endosymbiont [32] confounds this analysis such that we sought to evaluate this phenomenon with several subsequent datasets.
In the Plasmodium–human dataset, the aggregate number of read pairs aligning to each reference was >100 % until the seed length was set to >29 nt, when mapping drastically declined due to a likely over-stringent seed length. The aggregate number of read pairs decreased from 107.8 to 102.2 % when the seed length was increased from 18 to 23 nt (Fig. 1, Table 2). This problem of read pairs aligning to both references was resolved by combining the human and Plasmodium reference into one file (Fig. 1, Table 2). For all seed lengths mapped against this combined reference, the Plasmodium–human dataset consistently had 94.6–97.1 % of read pairs aligning (Fig. 1, Table 2). Aligning the Plasmodium–human data to the Plasmodium reference or the Plasmodium–human combined reference led to a consistent percentage of read pairs mapping across seed length up to 30 nt, indicating that increasing the seed length did not dramatically decrease the number of read pairs that were aligned for these seed lengths (Fig. 1, Table 2). Increasing the seed length beyond 30 nt led to a substantial decrease in the number of reads aligned, as expected (Table S1, available in the online Supplementary Material), which was only tested with this dataset.
Two further Plasmodium–human datasets were tested, ERR012739 and ERR015360, to ensure these observations were not limited to the original dataset selected. Parsing the taxonomy of best hits of 250 000 randomly selected first reads generated with a mega-blast search against NT, ERR012739 was estimated to contain 73 % Plasmodium reads and 18 % human reads, while ERR015360 was estimated to contain 60 % Plasmodium reads and 3 % human reads. All three datasets showed the same trends as the original dataset examined (Fig. 2), namely over-mapping of reads when mapped to separate references that could be largely eliminated by mapping to an aggregate reference and/or increasing the seed length to 23 nt.
Fig. 2.
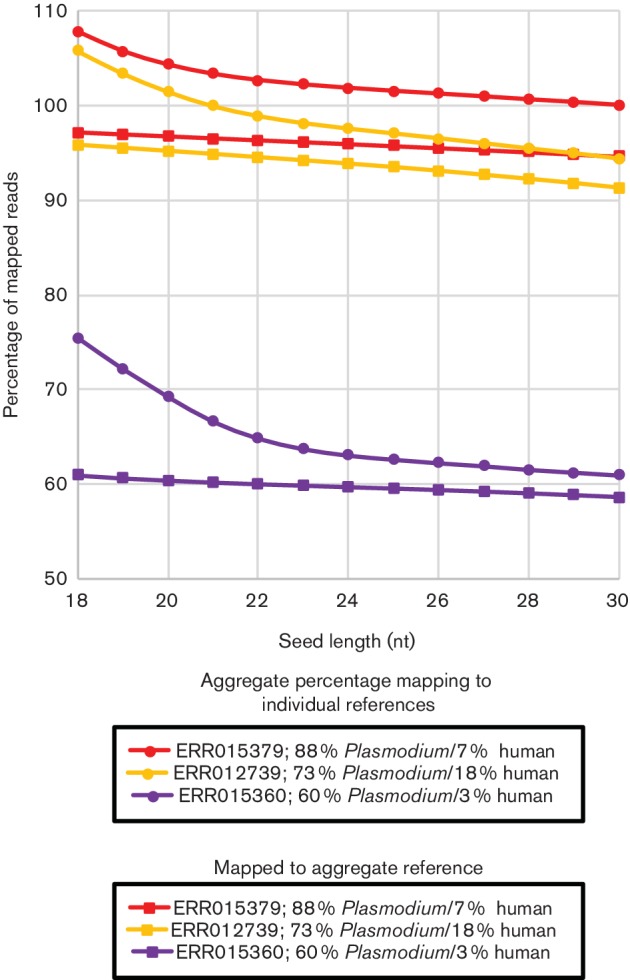
Percentage of mapped read pairs for different Plasmodium–human datasets. The percentage of mapped read pairs for all datasets against all references is shown for each seed length for three Plasmodium datasets containing varying levels of human sequence. ERR015379 is estimated to contain 88 % reads with best matches to P. falciparum reads and 7 % reads with best matches to human sequences. ERR012739 was estimated to contain 73 % Plasmodium reads and 18 % human reads. ERR015360 was estimated to contain 60 % Plasmodium reads and 3 % human reads. All three datasets showed the same trends as the original dataset examined, namely over-mapping of reads when mapped to separate references that could be largely eliminated by mapping to an aggregate reference and/or increasing the seed length to 23 nt.
