

Abstract
The etiology and pathogenesis of rheumatoid arthritis (RA) are influenced by environmental and genetic risk factors. Shared epitope-coding HLA-DRB1 alleles increase RA risk and severity; however, the underlying mechanisms of action remain unclear. In contrast, several other DRB1 alleles protect against RA. Additionally, genome-wide association studies suggest that RA associates with other, HLA and non-HLA, genes but the relative contributions of such risk loci to RA are incompletely understood. Future research challenges include integrating the epidemiological and genomic data into validated arthritogenic pathways, and determining the mechanisms of interaction between RA risk genes and environmental influences.
Keywords: HLA, rheumatoid arthritis, autoimmunity, shared epitope
Introduction
Rheumatoid arthritis (RA) is a common inflammatory disease in which both genetic and environmental factors play a role in disease development. Based on twin studies, the heritability of the disease was estimated at around 60%.1 Among all the genetic risk factors found to date, the human leukocyte antigen (HLA) locus is the most significant one. A particularly strong association between RA and HLA-DRB1 alleles that encode a HLA-DRβ chain containing a five amino acid sequence motif called the ‘shared epitope’ (SE) has long been documented.2 Here we review salient immunogenetic, clinical and mechanistic features of RA association with the HLA locus, focusing primarily on the SE.
HLA genes and their products
The immune system is composed of various cells that work together to protect the host against invading pathogens without harming its own tissues. Therefore, the host has to recognize which antigens are self and which are foreign. To discriminate between such self- and foreign antigens, the major histocompatibility complex (MHC) antigens, known in humans as HLA, have evolved. MHC molecules have the ability to recognize and present foreign antigens to the immune system, but at the same time disregard self-antigens. This ability to discriminate between self and foreign is established through a process called “MHC restriction”.3 During the development of T cells in the thymus, T cells that react to self-antigens are eliminated, while those that respond to foreign antigens that are presented by a self-HLA molecule are preserved. This selection results in CD4+ and CD8+ T cells that only respond to foreign antigens that are presented by self-HLA molecules. Despite their ability to selectively recognize and respond to foreign antigens, a number of HLA alleles has been found to confer susceptibility to various diseases, the majority of which involve dysregulated immunity or autoimmunity.
The HLA locus is located on the short arm of chromosome 6 and covers a 7.6 Mb region that contains over 250 highly polymorphic genes.4 The region is organized in three sub-regions: class I, class II and class III, which all have different functions. Both class I and II regions encode for glycoproteins that are expressed as cell surface receptors, whereas the class III region contains genes that encode for a variety of secreted immune system proteins, including complement factors and cytokines.
The class I region encodes for three main subsets of HLA molecules; HLA-A, HLA-B and HLA-C. Class I HLA molecules are composed of an HLA-coded heavy α-chain and an invariant light chain, beta-2 microglobulin (β2m), which is essential for functional expression of the HLA molecule at the cell surface. The α-chain is folded to form a peptide-binding cleft that is “closed” and can accommodate short antigenic peptides, typically 8-10 amino acids long. These class I molecules are expressed on all nucleated cells and specialize in presentation of intracellular antigens, including viral antigens, to cytotoxic (CD8+) T cells. Genes in the class II region encode for HLA-DR, HLA-DP and HLA-DQ molecules as well as a few other related proteins. Class II HLA molecules are composed of an α-chain and a β-chain, both coded by the HLA class II region. Unlike the class I molecules, the peptide-binding cleft of class II molecules are “open” which allows the accommodation of larger peptides of 15-20 amino acids long. Class II molecules are initially expressed on the cell surface of immune cells, in particular antigen presenting cells such as macrophages or dendritic cells, as well as B cells and activated T cells. These molecules present antigens from outside the cell to (CD4+) T cells which in turn stimulate B cells to produce antibodies towards that specific antigen. This results in an antigen specific immune response. After activation of the immune system, the HLA class II molecules can be expressed on other cells.
HLA-associated diseases
The HLA locus is a highly polymorphic region. Its high gene density, presence of clusters of genes with related functions, enormous polymorphism and a strong linkage disequilibrium (LD) between alleles, renders it difficult to unravel the comprehensive HLA functions. During the last several decades, various conditions such as infectious diseases5, cancer6 or autoimmune diseases7 have been found to be more prevalent in individuals that carry certain HLA alleles. The majority of the HLA associated diseases can be classified as autoimmune or immune-mediated disease and have been observed with merely HLA class I alleles (e.g. ankylosing spondylitis (AS))8 or merely class II alleles (e.g. seropositive RA).9 Additionally, some autoimmune diseases have been found to be influenced by both HLA class I and class II genes (e.g. diabetes mellitus type I).10 In addition to HLA alleles that predispose to disease, there are also a number of HLA alleles that are protective against disease.
How certain HLA alleles predispose to or protect against (autoimmune) diseases and what the underlying molecular mechanisms are is currently unclear. The hypotheses that have been proposed over the years commonly implicate atypical presentation of self-antigens11,12, an immune response to “altered self” antigens13 or cross reactivity with foreign or self- antigens.14,15 These hypotheses may seem plausible based on HLA function and their role in the immune response. However, despite their plausibility, the mechanistic and epidemiologic evidence that is currently available is difficult to reconcile with presentation of specific antigens being the underlying mechanism for HLA-disease association.
Several examples of HLA alleles are associated with more than one disease with completely different target tissues and pathogeneses, which defies the notion that antigen presentation should be specific for both the antigen and the presenting HLA molecule. Examples of such HLA alleles are HLA-DRB1*04:01 which is associated with both RA and type-1 diabetes16 and HLA-DQB1*03:02 which is associated with both type-1 diabetes and celiac disease.17,18 In addition, there are certain HLA alleles that predispose to one disease but protect against another, e.g. HLA-DRB1*04:02 has been found to confer susceptibility to pemphigus vulgaris (PV) but at the same time protect against RA (discussed below).
