

Abstract
Background
Cerebral tissue damage after an ischemic event can be exacerbated by inflammation and thrombosis. Elevated extracellular ATP and ADP levels are associated with cellular injury, inflammation and thrombosis. Ectonucleoside triphosphate diphosphohydrolase-1 (CD39), an enzyme expressed on the plasmalemma of leukocytes and endothelial cells, suppresses platelet activation and leukocyte infiltration by phosphohydrolyzing ATP/ADP. To investigate the effects of increased CD39 in an in vivo cerebral ischemia model, we developed a transgenic (TG) mouse expressing human CD39 (hCD39).
Methods
A floxed-stop sequence was inserted between the promoter and the hCD39 transcriptional start site, generating a mouse in which the expression of hCD39 can be controlled tissue-specifically, using Cre recombinase mice. We generated mice that express hCD39 globally or in myeloid-lineage cells only. Cerebral ischemia was induced by middle cerebral artery (MCA) occlusion. Infarct volumes were quantified by MRI after 48 hours.
Results
Both global and TG hCD39- and myeloid lineage (LysM) CD39-overexpressing mice (TG n=9, LysM n=6) demonstrated significantly smaller cerebral infarct volumes compared to wild type (WT) mice. Leukocytes from ischemic and contralateral hemispheres were analyzed by flow cytometry. While contralateral hemispheres had equal numbers of macrophages and neutrophils, ischemic hemispheres from TG mice had less infiltration (n=4). TG mice showed less neurologic deficit compared to WT mice (n=6).
Conclusions
This is the first report of transgenic overexpression of CD39 in mice imparting a protective phenotype following stroke, with reduced leukocyte infiltration, smaller infarct volumes, and decreased neurological deficit. CD39 overexpression, either globally or in myeloid lineage cells, quenches post-ischemic leukosequestration and reduces stroke-induced neurological injury.
Keywords: inflammation, CD39
Introduction
Ischemic events in the brain occur due to vessel occlusion by cholesterol-rich plaque or thrombus, which restricts blood flow. Cerebral ischemia can lead to neuronal death in those areas downstream of the occlusion, and a consequent deficit in cognitive function. Current tools used to mitigate tissue damage caused by acute thrombotic stroke involve the use of aspirin, or reperfusion by fibrinolytic treatment such as tissue plasminogen activator (rt-PA), but this must be administered within the first several hours of ischemia1, 2. Intravenously administered rt-PA, in fact, remains one of the very few therapies for acute ischemic stroke3, 4. Inflammation further contributes to the detrimental effects of cerebral ischemia, with neutrophils, macrophages, and T cells composing the major component of infiltrating cells5, 6. Further innovations into more specific and localized treatments capable of targeting inflammation and thrombosis apart from hemostasis are necessary.
One molecular target that is being considered for antithrombotic and anti-inflammatory therapeutic development includes ecto-apyrases, which limit inflammation and thrombosis by enzymatically degrading extracellular nucleotides7–12. Of these ecto-apyrases, CD39 (ENTPD1) is the dominant form expressed on the plasmalemma of various cell types, including endothelial cells and various leukocyte populations, and is responsible for dissipating the pro-inflammatory ATP and pro-thrombotic ADP to form AMP10, 13–15. CD39-mediated degradation of pro-thrombotic and pro-inflammatory signals acts locally,16 and may be less likely to increase hemorrhagic risk.
Here, we have employed a well-established photothrombotic middle cerebral artery occlusion (MCAO) model of cerebral ischemia17–20. Mice were injected with rose bengal dye, and the MCA permanently occluded by illuminating the vessel with a laser. This model was used because it results in the intraluminal development of a highly localized platelet-rich thrombus, without mechanical disruption of endothelium. This model was used to examine the effects of augmentation of endogenous CD39 expression in ischemic stroke. The MCA was chosen as the occlusion target since most ischemic strokes in humans occur in the MCA territory21, and the MCAO model described above was chosen for its high level of reproducibility. To test our hypothesis, we have developed a floxed CD39 transgenic mouse in which we create progeny which either globally or selectively overexpress CD39 in specific cell populations.
Materials and Methods
Animal studies
All procedures performed on animals were in accordance with institutional guidelines.
Generation of transgenic mice
C57Bl/6 mice (Jackson Laboratories) were used to generate mice that selectively expressed a human CD39 transgene in addition to their endogenous expression of murine CD39. The human gene was preferred over murine CD39 in order to retain the ability to differentiate between the transgene and the endogenous gene. The protein products of these two sequences both express all apyrase conserved regions (ACR) and have preserved apyrase activity22, 23. A backbone construct (pcall-2, a gift from the Lobe laboratory) was modified24, 25. A description of the final construct is as follows: a CAG promoter followed by two LoxP sequences flanking 3 repetitions of a polyA sequence, followed by the starting sequence and full-length human CD39 cDNA (Figure 1A). The sequence was purified and microinjected into fertilized eggs. Transgenic founders were identified via a specific PCR screen probing human CD39 DNA. Forward primer: 5′ ACA GGC GTG GTG CAT CAA GTA GAA 3′ and Reverse primer: 5′ CCT GGC ACC CTG GAA GTC AAA G -3′ (Figure 1B).
Figure 1.