Similar over-mapping of reads was again observed with a simulated dataset of a 21-member mock metagenomic community (Fig. 3). When mapped to the composite reference, 100 % of the reads mapped, as expected for a simulated dataset. However, even at a seed length of 30 nt, the aggregate percentage of reads mapping to each individual reference was still >104 % (Fig. 3), which may be due in part to multi-mapping of reads to the many highly similar rRNAs. Mapping to an aggregate reference and/or increasing the seed length to 23 nt improved the mapping specificity (Fig. 3). However, it is less clear that 23 nt was the ideal seed length as substantial improvements could be observed beyond a 23 nt seed length (Fig. 3). This difference may be due to using simulated sequence data, the high degree of similarity in regions of the genomes like the rRNA, or the complexity of the population. We investigated using real sequencing data from a mock community sequenced by the human microbiome project [33, 34], but only single end reads were available.
Fig. 3.

The difference in the percentage of mapped reads from that expected for simulated reads from a mock metagenomic community. Because of differences in the genome size, the percentage of reads mapped varied extensively, such that the difference between the observed percentage of mapped read pairs and the known percentage of mapped read pairs was interrogated for the simulated data from the mock metagenomic community. This value is shown for each seed length for a simulated dataset of 101 bp paired-end sequencing reads from a mock metagenomic community. Given that the data is simulated, the known percentage of mapped reads was calculated from the 8× sequencing depth and the relative fraction the genome contributes to the population for each organism. The aggregate percentage difference in mapping was plotted on the secondary y-axis on the right, while all individual percentage differences were plotted on the primary y-axis on the left.
CPU times and trends
The CPU time of the alignments was also interrogated, by measuring in triplicate using a single grid node with 48 CPUs and 95.6 GB memory, controlling for variation by limiting CPU usage to <40 % of the maximum and memory usage to <75 % of the maximum (Fig. 4). We could not control for other factors like caching and network traffic, but saw little variation in CPU time across replicates, which were not run simultaneously on the node. The alignment of the reads to the combined Brugia–Wolbachia reference database took similar CPU time relative to aligning to only the Brugia reference (Fig. 4, Table 3) demonstrating the benefit of using a combined reference when possible.
Fig. 4.

CPU time for all bwa-mem datasets. The mean and sd of CPU time in hours for each of three replicate alignments is plotted against each seed length for a subset of datasets. In the two dual-species data sets, the sequencing reads were aligned to the reference genome of each species separately, as well as a combined reference of both genomes. The 1000 Genomes dataset was only aligned to the human reference genome.
Table 3. The CPU time in hours for each replicate for a subset of datasets and references.
The CPU time in hours is provided for a subset of datasets for bwa-aln with default parameters and bwa-mem with 18–30 nt seed lengths. For bwa-aln, the CPU time reported is the sum of the CPU time for aligning read 1, the CPU time for aligning read 2 and the CPU time for running the sampe algorithm on those two alignments.