Furthermore, the most significant HLA-disease association to date has been found for a brain disorder (narcolepsy), which is not known to involve antigen presentation.19 Also, certain HLA molecules have been found to have functions unrelated to antigen presentation, including cognition20, olfaction and the activation of innate immune signaling (reviewed in 21).
In addition, some disease-associated alleles have been shown to demonstrate ‘cross-species susceptibility’, e.g. HLA-DRB1*04:01 associates with human RA and also confers susceptibility to inflammatory arthritis in mice.22
Moreover, despite extensive efforts to identify target antigens in autoimmune diseases, they have only been identified for a very small number of diseases. Also, presence of T cell clonality, a phenomenon that can be expected in case of the presence of a specific antigen has not been convincingly demonstrated in HLA associated diseases.
Lastly, RA disease severity has been shown to correlate with HLA allele dose, i.e. two allele copies confer greater susceptibility, severity and penetrance compared to one copy (discussed below). Such allele-dose impact on RA disease severity cannot be explained by antigen presentation-based hypotheses.
HLA-RA associations
The SE
RA association with HLA has been known since 196923, and the associations with specific DR4 allotypes were identified in the late 1970s.24,25 The term ‘shared epitope’2 was coined in the late 1980's following the discovery that the majority of RA patients share a 5 amino acid sequence motif (i.e. QKRAA, QRRAA, or RRRAA) in residues 70-74 of the DRβ chain, coded by several distinct DRB1 alleles in individuals expressing DR4 and non-DR4 allotypes. Recent genomic imputation analyses suggest that in addition to residues 70-74 in the α helical rim of DRβ chain, the classically defined “SE”, residues 11 and/or 13, located in the ‘floor’ of the HLA-DR groove, are also associated with RA susceptibility,26,27 suggesting that presentation of peptidic antigens may play a role in RA etiology. However, this imputation-based hypothesis awaits experimental validation and the relevance of these statistical data to RA etiology has recently been questioned.28 Additionally, despite several decades of research, the identity of putative arthritogenic peptides remains elusive. The findings of a recent study raise further doubts on the notion of specific antigen presentation by SE-expressing RA-associated DR molecules since a comparative analysis identified only negligible overlaps in the repertoires of peptides eluted from different SE-expressing HLA-DR molecules.29 The epidemiological and clinical aspects of RA genetics, including the SE, have been reviewed elsewhere.30,31
The SE not only confers a higher risk for RA, but also increases the likelihood of developing earlier disease onset, more severe bone erosions9,32-34 and anti-citrullinated protein antibodies (ACPA).35 SE-RA association and disease severity are gene-dose dependent. For example, in individuals carrying one SE-coding allele the odds ratio (OR) of developing joint damage compared to SE-negative individuals is 2.38. With two SE-coding alleles, the OR increases to 3.92.36 Furthermore, there is additional evidence of an allele-dose effect, in which early disease onset, severity of bone destruction and higher disease concordance among monozygotic twins all correlate with the number of SE-coding HLA-DRB1 alleles.9,33,34
It is worth to mention that not only inherited genes confer SE-RA association. Non-inherited maternal HLA antigens (NIMA) carrying SE motifs have been shown to confer RA risk in SE-negative individuals37, especially in younger onset RA in women.38 Thus minute amounts of maternal SE acquired during the fetal period is sufficient to determine RA susceptibility. This mechanism may account for some of the SE-negative subset of RA.
Ethnic and racial factors that affect SE-RA association
Considerable ethnic and racial stratification exist within the SE-positive RA subset in terms of association with specific SE-coding DRB1 alleles (reviewed in 39). For example, in European RA patients, DRB1*04:01; *04:04; *01:01, and *10:01 are the predominant SE-coding alleles, while in East Asians the most common SE-coding allele is DRB1*04:05. In Pima, Tlingit, Yakima and Chippewa Native American individuals a SE-coding allele DRB1*14:02 has been found to be a significant genetic risk factor for severe RA.40,41
RA is generally less common among Africans compared to populations of European descent42,43 and the frequency of SE-coding alleles in African Americans RA patients is approximately one third of that reported in European RA patients.44 Nonetheless, SE-coding DRB1 alleles are almost twice as common in African American RA patients, compared to healthy control subjects.44 This indicates that the SE is a significant risk factor in African Americans, albeit to a lesser extent than in Caucasians. Importantly, the prevalence of SE-coding alleles in African Americans correlates positively with the extent of estimated European ancestry, regardless of RA status, suggesting that genetic admixture between European and African American populations is responsible for introducing RA risk in the latter population.44
SE and ACPA
ACPA (or anti-cyclic citrullinated peptide (anti-CCP)) are a useful disease marker in RA. These antibodies associate with severe, erosive disease45,46 and, relevant to this review, they are strongly associated with SE-coding DRB1 alleles.35,47 Among different SE-positive RA patients, those carrying the DRB1*04 group of alleles display higher ACPA titers of compared to those carrying DRB1*01 alleles.48 An early study on RA patients in the Netherlands49 demonstrated an odds ratio (OR) of 3.3 for anti-CCP positivity in RA patients with a single SE-coding allele (DRB1*01, *04, *10). The OR in RA patients with two SE-coding alleles was 13.3, suggesting a SE gene-dose effect on anti-CCP positivity. In anti-CCP-negative European RA patients, on the other hand, a significant RA association (OR= 1.84) with the DR3 allotype was reported 50 In Japanese individuals, who rarely carry the DRB1*03:01 allele, a significant association with DR14 and DR8 was found.51 Thus, the ACPA-positive and –negative subsets of RA associate with different HLA-DRB1 alleles and should be therefore considered immunogenetically distinct diseases.
The strong association between SE-coding alleles and ACPA suggest a cause-effect relationship, although the precise underlying mechanism remains unknown. A common hypothesis states that the SE represents an obligatory amino acid sequence in the HLA-DR antigen presentation pocket that is critically necessary for antigen-specific presentation of citrullinated antigen-derived peptides to citrullinated protein-specific helper T cells, which help B cells to produce ACPA. According to this scenario, ACPA react against tissue citrullinated proteins which results in autoimmunity.52,53 While this hypothesis is plausible, it is difficult to reconcile with the findings that ACPA can be detected in RA patients, years before disease onset.54,55 Furthermore, ACPA in RA sera display promiscuous specificity against multiple citrullinated proteins which do not share sequence homology, such as vimentin56, a-enolase57, collagen type II58, or fibrin59, amongst other candidate self-proteins. Such promiscuity is inconsistent with the HLA-restricted antigen presentation concept.