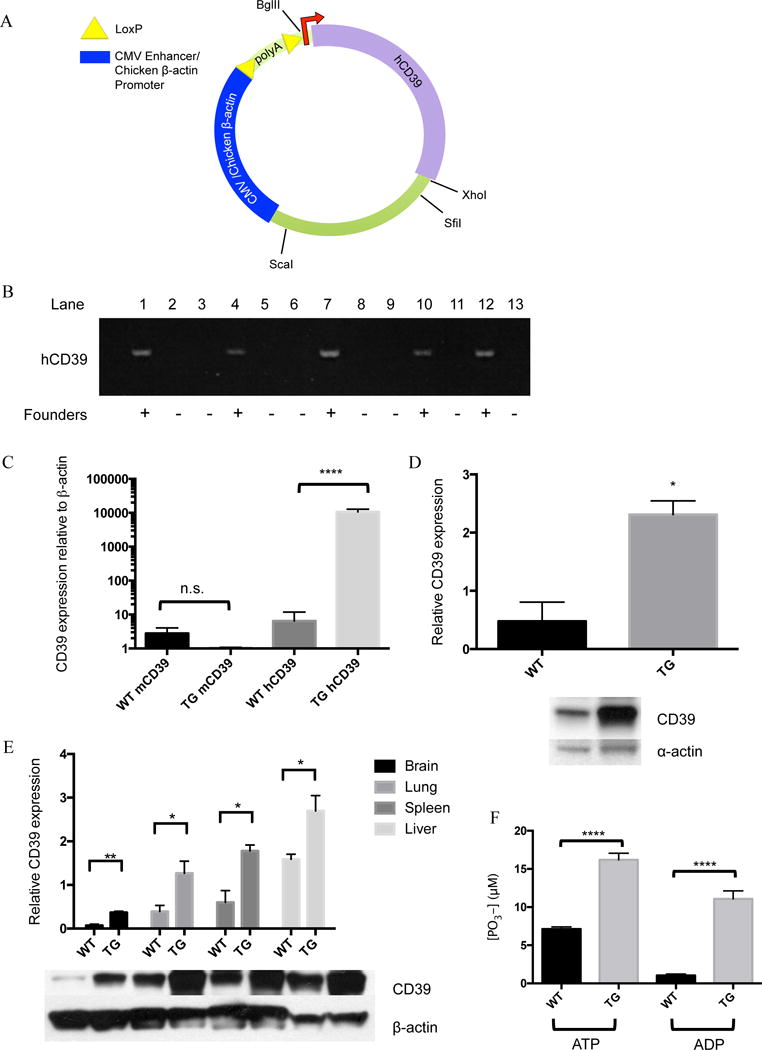
Transgenic mouse generation and confirmation of genotype and phenotype: A) A map of the plasmid used to generate CD39 transgenic mice. CD39 was inserted directly after the second loxp site. B) Transgenic mice were identified via PCR using primers probing for human CD39. Of 17 potential mice, 5 were identified as transgenic founders. C) Total RNA was isolated from bone marrow of naïve wild type and transgenic mice and probed with human or mouse CD39 primers in a qPCR assay. Results were normalized to mouse β-actin. D) Total protein was purified from hearts of naïve wild type and transgenic mice and probed with anti-CD39 antibodies via Western blot and normalized to α-actin expression. E) Total protein was purified from brains, lungs, livers and spleens of 3 naïve wild type and 3 transgenic mice and probed with anti-CD39 antibodies via Western blot and normalized to β-actin expression. F) Bone marrow was harvested from both wild type and transgenic mice, macrophages were grown, and phosphatase activity was measured. ATP or ADP was added as a substrate to live cells and free phosphate generation was quantified. * p < 0.05, ** p < 0.01, **** p < 0.0001.
Stable colonies were established and maintained before breeding to EIIA-Cre mice. EIIA-Cre mice have Cre expression driven by the EIIA promoter, and are often used when generating mice that express transgenes globally, including in the germ line26. Offspring of these breeding pairs generated mice expressing human CD39 globally. Mice carrying the floxed polyA sequence were subsequently bred to LysM-Cre mice (Jackson laboratories), so that offspring overexpress human CD39 in myeloid lineage cells, and are referred to here as LysM-Cre transgenic mice.
Middle cerebral artery occlusion (MCAO) model
Naïve male 8–13 week old C57Bl6/J mice were anesthetized using 2% isoflurane mixed with oxygen, and retro-orbitally administered rose bengal dye (1 mg/25 g body weight dissolved in saline). An incision was made above the left eye, exposing the temporalis muscle. A hole was drilled into the calvarium to visualize the MCA. The MCA was exposed to a 542-nm neon laser to form a thrombus, resulting in permanent focal ischemia. To confirm the occlusion, MCA bloodflow was monitored throughout the surgery using a laser Doppler flow probe (Transonic Systems Inc.). Occlusion was defined as 80% reduction in blood flow sustained for 10 minutes19. The skin was then closed, and mice were allowed to recover from surgery under a heat lamp and then returned to clean cages. Mice were sacrificed either 6 hours, 48 hours, or 7 days later for endpoint analyses.
Complete blood count (CBC)
Naïve male 8–13 week old C57Bl6/J mice, the same age range as all other mice used in other experiments described herein, were anesthetized using 2% isoflurane mixed with oxygen, and blood was drawn into tubes containing sodium citrate (Sigma). Blood was then analyzed using a HemaVet cell analyzer (Drew Scientific).
Heart rate and blood pressure measurements
Mice were acclimatized for two days before radio-telemetric heart rate and blood pressure measurements. Mice then underwent MCAO as described, and then blood pressures and heart rate measured again. Final measurements of blood pressure were taken at 7 or 9 days post MCAO.