| aln | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasmodium | 0.3±0.1 | 0.2±0 | 0.2±0 | 0.2±0 | 0.2±0 | 0.2±0 | 0.2±0 | 0.2±0 | 0.2±0 | 0.2±0 | 0.2±0 | 0.1±0 | 0.1±0 | 0.1±0 |
| Human | 0.9±0.1 | 1.7±0.1 | 1.1±0.1 | 0.8±0 | 0.7±0 | 0.6±0 | 0.5±0 | 0.5±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.3±0 | 0.3±0 |
| Human–Plasmodium | 1±0.1 | 0.7±0 | 0.6±0 | 0.6±0 | 0.5±0 | 0.5±0 | 0.5±0 | 0.5±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.3±0 | 0.3±0 |
| 1000 Genomes | 34.1±1 | 21.1±1.7 | 19.2±1.9 | 17.7±1.6 | 17.8±1.5 | 17.3±1.4 | 16.8±1 | 15.6±1.8 | 15.7±1.1 | 15.5±1 | 15.1±1.6 | 14.2±0.1 | 13.3±1.2 | 12.8±1.1 |
| Brugia | 1.3±0 | 1.1±0 | 1±0 | 0.9±0 | 0.9±0 | 0.9±0 | 0.8±0 | 0.8±0 | 0.8±0 | 0.8±0 | 0.7±0 | 0.8±0 | 0.8±0 | 0.7±0 |
| Wolbachia | 0.1±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 | 0.4±0 |
| Brugia– Wolbachia | 1.3±0 | 1.1±0.1 | 1±0 | 0.9±0.1 | 0.9±0 | 0.9±0 | 0.8±0 | 0.8±0 | 0.8±0 | 0.8±0 | 0.8±0 | 0.7±0 | 0.7±0 | 0.7±0 |
Alignments to the human reference genome had the most dramatic decrease in CPU time with increasing seed lengths for both the human–Plasmodium dataset and the 1000 Genomes dataset (Fig. 4). In the case of the 1000 Genomes dataset, the CPU time at a seed length of 18 nt was 21.1±1.7 h (Table 3). Increasing the seed length to 23 nt reduced the CPU time to 16.8±1.0 h, resulting in a 1.3-fold reduction or 4.3 h decrease in CPU time (Fig. 4, Table 3).
The Plasmodium–human dataset showed even more substantial improvements when aligning to the combined dataset, but most substantially between 18 and 20 nt seed lengths. For example, at a seed length of 18 nt, the alignment of this dataset to the human reference took 1.7±0.1 h, while the alignment to the combined reference only took 0.7±0 h, a 2.4-fold decrease (Table 3, Fig. 4). The seed length also plays a role as observed by the decrease in CPU hours with increasing seed length up to ~23 nt. An increase in seed length to 23 nt reduced the Plasmodium–human CPU time 3.4-fold in the alignments to just the human reference (Fig. 4, Table 3). Overall, improvements to the CPU time for both host–pathogen datasets plateaued by seed length of 23 nt (Fig. 4, Table 3). The decreasing CPU time needed as a function of seed length, combined with the over-mapping of reads against a single reference compared to a combined reference, suggests that using bwa-mem to generate poor alignments to the incorrect reference can lead to excessive use of computational resources. Increasing the seed length beyond 30 nt led to a sizable decrease in the CPU hours (Table S2), but this is likely a result of the undesirable decrease in the number of reads aligned (Table S1).
Comparing bwa-mem to bwa-aln
We also sought to compare the percentage of mapped read pairs aligned with bwa-aln and bwa-mem. We expected that bwa-mem would map more reads, since it could map reads with more sequencing errors, as well as greater sequence variation. Consistent with these expectations, in all cases tested, bwa-aln mapped a lower percentage of reads relative to bwa-mem (Table 2). Also, as expected, given the underlying algorithm, bwa-mem computed the alignments faster than bwa-aln when the majority of reads map to the reference genome (i.e. Plasmodium–human against the Plasmodium genome, Plasmodium–human against the combined reference, Brugia–Wolbachia against the Brugia genome, Brugia–Wolbachia against the combined reference, and 1000 Genomes against the human genome). Conversely, we anticipated that when the vast majority of reads did not map to the reference, bwa-mem would spend large amounts of compute time trying to incorrectly align them to the genome (i.e. Plasmodium–human against the human genome and Brugia– Wolbachia against the Wolbachia genome). As expected, these latter two cases were aligned more quickly with bwa-aln than bwa-mem at the lower seed lengths tested (Table 3).