In addition to the antigen specificity paradox discussed above, the hypotheses that postulate an effector role for ACPA in disease pathogenesis seem to dismiss a sizable body of literature implicating presence of polymorphisms in the peptidyl arginine deiminase 4 (PADI4) gene in certain populations60,61 and evidence for enzyme dysregulation and overabundance of citrullinated proteins in RA.62-64 These data suggest that citrullinated proteins, independent of ACPA, are at least partially accountable for RA pathogenesis. While definitive explanations of this paradox are absent, it is worth mentioning that the HLA Cusp Theory65,66 has previously proposed that in addition to antigen presentation, HLA-DR molecules may perform other, non-antigen presentation allele-specific functions, through the third allelic hypervariable region of the DRβ chain, as depicted graphically in Figure 1. Our group has demonstrated that the SE acts as a signal transduction ligand that leads to immune dysregulation and osteoclast activation, independent of its putative antigen presentation role.67-74 It remains to be seen if a non-antigen presentation mechanism, such as SE ligand-activated signaling could shed light on the SE-ACPA association paradox.
Figure 1. The SE ligand.
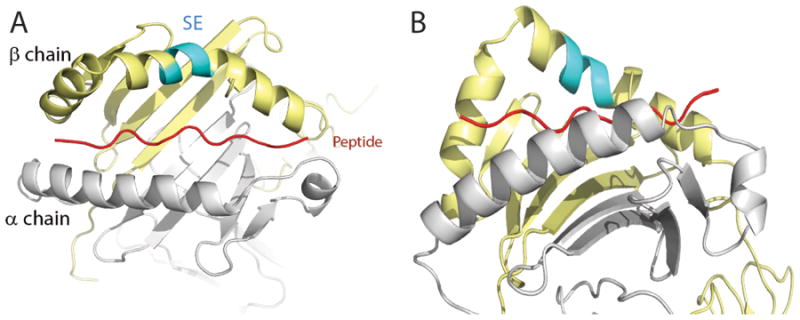
Crystal structure of HLA-DR4 (DRB1*04:01) molecule in a ‘top” (A) and ‘side’ (B) views. The DRα chain is colored in gray; the DRβ chain is shown in yellow and the groove peptide is shown in red. The SE (residues 70-74) is shown in cyan.
SE-environment interaction
Approximately two-thirds of RA risk is attributed to genetic factors, of which the SE is the most significant one. The remaining one third of disease susceptibility is attributed to non-genetic mechanisms that are most likely triggered by environmental factors. Over the years, ample evidence has been gathered to support the role of environmental factors in disease onset. For example, the disease is historically believed to have gained higher prevalence in the Old World concomitant with the Industrial Revolution; prevalence of the disease is higher in urban populations; the disease incidence varies by birth cohort; and most importantly, conclusive evidence has been established for an association between RA and exposure to cigarette smoke.75 Several studies have identified interactions between the SE and tobacco exposure.76-78 For example, a Swedish group reported a strong interaction between the SE and smoking in RA risk, particularly in SE homozygous individuals.77 Similar conclusions have been reached by a Danish study78, and a very large US survey has demonstrated strong interaction between heavy smoking and the SE in RA.76 In African Americans, the contribution of cigarette smoking to RA was found to be limited to those with more than 10 years of exposure, particularly among patients carrying SE-coding DRB1 alleles.79 Thus, there is compelling evidence for interaction between the SE and tobacco exposure in the etiology of RA in diverse populations.
The mechanistic basis of SE-smoking interaction in RA etiology and pathogenesis is unknown. A popular model for the etiology of RA suggests that the association between smoking and RA is due to the ability of the former to enhance SE-dependent immune reaction to citrullinated proteins, which, in turn trigger disease.80 According to this model, smoking increases the abundance of citrullinated proteins in the lung, which in SE-positive individuals may provide an antigenic stimulus for ACPA generation. More recently, another group has reported that SE-smoking interaction does not shape the reactivity of the ACPA response, suggesting that smoking activates antigen-nonspecific citrullination.81
Bacterial infection has long been proposed as an environmental etiologic factor for RA.82 Given that ACPA may be found years before disease onset in RA, it has been speculated that prior to RA, extra-articular infections may be the culprit. For example, recently, there has been a growing interest in the possible contribution of smoking-induced lung infection as an extra-articular source of disease-triggering infection. A recent study suggested that smoking-associated bronchiectasis might be an RA-triggering etiology due to enhanced production of ACPA in such individuals.83 Periodontal disease (PD), another extra-articular chronically infected site, has been proposed as a culprit in RA due to the known association between the two diseases84, and the fact that a well-studies PD-triggering bacterium, Porphyromonas gingivalis, expresses the bacterial PAD enzyme.85 In the context of this review, however, it should be pointed out that PD has been shown to associate with SE-coding alleles independent of RA.86,87 How this confounding factor affects the interpretation of studies focusing on the etiologic role of oral bacterial agents on RA etiology is currently unclear.