Quantitative Real Time-PCR (qRT-PCR)
qRT-PCR was used to quantify RNA levels. Organs were harvested from transgenic or age-matched wild type C57Bl/6 mice and then immediately stored in measured volumes of RNAlater (Qiagen) at −80°C until ready for RNA isolation. Samples were cut to a weight of 30 mg, and total RNA was isolated using RNAeasy kits (Qiagen). cDNA was made using cDNA synthesis kits (Applied Biosystems). Real time qPCR was carried out using a 7000 detection system (Applied Biosystems) with 2× Universal Mastermix and primers for human CD39 or mouse CD39 and mouse β-actin (Applied Biosystems). All data were normalized to β-actin.
Whole-cell protein isolation
Organs were harvested from transgenic or age-matched wild type C57Bl/6 mice and immediately homogenized with a handheld tissue homogenizer in ice-cold RIPA buffer (25 mM Tris-HCL, 150 mM NaCl, 0.1% SDS, 0.1% Triton-X100, with all reagents from Sigma) containing protease inhibitor (Roche). Samples were incubated on ice for 20 minutes before centrifugation on a desktop centrifuge for 10 minutes at 13,000 × g at 4°C. Resulting supernatants were transferred to new tubes. Protein concentrations of total protein obtained from tissue lysates were determined via a colorimetric protein assay based on the Bradford method (Bio-Rad). Protein samples were flash frozen in aliquots and stored at −80°C.
Western blot assay
Total protein was quantified and added to 4× sample buffer (Invitrogen), boiled for 3 minutes at 100°C, separated by 10% SDS-PAGE, and electrophoretically-transferred onto PVDF membranes (Invitrogen). Membranes were probed with mouse monocolonal anti-CD39 IgG1 antibodies (Abcam) and HRP-conjugated anti-mouse antibodies (Sigma), and then detected using the enhanced chemiluminescence (ECL) detection system (Amersham Biosciences). Blots were then washed and probed with mouse monoclonal α- or β-actin IgG1 conjugated to peroxidase (Sigma), and then detected using enhanced chemiluminescence.
Multiplex cytokine assay
Ischemia was induced in age-matched male wild type and transgenic mice, and 6 hours later, ipsilateral (ischemic) and contralateral cerebral hemispheres were harvested. Total protein was immediately collected by homogenizing tissue in ice-cold RIPA buffer (without ionic detergent) using a handheld homogenizer. Samples were snap frozen in liquid nitrogen until just prior to initiation of the assay. Samples were gently thawed on ice, and then centrifuged at 13,000 × g on a desktop centrifuge at 4° C for 10 minutes. Supernatants were collected into fresh microcentrifuge tubes. A 96-well filter plate provided in the mouse cytokine panel kit (Invitrogen) was prepared according to manufacturer instructions. Briefly, the filters were incubated with wash buffer, and beads were sonicated and evenly distributed into each well. Duplicate standards were used, and total protein in samples was measured for normalization. After incubation with a biotinylated antibody and streptavidin, a Luminex 100 was used to read the plate.
Magnetic resonance imaging (MRI)
48 hours after inducing cerebral ischemia, prior to brain harvest, mice were anaesthetized with a mixture of 2% isoflurane and air, and placed in a 7.0 T Varian MR scanner (183-mm horizontal bore). Body temperature was maintained at 37°C with heated air. A double-tuned volume radio-frequency coil was used to scan the heads of the mice, and axial T2-weighted images were acquired using a spin-echo sequence. Repetition time/effective echo time was 4000/40 ms, field size was 30 × 30 mm, matrix was 128 × 128, slice thickness was 0.5 mm, slice spacing was 0 mm, and 25 slices were taken for each brain. Infarct volumes were then calculated under blinded conditions.
Neurologic deficit scoring
A 5-point scoring system was employed to assess mice 48 hours after MCAO27. 1: normal function; 2: flexion of the torso and contralateral forelimb upon lifting of the animal by the tail; 3: circling to the contralateral side with normal posture at rest; 4: leaning to the contralateral side at rest; 5: no spontaneous motor activity.
Flow cytometry
48 hours after MCAO, brains were harvested. The cerebellum was excluded, and then hemispheres were separated into individual dishes. Brains were washed with sterile PBS on ice, and then minced with scissors. Samples were then transferred into gentleMACS tubes (Miltenyi) and cells were dissociated using a gentleMACS dissociator (Miltenyi). Samples were then passed through a sterile 18 g syringe ten times in order to obtain a single-cell suspension. Red blood cells were lysed by incubating samples with ACK (Ammonium-Chloride-Potassium) lysis buffer (Invitrogen) for 6 minutes at room temperature in the dark. Samples were then washed and then mixed with a 30% Percoll solution, and centrifuged at 1,400 × g at 4°C for 10 minutes to separate myelin. Cells were then washed twice in FACS buffer (0.5% FBS, 0.1% sodium azide in PBS), and then 100,000 cells were distributed into each tube for antibody staining. Each experiment was performed with isotype controls. Cells were blocked for 30 minutes on ice with Fc-block (BD Biosciences), and then fluorescently-labeled antibodies (F4/80 (FITC), CD45 (PE), Ly6G (FITC); BD Biosciences) were added for 30 minutes on ice in the dark. Samples were washed twice in FACS buffer, and kept in the dark, on ice, until ready for analysis using a Becton Dickenson FACScalibur.