The increase in the percentage of reads mapping with bwa-mem at a seed length of 18 nt led to an increase in both the sequencing read depths, as well as the coverage. There are 12 127 496 positions in the human genome that had 1× sequencing depth with a seed length of 18 nt using bwa-mem with a maximum sequencing depth of 7990× with the default parameters of samtools mpileup, which would discard positions with >8000× sequencing depth. In contrast, 5 378 722 positions in the human genome had 1× sequencing depth with bwa-aln/sampe with a maximum sequencing depth 919×. Therefore, there was >8-fold higher maximum sequencing depth and >2-fold more positions with coverage.
The Plasmodium reads mapping to the human genome with a seed length of 18 nt were not evenly distributed throughout the human genome. While reads mapped to all of the chromosomes evenly, three non-chromosomal fasta entries had reads that were significantly over-represented relative to the length of the sequence (χ2, P<0.00005), including the unplaced genomic contig GL000220.1 with 69 056 reads in 161 802 bp, the unplaced genomic contig GL000226.1 with 16 735 reads in 15 008 bp, and the decoy sequence contig hs37d5 with 569 969 reads in 35 477 943 bp.
We further wanted to examine whether high-quality matches of Plasmodium reads to the human genome reference enabled alignment of related poorly mapping reads through any of bwa’s processes like chaining, filtering chaining, and seed extension. We used the 10.2 % of Plasmodium reads aligning to the human reference with bwa-aln as our proxy for high-quality matches. Those reads and their mates were removed from the fastq files and these modified human– Plasmodium fastq files were realigned to the human genome using bwa-mem for all seed lengths tested. We found that the percentage of reads mapping to the human reference with bwa-mem was the same from both the filtered fastq files and the initial fastq files after accounting for the removal of the 10.2 % of the reads that correctly aligned to Plasmodium (Table S3).
There was no substantial difference between mapping to the Plasmodium genome with bwa-aln/sampe and bwa-mem. There were 16 995 093 Plasmodium positions with >5× coverage in the bwa-mem alignment with a seed length of 18 nt, while there were 16 698 169 Plasmodium positions with >5× coverage in the bwa- aln/sampe alignment. When modified fastq files were made by removing reads identified as human using bwa-aln/sampe and those modified files were mapped with bwa-aln/sampe back to the Plasmodium genome, 16 698 614 Plasmodium positions still had >5× coverage, which is remarkably similar to the 16 698 169 Plasmodium positions with >5× coverage when mapping all reads to the Plasmodium genome without filtering. However, when modified fastq files were made after removing reads identified as human using bwa-mem and those modified files were mapped with bwa-aln/sampe back to the Plasmodium genome, only 14 214 800 Plasmodium positions still had >5× coverage, which is only 85 % of the positions with coverage in the Plasmodium genome using all reads and bwa- aln/sampe. Therefore, the excess removal of reads based on their alignment with bwa-mem at a seed length of 18 nt may result in a loss of coverage of ~2 000 000 Plasmodium positions that could lead to an inability to call SNPs and other important genetic variants.
Discussion
Effects on multi-species genome sequencing datasets
New algorithms are making remarkable advances to expand the complexity of alignments to best capture all the genomic variation in a single organism [1, 2, 35–38]. These improved alignments are essential for better identification of SNPs, copy number variants and instances of structural variation. However, there is the potential for problems to arise when these algorithms are applied in untested scenarios, like multi-species sequencing projects. In these cases, one of the vital steps is properly attributing the reads to each organism.
Numerous studies have reported transcriptomics-based gene expression differences of two organisms within a single sample [39–50]. For the gene expression differences to be accurately obtained, the reads from both organisms need to map uniquely to the appropriate reference. The results presented here for Brugia– Wolbachia and Plasmodium–human read mappings suggests this may not occur all the time. Plasmodium reads map to the human genome and Brugia reads map to the Wolbachia genome, which could result in transcripts appearing to be transcribed that are not. It also could result in erroneous assessment of the level of differential gene expression.