Protective DRB1 alleles
While the QKRAA, QRRAA or RRRAA sequences in position 70-74 of the DRβ chain have been shown to increase RA risk, alleles that code for D instead of Q or R in position 7088,89, particularly the 70-DERAA-74 sequence which is coded by several alleles, including DRB1*01:03, DRB1*13:01, DRB1*13:02 or DRB1*04:02 exert a protective effect.90,91 Interestingly, diametrically opposite of the pro-RA effect of SE-expressing NIMA37,38, DERAA-expressing NIMA have been shown to protect against the disease.92
The mechanism of this allele-based protection is unknown, although it was recently proposed that this association is due to cross-reactivity between citrullinated-vinculin and microbial proteins, due to presentation of the DERAA sequence by DQB alleles that are in linkage disequilibrium with SE-coding DRB1 alleles.15 Nonetheless, besides the fact that this intriguing hypothesis awaits experimental validation, it also appears to contradict published data from the same group which indicate that DERAA-expressing alleles are protective against RA independent of a SE-coding DRB1 allele90 and the dominant protective effect of a DERAA-expressing transgene on CIA development independent of any DQ molecules.93 Moreover, this hypothesis does not explain why exposure of SE-positive haplotypes to the DERAA sequence, ubiquitously expressed by microbial proteins, cannot mount a similar immune protective effect in individuals without DERAA-coding alleles. Finally, DERAA-coding DRB1 alleles have been shown to be protective against several other autoimmune diseases besides RA94,95, suggesting an antigen-nonspecific modulatory effect, rather than antigen presentation-based mechanism.
The 70-DERAA-74 sequence coded by HLA-DRB1*0402, as well as by several other alleles, exerts a dominant protective effect in RA90,91, presence of one DERAA-coding HLA-DRB1 allele provides protection against RA even in the presence of predisposing SE-positive alleles.90
However, this allele has been shown to predispose to pemphigus vulgaris (PV), an potentially lethal autoimmune disease that is characterized by blistering of the skin and the mucosal membranes. Although this disease is relatively rare, it is associated with considerable morbidity and mortality.96 Little is currently known about the mechanistic basis of the association of HLA-DRB1*04:02 with PV, but some have postulated that this allele binds and allows presentation of desmoglein 3, an identified auto-antigen in PV. However, the evidence supporting this hypothesis is ambiguous. The dual role of HLA-DRB1*04:02 in HLA-disease association, being protective in RA and at the same time being a genetic risk factor for PV, is currently not understood.
Non-SE-coding HLA alleles in RA
As mentioned above, while ACPA-positive RA strongly associates with SE-coding DRB1 alleles, the ACPA-negative subset has been shown to associate with other, non-SE-coding HLA alleles, such as DRB1*03:01, DR14 or DR8. In East Asians, associations of RA with a homozygous non-SE-coding allele DRB1*09:0197, or a heterozygous combination ofDRB1*04:05/09:01 alleles98 have been anecdotally reported. While the prevalence and mechanistic basis of these associations remain to be better explored, it is clear that there is more to learn about the stringency of the SE motif as a RA genetic risk factor.
The DR and DQ loci are inherited in strong linkage disequilibrium. It is therefore not surprising that both loci are statistically found to associate with RA. RA-associated DRB1 alleles have been particularly demonstrated in haplotypes with certain DQ loci. For example, the SE-coding DRB1*04:01 has been found in haplotypes that include DQA1*03-DQB1*03:01 or DQA1*03-DQB1*03:02 alleles, the SE-coding DRB1*04:04 has been shown to form haplotypes with DQA1*03-DQB1*03:02.99 However, while extensive evidence exists to substantiate a direct role of the DRB1 locus in RA, evidence to support such role for the DQ locus remains to be conclusively shown.
Associations with non-HLA genes
With the advent of GWAS technologies over the past decade, the field has seen major expansion in the number of single nucleotide polymorphism (SNP) sites that identify RA susceptibility gene candidates. A large meta-analysis of multiple independent GWAS data sets100, covering over 100,000 subjects of European or Asian ancestries, identified 101 RA risk loci corresponding to 98 biological candidate genes.
Among the SNPs identified there are many that involve immune system genes and/or known targets of approved therapy.100 For example, a missense variant of protein tyrosine phosphatase nonreceptor 22, coded by PTPN22 that introduces an R620W substitution is associated with RA as well as with many other autoimmune diseases in Caucasian patients.101 This variant has been shown to affect immune responses relevant to autoimmunity.102 Another important RA-associated polymorphism involves PADI4 103, the gene that codes for the citrullinating enzyme PAD4. The risk variant is associated with increased PADI4 transcription stability, and has been shown to associate with RA primarily in Asians. An interesting SNP association has been reproducibly found in the tumor necrosis factor-a protein 3 (TNFAIP3) locus.104 The variant leads to impaired A20, an enzyme that inhibits NF-kB activity. As a result, NF-kB signaling is enhanced, and in mice with ablation of Tnfaip3 there is spontaneous inflammatory polyarthritis.105 Additional information about GWAS-based RA association data is discussed elsewhere.100,106-108
Finally, it should be added that the contribution of the HLA locus to RA susceptibility is far higher than any of the known non-HLA loci, with the DRB1-associated risk alone being greater than the aggregate contribution of all known non-HLA risk loci. Additionally, even when the genetic contributions of the entire list of known HLA and non-HLA risk loci are compiled, there is still a large percentage of heritability that remains unaccounted for. The research challenges in the coming years will be to fill in the missing hereditability gaps, address the role of gene-environment and gene-gene interactions, and validate the functional roles of the SE and a myriad of GWAS-based RA risk loci.
Key points.
Certain human leukocyte antigen (HLA) alleles have been found to be associated with immune mediated or autoimmune diseases, but the underlying mechanisms are largely unknown
Rheumatoid arthritis (RA) strongly associates with HLA-DRB1 alleles that encode a sequence motif called ‘shared epitope’ (SE) and there is variability in the strength of RA-SE association among ethnic and racial populations
The SE shows interaction with environmental factors (tobacco exposure) and together significantly amplify the disease risk
In contrast to RA risk-conferring SE-coding alleles, there are several other DRB1 alleles that protect against the disease
Genome-wide association studies discovered many non-HLA RA risk loci, but their aggregate contribution to RA risk is outweighed by that of the SE
Footnotes
Disclosure: The authors have no known or potential commercial or financial conflicts of interest.