Immunostaining
Whole brains of mice were harvested 6 hours, 48 hours, or 7 days after MCA occlusion, perfused with PBS, and embedded in Tissue-Tek OCT (Sakura Finetek). They were then frozen at −80° C prior to sectioning. Coronal sections were cut to a thickness of 10 μM and mounted on glass slides. Sections were kept at −80° C until staining. Slides were allowed to warm briefly before fixation in ice-cold acetone for 10 minutes. Slides were then stained using the Vectastain Elite kit for mouse-derived antibodies, and then the signal amplified using a tyramide signal amplification kit (Perkin-Elmer). CD39, P2X7 receptors, TNFα, and Iba-1 were detected using primary monoclonal antibodies (Abcam), and DAPI was used as a counter stain.
Isolation of bone marrow derived macrophages
Bone marrow macrophages were harvested from globally transgenic mice and wild type C57Bl/6 mice. Briefly, following humane euthanasia of mice, femurs were isolated, sterilized, and both ends of the bone severed. Femurs were flushed through with sterile media (DMEM-F12, 10% FBS, 10 mM glutamine), and then spun down in at 190 × g for 10 minutes. Cells were suspended in cell culture medium and grown with GM-CSF overnight. All media and floating cells were transferred and incubated for 3 days. More GM-CSF was added to media, after which cells were incubated for 4 more days. All cells were then split into appropriate experimental wells and no further passaging was performed.
Malachite green assay to measure apyrase activity
Bone marrow macrophages were harvested from wild type and CD39 globally-transgenic mice and plated in 24-well plates (Corning). Cells were washed twice in PBS before the addition of ATP or ADP. Supernatants were collected and transferred to a flat-bottom 96-well plate. Standards were run alongside samples, and were prepared using serial dilutions of a solution of known concentration of phosphate into the same background buffer as the samples being measured. Finally, the working solution for the malachite green assay was added (Bioassay Systems). Plates were incubated for 25 minutes before reading O.D. measurements at 620 nm.
Statistics
Figures and tables comparing two groups were analyzed by a Student’s t-test. Data are shown as mean +/− SEM with the exception of Supplemental Figure 1 which is presented as mean +/− SD. A mixed effect linear regression model was used to compare groups of animals for experiments in which the contralateral hemisphere was used as the control for the ipsilateral (stroked) hemisphere. Differences were considered significant if p was <0.05, except for the multiplex protein array analysis. For the multiplex protein array analysis, using the Bonferonni correction, differences were considered significant if p was < 0.0025.
Results
Identification of transgenic mice
Conditional transgenic CD39 mice were generated in order to investigate the protective nature of CD39 in the event of cerebral ischemia. A backbone containing a floxed-stop sequence in front of the start codon (Figure 1A) was used to generate transgenic mice in a pure C57Bl/6 background. Five transgenic founders were identified using a PCR screen for the inserted transgene (Figure 1B). Mice were then bred to EIIA-cre mice with a C57bl/6 background, and transgenic offspring expressing human CD39 were identified using a second PCR screen. Bone marrow was isolated from these mice in order to measure human CD39 mRNA expression in tissue. Transgenic mice expressed human CD39 mRNA, and endogenous levels of murine CD39 were no different from wild type C57BL/6 mice (Figure 1C). Western blots comparing tissue of wild type mice to transgenic mice shows that CD39 is ubiquitously over-expressed across a panel of different organ tissues. (Figure 1D and 1E). To confirm the functional significance vis-à-vis enzymatic (apyrase) activity of overexpressed CD39, a malachite green assay was performed (Figure 1F) on bone marrow derived macrophages of wild type and transgenic mice. This assay measures generation of inorganic phosphate from precursor substrates. These results demonstrate that phosphohydrolysis of ATP and ADP are significantly elevated in samples containing bone marrow-derived macrophages from transgenic mice compared to wild type mice. Complete blood counts were also taken (Table 1) from wild type and transgenic mice in order to determine whether any downstream differences could be accounted for by decreased platelet or leukocyte counts at baseline. These results show that across all parameters measured, there are no significant differences between the two groups. Similarly, heart rates and blood pressure were measured in WT, TG and LysM mice before and after MCAO, and there were no differences between these groups at any time point (Supplemental Figure 1).
Table 1. Complete blood counts for wild type and transgenic mice.
Complete blood counts were measured for wild type and transgenic mice (n=6), and transgenic mice do not differ from wild type mice in any parameter. Values are averages ± SEM and p-values determined by t-test.