This may also present problems in mapping-based metagenomics analyses, and our results suggest that using a combined reference and increasing the seed length may lead to improvements. The exact seed length is not clear, and further work using real sequencing data from mock communities, as opposed to simulated data, may be necessary to resolve the matter.
In parasite and pathogen sequencing projects that require the direct isolation of nucleic acids from a human patient, sequencing reads are often screened prior to deposition in public repositories, like the SRA [51], to remove human sequences. Theoretically, mapping-based algorithms, like bwa, provide fast and efficient mechanisms for removing such human reads from parasite and pathogen sequencing projects. However, the results presented here suggest that too many sequences may be erroneously targeted for removal. Specifically, only 85 % of the positions have coverage in the Plasmodium genome after removing ‘human’ reads with bwa-mem with a seed length of 18 nt relative to removing ‘human’ reads with bwa- aln/sampe.
Lastly, pipelines like PathSeq [52] and LGTSeek [24] use aligners to identify the putative provenance of reads, assigning a taxonomy for reads and/or read pairs. PathSeq identifies microbial reads in samples, in order to identify novel microbe–disease associations [52]. LGTSeek identifies read pairs with discordant taxonomy, like bacteria-eukaryote read pairs in animal and human genomes that may indicate bacteria-animal LGT[24]. In this case, a stringent mapping assignment is needed in order to assign the most appropriate taxonomy. The results from both the Brugia– Wolbachia and the Plasmodium–human datasets suggest that this may not be the case with bwa-mem without further optimization. While we focus on differences between bwa-aln/sampe and bwa-mem, it is important that any aligner be thoroughly tested when used in these scenarios for the particular organisms under study.
Solutions
Given the results presented here, one solution that should be employed whenever possible is to include the genomes of all expected organisms in a single reference database. Doing so increased the specificity of the alignments and decreased computation times. This strategy has already been used in human genome sequencing projects that include the Broad Institute’s ‘decoy’ genome sequence in their reference genome. The ‘decoy’ genome sequence contains sequences frequently encountered in human whole-genome sequencing projects that are otherwise not in the reference [5].
In some studies, however, not all reference organisms are known. For example, when running algorithms like PathSeq [52] or LGTSeek [24] to identify instances of ‘foreign’ DNA, the potential sources are unknown and are the focus of the study. Additionally, if a sequencing library or run is contaminated, the researcher may not be aware of the presence of sequence reads from additional organisms that could be leading to an increase in computation times. In these cases, the specificity would be improved and the CPU times decreased by increasing the seed length. In this study, a seed length of 23 nt was ideal since lower seed lengths increased computation times and off-target mappings, while higher seed lengths could reduce sensitivity with no improvement to computation times or off-target mappings. It is not clear if this is the universal ideal seed length that would be applicable for all scenarios. The ideal seed length may be a function of the evolutionary distances and similarity between the organisms being sequenced concomitantly. However, we have demonstrated that the use of simulated datasets constructed from the reference datasets may be useful in informing these decisions and identifying what further experimental controls may be needed.
While much of our analysis focuses on a seed length of 18 nt, it is important to remember that the default seed length for bwa-mem is 19 nt. We initially identified this problem with a seed length of 19 nt; however, it is much more apparent and easier to interrogate at a seed length of 18 nt. Lastly, there may be scenarios that warrant the use of legacy aligners, like bwa- aln, which may be less sensitive, but have a higher degree of specificity.