References
- 1.MacGregor AJ, Snieder H, Rigby AS, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43:30–37. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 3.Zinkernagel RM, Doherty PC. The discovery of MHC restriction. Immunol Today. 1997;18:14–17. doi: 10.1016/s0167-5699(97)80008-4. [DOI] [PubMed] [Google Scholar]
- 4.Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet. 2013;14:301–323. doi: 10.1146/annurev-genom-091212-153455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaslow RA, Shaw S. The role of histocompatibility antigens (HLA) in infection. Epidemiol Rev. 1981;3:90–114. doi: 10.1093/oxfordjournals.epirev.a036241. [DOI] [PubMed] [Google Scholar]
- 6.Gill TJ., 3rd Role of the major histocompatibility complex region in reproduction, cancer, and autoimmunity. Am J Reprod Immunol. 1996;35:211–215. doi: 10.1111/j.1600-0897.1996.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 7.Gough SC, Simmonds MJ. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr Genomics. 2007;8:453–465. doi: 10.2174/138920207783591690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reveille JD. Major histocompatibility genes and ankylosing spondylitis. Best Pract Res Clin Rheumatol. 2006;20:601–609. doi: 10.1016/j.berh.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Mattey DL, Hassell AB, Dawes PT, et al. Independent association of rheumatoid factor and the HLA-DRB1 shared epitope with radiographic outcome in rheumatoid arthritis. Arthritis Rheum. 2001;44:1529–1533. doi: 10.1002/1529-0131(200107)44:7<1529::AID-ART275>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 10.Erlich H, Valdes AM, Noble J, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084–1092. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nepom GT, Kwok WW. Molecular basis for HLA-DQ associations with IDDM. Diabetes. 1998;47:1177–1184. doi: 10.2337/diab.47.8.1177. [DOI] [PubMed] [Google Scholar]
- 12.Ridgway WM, Fathman CG. The association of MHC with autoimmune diseases: understanding the pathogenesis of autoimmune diabetes. Clin Immunol Immunopathol. 1998;86:3–10. doi: 10.1006/clin.1997.4449. [DOI] [PubMed] [Google Scholar]
- 13.Yin L, Dai S, Clayton G, et al. Recognition of self and altered self by T cells in autoimmunity and allergy. Protein Cell. 2013;4:8–16. doi: 10.1007/s13238-012-2077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oldstone MB. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12:1255–1265. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Heemst J, Jansen DT, Polydorides S, et al. Crossreactivity to vinculin and microbes provides a molecular basis for HLA-based protection against rheumatoid arthritis. Nat Commun. 2015;6:6681. doi: 10.1038/ncomms7681. [DOI] [PubMed] [Google Scholar]
- 16.Tait BD, Drummond BP, Varney MD, Harrison LC. HLA-DRB1*0401 is associated with susceptibility to insulin-dependent diabetes mellitus independently of the DQB1 locus. Eur J Immunogenet. 1995;22:289–297. doi: 10.1111/j.1744-313x.1995.tb00245.x. [DOI] [PubMed] [Google Scholar]
- 17.Sabbah E, Savola K, Kulmala P, et al. Disease-associated autoantibodies and HLA-DQB1 genotypes in children with newly diagnosed insulin-dependent diabetes mellitus (IDDM). The Childhood Diabetes in Finland Study Group. Clin Exp Immunol. 1999;116:78–83. doi: 10.1046/j.1365-2249.1999.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Setty M, Hormaza L, Guandalini S. Celiac disease: risk assessment, diagnosis, and monitoring. Mol Diagn Ther. 2008;12:289–298. doi: 10.1007/BF03256294. [DOI] [PubMed] [Google Scholar]
- 19.Nishino S, Okuro M, Kotorii N, et al. Hypocretin/orexin and narcolepsy: new basic and clinical insights. Acta Physiol (Oxf) 2010;198:209–222. doi: 10.1111/j.1748-1716.2009.02012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shepherd CE, Piguet O, Broe GA, et al. Histocompatibility antigens, aspirin use and cognitive performance in non-demented elderly subjects. J Neuroimmunol. 2004;148:178–182. doi: 10.1016/j.jneuroim.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms. Adv Immunol. 2009;101:27–79. doi: 10.1016/S0065-2776(08)01002-X. [DOI] [PubMed] [Google Scholar]
- 22.Taneja V, Behrens M, Mangalam A, Griffiths MM, Luthra HS, David CS. New humanized HLA-DR4-transgenic mice that mimic the sex bias of rheumatoid arthritis. Arthritis Rheum. 2007;56:69–78. doi: 10.1002/art.22213. [DOI] [PubMed] [Google Scholar]
- 23.Astorga GP, Williams RC., Jr Altered reactivity in mixed lymphocyte culture of lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum. 1969;12:547–554. doi: 10.1002/art.1780120602. [DOI] [PubMed] [Google Scholar]
- 24.McMichael AJ, Sasazuki T, McDevitt HO, Payne RO. Increased frequency of HLA-Cw3 and HLA-Dw4 in rheumatoid arthritis. Arthritis Rheum. 1977;20:1037–1042. doi: 10.1002/art.1780200501. [DOI] [PubMed] [Google Scholar]
- 25.Stastny P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med. 1978;298:869–871. doi: 10.1056/NEJM197804202981602. [DOI] [PubMed] [Google Scholar]
- 26.Kim K, Jiang X, Cui J, et al. Interactions between amino acid-defined major histocompatibility complex class II variants and smoking in seropositive rheumatoid arthritis. Arthritis Rheumatol. 2015;67:2611–2623. doi: 10.1002/art.39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raychaudhuri S, Sandor C, Stahl EA, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Heemst J, Huizinga TJ, van der Woude D, Toes RE. Fine-mapping the human leukocyte antigen locus in rheumatoid arthritis and other rheumatic diseases: identifying causal amino acid variants? Curr Opin Rheumatol. 2015;27:256–261. doi: 10.1097/BOR.0000000000000165. [DOI] [PubMed] [Google Scholar]
- 29.Scholz E, Mestre-Ferrer A, Daura X, et al. A Comparative Analysis of the Peptide Repertoires of HLA-DR Molecules Differentially Associated With Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68:2412–2421. doi: 10.1002/art.39736. [DOI] [PubMed] [Google Scholar]