| WT | TG | p-value (t-test) |
|
|---|---|---|---|
| White blood cell (K/μL) | 2.29 ± .63 | 2.95 ± .99 | ns |
| Neutrophils (K/μL) | 0.15 ± 0.05 | 0.4 ± 0.19 | ns |
| Lymphocytes (K/μL) | 2.08 ± .6 | 2.5 ± .79 | ns |
| Monocyte (K/μL) | 0.22 ± 0.02 | 0.042 ± 0.01 | ns |
| Eosinophil (K/μL) | 0.005 ± 0.00 | 0.01 ± 0.00 | ns |
| Basophil (K/μL) | 0.002 ± 0.00 | 0.003 ± 0.00 | ns |
| Red blood cell (M/μL) | 7.23 ± .50 | 7.49 ± .49 | ns |
| Hemoglobin (g/dL) | 9.93 ± .81 | 10.067 ± .62 | ns |
| Hematocrit (decimal fraction) | .36 ± .03 | .37 ± .02 | ns |
| Mean corpuscular volume (fL) | 49.7 ± .52 | 48.72 ± 0.29 | ns |
| Mean corpuscular hemoglobin (pg) | 13.65 ± 0.3 | 13.47 ± 0.18 | ns |
| Mean corpuscular hemoglobin concentration (g/dL) | 27.47 ± .46 | 27.63 ± .42 | ns |
| Red cell distribution width (%) | 15.45 ± 0.24 | 16.57 ± .49 | ns |
| Platelets (K/μL) | 246.67 ± 47.19 | 262.33 ± 60.99 | ns |
CD39 mice are protected from effects of ischemia
Forty-eight hours after the induction of cerebral ischemia, brains were harvested, and ischemic hemispheres were compared to contralateral hemispheres using flow cytometry. Recruitment of macrophages was drastically suppressed in ischemic hemispheres of transgenic mice (Figure 2A). Similarly, recruitment of neutrophils was also suppressed in the ischemic hemispheres of transgenic mice (Figure 2B). Cell counts for total leukocytes, macrophages and neutrophils in the non-stroke (contralateral) hemisphere did not differ between wild type and transgenic mice, but there was significantly less inflammation in transgenic ischemic hemispheres compared to wild type ischemic hemispheres (Figure 2C). The suppression of inflammation in ischemic hemispheres of transgenic mice compared to wild type mice corresponded to smaller cerebral infarct volumes. In terms of microglia, it seems that EIIA-Cre recombination is low in microglia in comparison to bone marrow derived macrophages (Supplemental Figure 2), which may indicate that differences observed between wild type and transgenic infarcted brains is driven largely by infiltrating cells as opposed to resident cells. Magnetic resonance imaging (MRI) scans obtained from transgenic mice 48 hours after induction of ischemia demonstrated 20% reduced cerebral infarct volumes compared to wild type mice. (Figures 3A, B, C). Mice were scored according to a 5-point neurological deficit scale 48 hours after MCAO. CD39 transgenic mice exhibited less neurological deficit than their wild type controls (Figure 3D).
Figure 2.
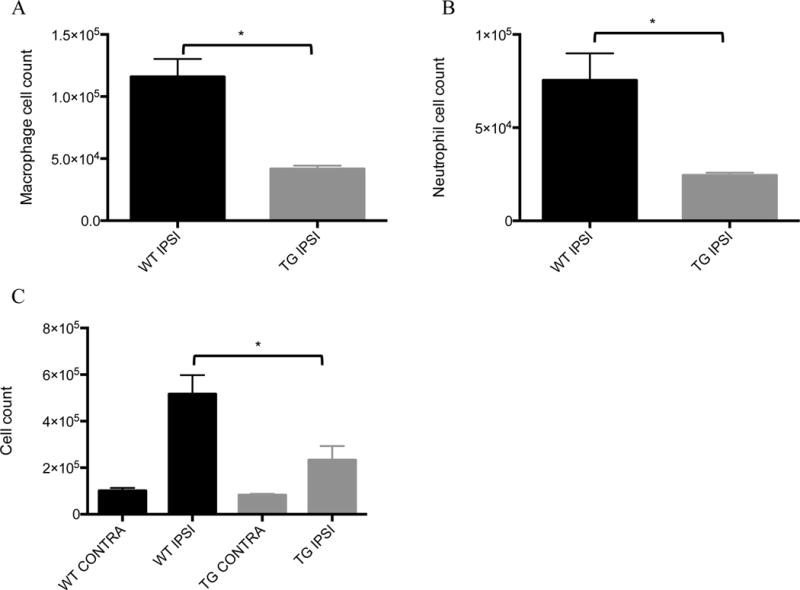
Myeloid cell infiltration after stroke: Middle cerebral arteries of wild type and transgenic mice were occluded, and then 48 hours later, left and right cerebral hemispheres were harvested for further analysis by flow cytometry. Cells were then stained with A) F4/80 (FITC) and CD45 (PE) to identify macrophages and B) Ly6g (FITC) and CD45 (PE) to identify neutrophils. Data were analyzed using a t-test with Welch’s modification. 4 mice were analyzed for each group. C) Leukocytes were purified of myelin and cellular debris, and total leukocyte counts were obtained. Data were analyzed using a mixed effect linear regression model. 6 mice were analyzed for each group. * p < 0.05.
Figure 3.