Computational resource optimization
Analysis of genomic data can be computationally intensive, leading to long CPU times and high analysis costs. Our analysis suggests that almost all datasets can benefit from an increase in the seed length to reduce CPU time and associated costs. However, this decrease in computation time may affect the ability to identify ‘split reads’, where a read matches two different locations in the genome, suggesting structural variation in genomics projects and splicing in transcriptomics projects. As such, further work may be needed by specialists in a field (e.g. structural genomic scientists and annotation specialists) to demonstrate how changing the seed length may or may not affect the validity of structural variation and gene structure predictions. Split reads in RNASeq projects of dual-species projects need to be handled with care, balancing the sensitivity of detecting splice junctions with the specificity of correct mapping. At a minimum, this can be dealt with in multi-species datasets by aligning to a combined reference that contains all known members of the consortium sequenced.
Conclusion
As sequencing technologies continue to advance and sequencing becomes less expensive, the tools to analyze these sequences need to be optimized for the intended study. Here, bwa-mem was optimized to reduce alignment time and increase the specificity of alignments. While this optimization might not be necessary for all datasets, we found it to be particularly helpful in sequencing data obtained simultaneously from multiple organisms. In these cases, there was one dominant sequence source (i.e. Plasmodium or Brugia) and one minority sequence source (i.e. human or Wolbachia). Increasing the seed length reduced the number of read pairs aligning, incorrectly, to the reference genome from the minority sequence source (human or Wolbachia). Overall, increasing the seed length to 23 nt and combining both organisms into one reference genome can reduce the CPU time, sometimes quite substantially, with more specificity in the mapping, as was the case with the Plasmodium–human dataset. In some situations, the use of legacy aligners may also be warranted. Less substantial, but still possibly significant, improvements to CPU time upon changing the seed length were found in all cases. This suggests using a 23 nt seed length by default may be desirable.
Data bibliography
1000 Genomes Project. Wellcome Trust Sanger Institute. Sequence Read Archive – ERR022446 (2017).
Wellcome Trust Sanger Institute. Sequence Read Archive – ERR015379 (2017).
Wellcome Trust Sanger Institute. Sequence Read Archive – ERR012739 (2017).
Wellcome Trust Sanger Institute. Sequence Read Archive – ERR015360 (2017).
University of Maryland Institute for Genome Sciences. Sequence Read Archive – SRR5188379 (2017).
University of Maryland Institute for Genome Sciences. Dryad – doi:10.5061/dryad.m1m0p (2017).
Funding information
This work was funded by the National Institute of Allergy and Infectious Diseases U19AI110820 to CMF, DAR, JCS, and JCDH (U19AI110820), the National Institutes of Health through an NIH Director’s Transformative Research Award (1-R01-CA206188) to JCDH, and the National Science Foundation Advances in Biological Informatics (ABI-1457957) to JCDH.
Acknowledgements
The results published here are in part based upon data generated by the 1000 Genomes Project. Information about the 1000 Genomes Project and the International Genome Sample Resource that has been continuing to support the 1000 Genomes Project can be found at “http://www.internationalgenome.org/”. We would like to thank Dr. Jeremy Foster and Dr. Silvia Libro at New England Biolabs for kindly providing the Brugia malayi DNA used for sequencing.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
References
- 1.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013;3:13033997. [Google Scholar]
- 3.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–145. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akbani R, Akdemir Kadir C, Aksoy BA, Albert M, Ally A, et al. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agrawal N, Akbani R, Aksoy BA, Ally A, Arachchi H, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riley DR, Sieber KB, Robinson KM, White JR, Ganesan A, et al. Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples. PLoS Comput Biol. 2013;9:e1003107. doi: 10.1371/journal.pcbi.1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar N, Lin M, Zhao X, Ott S, Santana-Cruz I, et al. Efficient enrichment of bacterial mRNA from host-bacteria total RNA samples. Sci Rep. 2016;6:34850. doi: 10.1038/srep34850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manske M, Miotto O, Campino S, Auburn S, Almagro-Garcia J, et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. 