- 30.Furukawa H, Oka S, Shimada K, Hashimoto A, Tohma S. Human leukocyte antigen polymorphisms and personalized medicine for rheumatoid arthritis. J Hum Genet. 2015;60:691–696. doi: 10.1038/jhg.2015.36. [DOI] [PubMed] [Google Scholar]
- 31.Kurko J, Besenyei T, Laki J, Glant TT, Mikecz K, Szekanecz Z. Genetics of rheumatoid arthritis - a comprehensive review. Clin Rev Allergy Immunol. 2013;45:170–179. doi: 10.1007/s12016-012-8346-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez-Gay MA, Garcia-Porrua C, Hajeer AH. Influence of human leukocyte antigen-DRB1 on the susceptibility and severity of rheumatoid arthritis. Semin Arthritis Rheum. 2002;31:355–360. doi: 10.1053/sarh.2002.32552. [DOI] [PubMed] [Google Scholar]
- 33.Plant MJ, Jones PW, Saklatvala J, Ollier WE, Dawes PT. Patterns of radiological progression in early rheumatoid arthritis: results of an 8 year prospective study. J Rheumatol. 1998;25:417–426. [PubMed] [Google Scholar]
- 34.Weyand CM, Goronzy JJ. Disease mechanisms in rheumatoid arthritis: gene dosage effect of HLA-DR haplotypes. J Lab Clin Med. 1994;124:335–338. [PubMed] [Google Scholar]
- 35.Huizinga TW, Amos CI, van der Helm-van Mil AH, et al. Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum. 2005;52:3433–3438. doi: 10.1002/art.21385. [DOI] [PubMed] [Google Scholar]
- 36.Marotte H, Pallot-Prades B, Grange L, et al. The shared epitope is a marker of severity associated with selection for, but not with response to, infliximab in a large rheumatoid arthritis population. Ann Rheum Dis. 2006;65:342–347. doi: 10.1136/ard.2005.037150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Horst-Bruinsma IE, Hazes JM, Schreuder GM, et al. Influence of non-inherited maternal HLA-DR antigens on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 1998;57:672–675. doi: 10.1136/ard.57.11.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guthrie KA, Tishkevich NR, Nelson JL. Non-inherited maternal human leukocyte antigen alleles in susceptibility to familial rheumatoid arthritis. Ann Rheum Dis. 2009;68:107–109. doi: 10.1136/ard.2008.092312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plenge RM. Recent progress in rheumatoid arthritis genetics: one step towards improved patient care. Curr Opin Rheumatol. 2009;21:262–271. doi: 10.1097/BOR.0b013e32832a2e2d. [DOI] [PubMed] [Google Scholar]
- 40.Ferucci ED, Templin DW, Lanier AP. Rheumatoid arthritis in American Indians and Alaska Natives: a review of the literature. Semin Arthritis Rheum. 2005;34:662–667. doi: 10.1016/j.semarthrit.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Nelson JL, Boyer G, Templin D, et al. HLA antigens in Tlingit Indians with rheumatoid arthritis. Tissue Antigens. 1992;40:57–63. doi: 10.1111/j.1399-0039.1992.tb01960.x. [DOI] [PubMed] [Google Scholar]
- 42.Brighton SW, de la Harpe AL, van Staden DJ, Badenhorst JH, Myers OL. The prevalence of rheumatoid arthritis in a rural African population. J Rheumatol. 1988;15:405–408. [PubMed] [Google Scholar]
- 43.Silman AJ, Ollier W, Holligan S, et al. Absence of rheumatoid arthritis in a rural Nigerian population. J Rheumatol. 1993;20:618–622. [PubMed] [Google Scholar]
- 44.Hughes LB, Morrison D, Kelley JM, et al. The HLA-DRB1 shared epitope is associated with susceptibility to rheumatoid arthritis in African Americans through European genetic admixture. Arthritis Rheum. 2008;58:349–358. doi: 10.1002/art.23166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mustila A, Korpela M, Haapala AM, et al. Anti-citrullinated peptide antibodies and the progression of radiographic joint erosions in patients with early rheumatoid arthritis treated with FIN-RACo combination and single disease-modifying antirheumatic drug strategies. Clin Exp Rheumatol. 2011;29:500–505. [PubMed] [Google Scholar]
- 46.van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibodies: the past, the present and the future. Nat Rev Rheumatol. 2011;7:391–398. doi: 10.1038/nrrheum.2011.76. [DOI] [PubMed] [Google Scholar]
- 47.Lundstrom E, Kallberg H, Alfredsson L, Klareskog L, Padyukov L. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum. 2009;60:1597–1603. doi: 10.1002/art.24572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Snir O, Widhe M, von Spee C, et al. Multiple antibody reactivities to citrullinated antigens in sera from patients with rheumatoid arthritis: association with HLA-DRB1 alleles. Ann Rheum Dis. 2009;68:736–743. doi: 10.1136/ard.2008.091355. [DOI] [PubMed] [Google Scholar]
- 49.van Gaalen FA, van Aken J, Huizinga TW, et al. Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum. 2004;50:2113–2121. doi: 10.1002/art.20316. [DOI] [PubMed] [Google Scholar]
- 50.Verpoort KN, van Gaalen FA, van der Helm-van Mil AH, et al. Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum. 2005;52:3058–3062. doi: 10.1002/art.21302. [DOI] [PubMed] [Google Scholar]
- 51.Terao C, Ohmura K, Ikari K, et al. ACPA-negative RA consists of two genetically distinct subsets based on RF positivity in Japanese. PLoS One. 2012;7:e40067. doi: 10.1371/journal.pone.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amara K, Steen J, Murray F, et al. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med. 2013;210:445–455. doi: 10.1084/jem.20121486. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Scally SW, Petersen J, Law SC, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. 2013;210:2569–2582. doi: 10.1084/jem.20131241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 55.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 56.Vossenaar ER, Despres N, Lapointe E, et al. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res Ther. 2004;6:R142–150. doi: 10.1186/ar1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kinloch A, Tatzer V, Wait R, et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1421–1429. doi: 10.1186/ar1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burkhardt H, Sehnert B, Bockermann R, Engstrom A, Kalden JR, Holmdahl R. Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur J Immunol. 2005;35:1643–1652. doi: 10.1002/eji.200526000. [DOI] [PubMed] [Google Scholar]