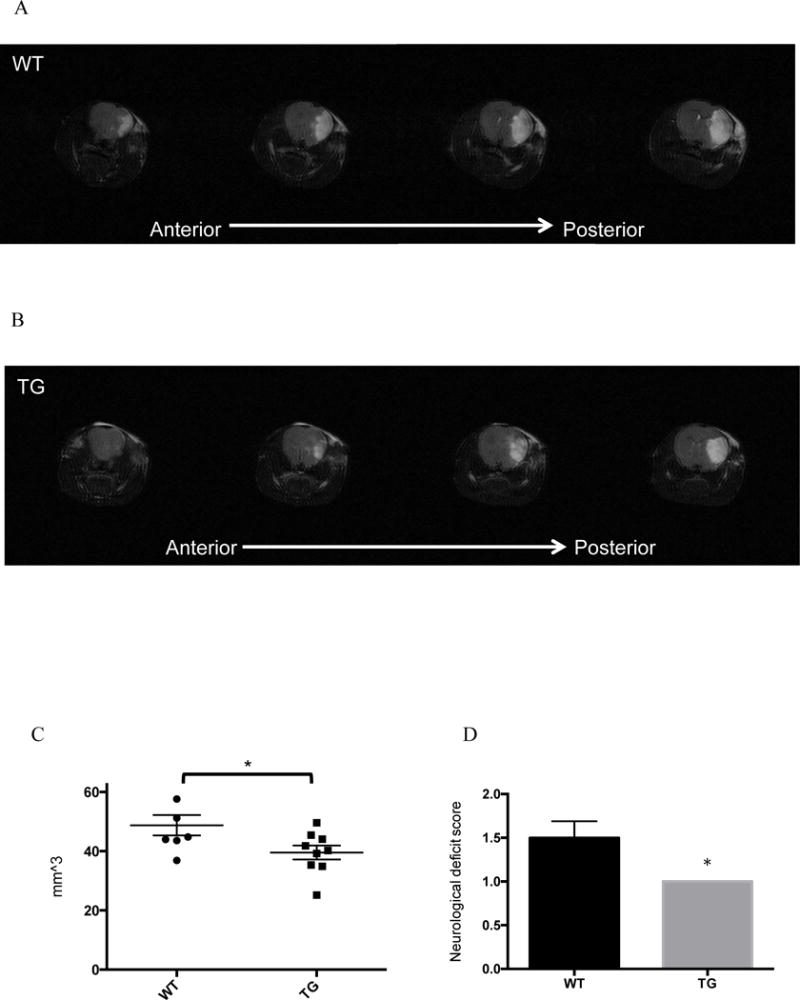
Cerebral infarct volumes after stroke: Middle cerebral arteries of wild type and transgenic mice were occluded, and for 48 hours, and then 48 hours later mice were anesthetized in order to image brains via MRI. Scans of wild type (A) and transgenic (B) mouse brains are shown in coronal slices moving from anterior to posterior sections, where white areas indicate edematous tissue. C) Cerebral infarct volumes were calculated by integrating infarct area across all slices. Central horizontal bars indicate mean and flanking bars represents SEM. * p < 0.05. D) Mice were scored for neurological deficit 48 hours after MCAO. Higher scores indicate higher levels of neurological impairment. Transgenic mice all scored lowest on the scale with a value of 1. 6 WT mice and 9 transgenic mice were analyzed.
CD39 overexpression leads to differential cytokine expression following cerebral ischemia
Six hours after inducing cerebral ischemia, brains were harvested and contralateral and ipsilateral hemispheres were isolated separately, and homogenized in order to analyze cytokine expression via a multiplex protein array (Figure 4 and Supplemental Figure 3). Cytokine/chemokine expression levels in contralateral hemispheres of wild type and transgenic mice were not different from each other. However, ischemic hemispheres of wild type mice showed elevated levels of pro-inflammatory cytokines and chemokines. Classically designated anti-inflammatory cytokines showed mixed patterns of expression. IL-4 showed no significant differences between any of the hemispheres, whereas wild type ipsilateral hemispheres showed greater IL-5 expression compared to transgenic ipsilateral hemispheres. IL-6 was also more highly expressed in ischemic wild type cerebral hemispheres. TNF-α was elevated in wild type ipsilateral (ischemic) hemispheres and conversely suppressed in transgenic mice. TNF-α expression across both ischemic and non-ischemic hemispheres in wild type and transgenic mice was visualized (Figure 5A). TNF-α expression in the ischemic hemisphere of transgenic mice was lower relative to the wild type mouse. CD39 was elevated in the transgenic mouse compared to wild type mice, as expected.
Figure 4.
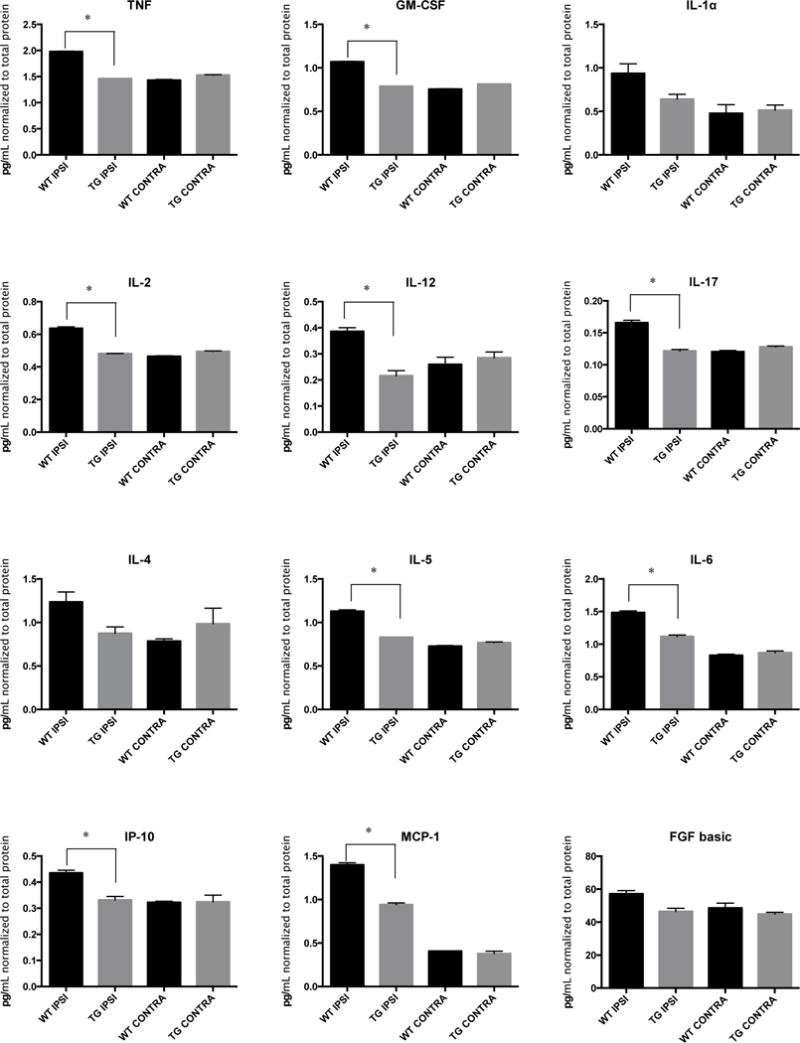
Cytokine profiles in brains after stroke: Middle cerebral arteries of wild type and transgenic mice were occluded, and 6 hours later, left and right cerebral hemispheres were harvested and total protein purified. A panel of cytokine levels was measured using a multiplex bead assay. Cytokine levels in ischemic (ipsilateral) hemispheres were compared to contralateral hemispheres. n = 3 mice for each group. Data was analyzed using a mixed effect linear regression model with a Bonferroni correction. * p < 0.0025.