2012;487:375–379. doi: 10.1038/nature11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oyola SO, Manske M, Campino S, Claessens A, Hamilton WL, et al. Optimized whole-genome amplification strategy for extremely AT-biased template. DNA Res. 2014;21:661–671. doi: 10.1093/dnares/dsu028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics. 2012;28:593–594. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, et al. Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007;317:1756–1760. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster J, Ganatra M, Kamal I, Ware J, Makarova K, et al. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005;3:e121. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner MJ, Hall N, Fung E, White O, Berriman M, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ioannidis P, Johnston KL, Riley DR, Kumar N, White JR, et al. Extensively duplicated and transcriptionally active recent lateral gene transfer from a bacterial Wolbachia endosymbiont to its host filarial nematode Brugia malayi. BMC Genomics. 2013;14:639. doi: 10.1186/1471-2164-14-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Human Microbiome Project Consortium A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. NatMeth. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee WP, Stromberg MP, Ward A, Stewart C, Garrison EP, et al. MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS One. 2014;9:e90581. doi: 10.1371/journal.pone.0090581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphrys MS, Creasy T, Sun Y, Shetty AC, Chibucos MC, et al. Simultaneous transcriptional profiling of bacteria and their host cells. PLoS One. 2013;8:e80597. doi: 10.1371/journal.pone.0080597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vannucci FA, Foster DN, Gebhart CJ. Laser microdissection coupled with RNA-seq analysis of porcine enterocytes infected with an obligate intracellular pathogen (Lawsonia intracellularis) BMC Genomics. 2013;14:421. doi: 10.1186/1471-2164-14-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rienksma RA, Suarez-Diez M, Mollenkopf HJ, Dolganov GM, Dorhoi A, et al. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genomics. 2015;16:34. doi: 10.1186/s12864-014-1197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avraham R, Haseley N, Brown D, Penaranda C, Jijon HB, et al. Pathogen cell-to-cell variability drives heterogeneity in host immune responses. Cell. 2015;162:1309–1321. doi: 10.1016/j.cell.2015.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mavromatis CH, Bokil NJ, Totsika M, Kakkanat A, Schaale K, et al. The co-transcriptome of uropathogenic Escherichia coli-infected mouse macrophages reveals new insights into host-pathogen interactions. Cell Microbiol. 2015;17:730–746. doi: 10.1111/cmi.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aprianto R, Slager J, Holsappel S, Veening JW. Time-resolved dual RNA-seq reveals extensive rewiring of lung epithelial and pneumococcal transcriptomes during early infection. Genome Biol. 2016;17:198. doi: 10.1186/s13059-016-1054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westermann AJ, Förstner KU, Amman F, Barquist L, Chao Y, et al. Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature. 2016;529:496–501. doi: 10.1038/nature16547. [DOI] [PubMed] [Google Scholar]
- 46.Baddal B, Muzzi A, Censini S, Calogero RA, Torricelli G, et al. Dual RNA-seq of nontypeable Haemophilus influenzae and host cell transcriptomes reveals novel insights into host-pathogen cross talk. MBio. 2015;6:e01765-15. doi: 10.1128/mBio.01765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tierney L, Linde J, Müller S, Brunke S, Molina JC, et al. An interspecies regulatory Network inferred from simultaneous RNA-seq of Candida albicans invading innate immune cells. Front Microbiol. 2012;3:85. doi: 10.3389/fmicb.2012.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chibucos MC, Soliman S, Gebremariam T, Lee H, Daugherty S, et al. An integrated genomic and transcriptomic survey of mucormycosis-causing fungi. Nat Commun. 2016;7:12218. doi: 10.1038/ncomms12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bruno VM, Shetty AC, Yano J, Fidel PL, Noverr MC, et al. Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the NLRP3 inflammasome. MBio. 2015;6:e00182-15. doi: 10.1128/mBio.00182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Shetty AC, Schwartz JA, Bradford LL, Xu W, et al. New signaling pathways govern the host response to C. albicans infection in various niches. Genome Res. 2015;25:679–689. doi: 10.1101/gr.187427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.National Center for Biotechnology Information Sequence Read Archive Overview [updated January 10 2017] 2017. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=announcement
- 52.Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, et al. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.