- 59.Masson-Bessiere C, Sebbag M, Girbal-Neuhauser E, et al. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J Immunol. 2001;166:4177–4184. doi: 10.4049/jimmunol.166.6.4177. [DOI] [PubMed] [Google Scholar]
- 60.Ikari K, Kuwahara M, Nakamura T, et al. Association between PADI4 and rheumatoid arthritis: a replication study. Arthritis Rheum. 2005;52:3054–3057. doi: 10.1002/art.21309. [DOI] [PubMed] [Google Scholar]
- 61.Lee YH, Rho YH, Choi SJ, Ji JD, Song GG. PADI4 polymorphisms and rheumatoid arthritis susceptibility: a meta-analysis. Rheumatol Int. 2007;27:827–833. doi: 10.1007/s00296-007-0320-y. [DOI] [PubMed] [Google Scholar]
- 62.Andrade F, Darrah E, Gucek M, Cole RN, Rosen A, Zhu X. Autocitrullination of human peptidyl arginine deiminase type 4 regulates protein citrullination during cell activation. Arthritis Rheum. 2010;62:1630–1640. doi: 10.1002/art.27439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giles JT, Fert-Bober J, Park JK, et al. Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Res Ther. 2012;14:R39. doi: 10.1186/ar3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki A, Yamada R, Yamamoto K. Citrullination by peptidylarginine deiminase in rheumatoid arthritis. Ann N Y Acad Sci. 2007;1108:323–339. doi: 10.1196/annals.1422.034. [DOI] [PubMed] [Google Scholar]
- 65.de Almeida DE, Holoshitz J. MHC molecules in health and disease: At the cusp of a paradigm shift. Self Nonself. 2011;2:43–48. doi: 10.4161/self.2.1.15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holoshitz J. The quest for better understanding of HLA-disease association: scenes from a road less travelled by. Discov Med. 2013;16:93–101. [PMC free article] [PubMed] [Google Scholar]
- 67.De Almeida DE, Ling S, Pi X, Hartmann-Scruggs AM, Pumpens P, Holoshitz J. Immune dysregulation by the rheumatoid arthritis shared epitope. J Immunol. 2010;185:1927–1934. doi: 10.4049/jimmunol.0904002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fu J, Ling S, Liu Y, et al. A small shared epitope-mimetic compound potently accelerates osteoclast-mediated bone damage in autoimmune arthritis. J Immunol. 2013;191:2096–2103. doi: 10.4049/jimmunol.1203231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holoshitz J, Ling S. Nitric oxide signaling triggered by the rheumatoid arthritis shared epitope: a new paradigm for MHC disease association? Ann N Y Acad Sci. 2007;1110:73–83. doi: 10.1196/annals.1423.009. [DOI] [PubMed] [Google Scholar]
- 70.Holoshitz J, Liu Y, Fu J, et al. An HLA-DRB1-coded signal transduction ligand facilitates inflammatory arthritis: a new mechanism of autoimmunity. J Immunol. 2013;190:48–57. doi: 10.4049/jimmunol.1202150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ling S, Lai A, Borschukova O, Pumpens P, Holoshitz J. Activation of nitric oxide signaling by the rheumatoid arthritis shared epitope. Arthritis Rheum. 2006;54:3423–3432. doi: 10.1002/art.22178. [DOI] [PubMed] [Google Scholar]
- 72.Ling S, Li Z, Borschukova O, Xiao L, Pumpens P, Holoshitz J. The rheumatoid arthritis shared epitope increases cellular susceptibility to oxidative stress by antagonizing an adenosine-mediated anti-oxidative pathway. Arthritis Res Ther. 2007;9:R5. doi: 10.1186/ar2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ling S, Pi X, Holoshitz J. The rheumatoid arthritis shared epitope triggers innate immune signaling via cell surface calreticulin. J Immunol. 2007;179:6359–6367. doi: 10.4049/jimmunol.179.9.6359. [DOI] [PubMed] [Google Scholar]
- 74.Naveh S, Tal-Gan Y, Ling S, Hoffman A, Holoshitz J, Gilon C. Developing potent backbone cyclic peptides bearing the shared epitope sequence as rheumatoid arthritis drug-leads. Bioorg Med Chem Lett. 2012;22:493–496. doi: 10.1016/j.bmcl.2011.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klareskog L, Padyukov L, Lorentzen J, Alfredsson L. Mechanisms of disease: Genetic susceptibility and environmental triggers in the development of rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006;2:425–433. doi: 10.1038/ncprheum0249. [DOI] [PubMed] [Google Scholar]
- 76.Karlson EW, Chang SC, Cui J, et al. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Annals of the Rheumatic Diseases. 2010;69:54–60. doi: 10.1136/ard.2008.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50:3085–3092. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- 78.Pedersen M, Jacobsen S, Garred P, et al. Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis: a nationwide case-control study in Denmark. Arthritis Rheum. 2007;56:1446–1453. doi: 10.1002/art.22597. [DOI] [PubMed] [Google Scholar]
- 79.Mikuls TR, Sayles H, Yu F, et al. Associations of cigarette smoking with rheumatoid arthritis in African Americans. Arthritis Rheum. 2010;62:3560–3568. doi: 10.1002/art.27716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klareskog L, Stolt P, Lundberg K, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54:38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- 81.Willemze A, van der Woude D, Ghidey W, et al. The interaction between HLA shared epitope alleles and smoking and its contribution to autoimmunity against several citrullinated antigens. Arthritis Rheum. 2011;63:1823–1832. doi: 10.1002/art.30409. [DOI] [PubMed] [Google Scholar]