Figure 5.
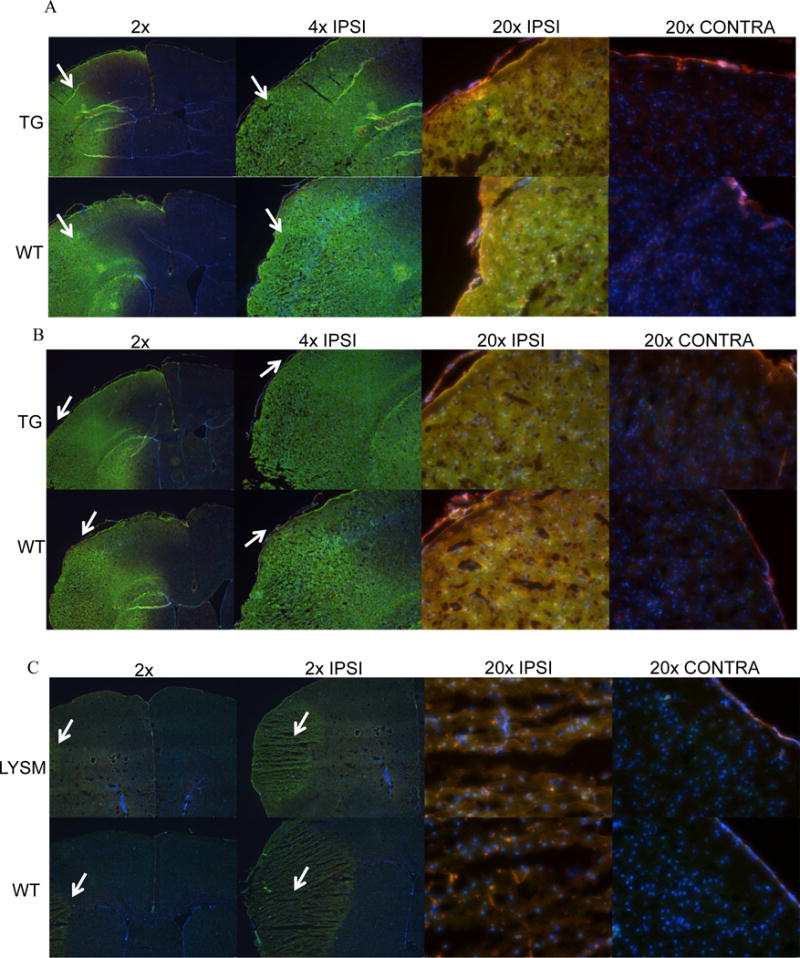
Expression of TNF-α and P2X7 receptor in brains after stroke: Middle cerebral arteries (MCA) of wild type and transgenic mice were occluded and were later frozen in OCT at either 6 or 48 hours after MCA occlusion. Coronal sections of transgenic and wild type mouse brains were stained for A) TNF-α shown in green and CD39 shown in red 6 hours after occlusion, B) P2X7 receptor shown in green and CD39 shown in red 6 hours after occlusion, and C) P2X7 receptor shown in green and CD39 shown in red 48 hours after occlusion in LysM-Cre/CD39 transgenic mice. DAPI was used as a counter stain in all panels.
Since ATP is a substrate for both the P2Y and P2X families of purinergic receptors, but the P2X7 receptor is specifically activated at the high concentrations of extracellular ATP (greater than 100 μM) present in points of inflammation and damaged tissue28, 29, we next examined the expression of P2X7 in this model. Figure 5B shows the distribution of P2X7 receptors after stroke. P2X7 receptor is widely expressed in macrophages and other circulating leukocytes30, and it was seen in the ischemic hemispheres of both mice, whereas non-ischemic hemispheres appeared unaffected. Figure 5C shows coronal sections of tissue-specific, LysM-Cre/CD39 transgenic mice. These mice overexpress CD39 in a tissue-specific manner in myeloid lineage cells only. After 48 hours of stroke, the area affected by MCAO was much smaller in LysM-Cre transgenic mouse brains. Notably, there was no difference in CD39 expression between the contralateral hemispheres of wild type and LysM-Cre transgenic mice.
MRI scans were performed on LysM-Cre transgenic mice 48 hours after MCAO (Figure 6A). As with globally CD39-overexpressing transgenic mice, CD39-LysM-Cre transgenic mice were protected. LysM-Cre transgenic mice exhibited smaller cerebral infarct volumes compared to wild type mice (Figure 6B). To assess the longer-term impact of myeloid-cell restricted overexpression of CD39 on cerebral infarct volumes, both WT and CD39-LysM Cre transgenic mice were subjected to MCAO and their brains analyzed 1 week later (Figure 6C and 6D). The protective effect of myeloid overexpression of CD39 was also observed one week after MCAO, where LysM-Cre transgenic mice exhibited smaller cerebral infarct volumes compared to wild type mice.