- 82.Carty SM, Snowden N, Silman AJ. Should infection still be considered as the most likely triggering factor for rheumatoid arthritis? Ann Rheum Dis. 2004;63(Suppl 2):ii46–ii49. doi: 10.1136/ard.2004.028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quirke AM, Perry E, Cartwright A, et al. Bronchiectasis is a Model for Chronic Bacterial Infection Inducing Autoimmunity in Rheumatoid Arthritis. Arthritis Rheumatol. 2015;67:2335–2342. doi: 10.1002/art.39226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Detert J, Pischon N, Burmester GR, Buttgereit F. The association between rheumatoid arthritis and periodontal disease. Arthritis Res Ther. 2010;12:218. doi: 10.1186/ar3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McGraw WT, Potempa J, Farley D, Travis J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect Immun. 1999;67:3248–3256. doi: 10.1128/iai.67.7.3248-3256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonfil JJ, Dillier FL, Mercier P, et al. A “case control” study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis. Identification of types and subtypes using molecular biology (PCR.SSO) J Clin Periodontol. 1999;26:77–84. doi: 10.1034/j.1600-051x.1999.260203.x. [DOI] [PubMed] [Google Scholar]
- 87.Marotte H, Farge P, Gaudin P, Alexandre C, Mougin B, Miossec P. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann Rheum Dis. 2006;65:905–909. doi: 10.1136/ard.2005.036913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mackie SL, Taylor JC, Martin SG, et al. A spectrum of susceptibility to rheumatoid arthritis within HLA-DRB1: stratification by autoantibody status in a large UK population. Genes Immun. 2012;13:120–128. doi: 10.1038/gene.2011.60. [DOI] [PubMed] [Google Scholar]
- 89.Shadick NA, Heller JE, Weinblatt ME, et al. Opposing effects of the D70 mutation and the shared epitope in HLA-DR4 on disease activity and certain disease phenotypes in rheumatoid arthritis. Ann Rheum Dis. 2007;66:1497–1502. doi: 10.1136/ard.2006.067603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van der Helm-van Mil AH, Huizinga TW, Schreuder GM, Breedveld FC, de Vries RR, Toes RE. An independent role of protective HLA class II alleles in rheumatoid arthritis severity and susceptibility. Arthritis Rheum. 2005;52:2637–2644. doi: 10.1002/art.21272. [DOI] [PubMed] [Google Scholar]
- 91.van der Woude D, Lie BA, Lundstrom E, et al. Protection against anti-citrullinated protein antibody-positive rheumatoid arthritis is predominantly associated with HLA-DRB1*1301: a meta-analysis of HLA-DRB1 associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in four European populations. Arthritis Rheum. 2010;62:1236–1245. doi: 10.1002/art.27366. [DOI] [PubMed] [Google Scholar]
- 92.Feitsma AL, van der Helm-van Mil AH, Huizinga TW, de Vries RR, Toes RE. Protection against rheumatoid arthritis by HLA: nature and nurture. Ann Rheum Dis. 2008;67(Suppl 3):iii61–63. doi: 10.1136/ard.2008.098509. [DOI] [PubMed] [Google Scholar]
- 93.Taneja V, Taneja N, Behrens M, et al. HLA-DRB1*0402 (DW10) transgene protects collagen-induced arthritis-susceptible H2Aq and DRB1*0401 (DW4) transgenic mice from arthritis. J Immunol. 2003;171:4431–4438. doi: 10.4049/jimmunol.171.8.4431. [DOI] [PubMed] [Google Scholar]
- 94.Bettencourt A, Carvalho C, Leal B, et al. The Protective Role of HLA-DRB1( *)13 in Autoimmune Diseases. J Immunol Res. 2015;2015:948723. doi: 10.1155/2015/948723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cruz-Tapias P, Perez-Fernandez OM, Rojas-Villarraga A, Rodriguez-Rodriguez A, Arango MT, Anaya JM. Shared HLA Class II in Six Autoimmune Diseases in Latin America: A Meta-Analysis. Autoimmune Dis. 2012;2012:569728. doi: 10.1155/2012/569728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris--incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180. doi: 10.1136/bmj.a180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wakitani S, Imoto K, Murata N, Toda Y, Ogawa R, Ochi T. The homozygote of HLA-DRB1*0901, not its heterozygote, is associated with rheumatoid arthritis in Japanese. Scand J Rheumatol. 1998;27:381–382. doi: 10.1080/03009749850154447. [DOI] [PubMed] [Google Scholar]
- 98.Lee HS, Lee KW, Song GG, Kim HA, Kim SY, Bae SC. Increased susceptibility to rheumatoid arthritis in Koreans heterozygous for HLA-DRB1*0405 and *0901. Arthritis Rheum. 2004;50:3468–3475. doi: 10.1002/art.20608. [DOI] [PubMed] [Google Scholar]
- 99.Zanelli E, Breedveld FC, de Vries RR. HLA class II association with rheumatoid arthritis: facts and interpretations. Hum Immunol. 2000;61:1254–1261. doi: 10.1016/s0198-8859(00)00185-3. [DOI] [PubMed] [Google Scholar]
- 100.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zheng J, Ibrahim S, Petersen F, Yu X. Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes Immun. 2012;13:641–652. doi: 10.1038/gene.2012.46. [DOI] [PubMed] [Google Scholar]
- 102.Zheng J, Petersen F, Yu X. The role of PTPN22 in autoimmunity: learning from mice. Autoimmun Rev. 2014;13:266–271. doi: 10.1016/j.autrev.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 103.Suzuki A, Yamada R, Chang X, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 104.Thomson W, Barton A, Ke X, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matmati M, Jacques P, Maelfait J, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. 2011;43:908–912. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- 106.Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest. 2000;105:625–631. doi: 10.1172/JCI8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Diogo D, Okada Y, Plenge RM. Genome-wide association studies to advance our understanding of critical cell types and pathways in rheumatoid arthritis: recent findings and challenges. Curr Opin Rheumatol. 2014;26:85–92. doi: 10.1097/BOR.0000000000000012. [DOI] [PubMed] [Google Scholar]
- 108.Kochi Y, Suzuki A, Yamamoto K. Genetic basis of rheumatoid arthritis: a current review. Biochem Biophys Res Commun. 2014;452:254–262. doi: 10.1016/j.bbrc.2014.07.085. [DOI] [PubMed] [Google Scholar]