Figure 6.
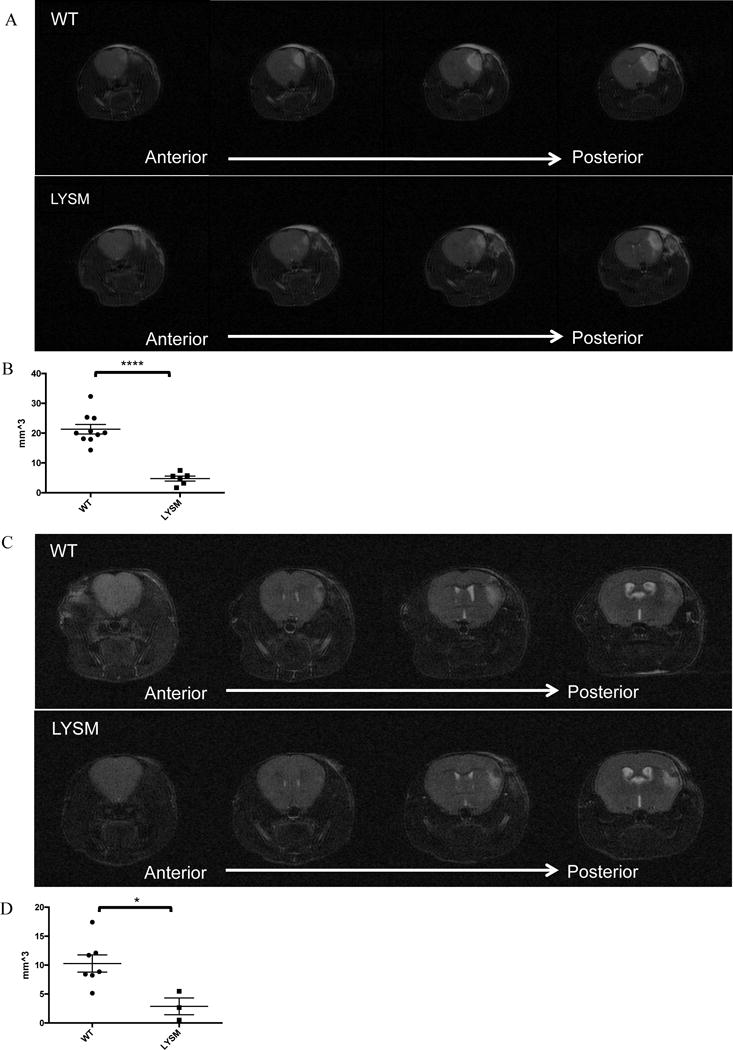
Cerebral infarct volumes after stroke in LysM-Cre transgenic mice: Middle cerebral arteries of wild type and LysM-Cre transgenic mice were occluded, and 48 hours later, mice were anesthetized in order to image brains via MRI. A) Scans of wild type and LysM-Cre transgenic mouse brains are shown in coronal slices moving from anterior to posterior sections, where white areas indicate edematous tissue. B) Cerebral infarct volumes were calculated by integrating infarct area across all slices. 10 WT animals and 6 LysM mice were analyzed. ****p<0.0001. C) MCA of wild type and LysM-Cre transgenic mice were occluded, and after 7 days, were imaged by MRI. D) Cerebral infarct volumes were calculated by integrating infarct area across all slices. 7 WT animals and 3 LysM mice were analyzed. * p < 0.05. Central horizontal bars indicate mean and flanking bars represents SEM.
Discussion
Though some effective fibrinolytic and anti-platelet treatments exist which promote reperfusion in ischemic brain tissue, alternative approaches remain a priority, as current treatments are limited and are associated with complications such as an increased risk of intracerebral hemorrhage. Treatment with fibrinolytics is confined to a narrow time frame in which the neurologic benefit outweighs the risk of bleeding, a maximum of only 4.5 hours from symptom onset31. As a result, only a small portion of those with ischemic stroke benefit from this intervention1. Reperfusion injury to the ischemic region is a well-known risk of thrombolytic treatment and can result in cellular death, edema, and necrosis, and may exacerbate tissue damage32–34. Increased leukocyte infiltration and release of reactive oxygen species (ROS), as well as increased platelet activation contribute to this injury34, 35. Therefore, the search for more selective methods of controlling the brain tissue response to ischemia continues, wherein anti-thrombotic and anti-inflammatory mechanisms can be employed in a cell-specific and directed manner. The model employed in this study investigates the actions of CD39 in the absence of reperfusion, and thus aims to demonstrate the efficacy of CD39 overexpression on limiting inflammation and infarct size due to ischemia.
Previous work has readily established CD39 (ENTPDase1) as a strong mediator of thrombosis and inflammation12, 36–38 ATP and ADP, potent triggers of inflammation and thrombosis, respectively, can be present at high concentrations in localized areas of tissue damage. It is this initial pro-inflammatory signal of ATP, which occurs with even minor perturbations of the cellular membrane, that the activity of CD39 is positioned to dissipate. The apyrase activity of CD39 cleaves ATP and ADP to AMP, and the co-expressed partner enzyme of CD39, CD73, completes the cleavage of AMP to adenosine, which modulates inflammation and may protect against ischemic injury39–42. Combined with our work which uses a permanent occlusion model, this suggests that CD39 or its downstream signaling pathway has a high therapeutic potential to limit ischemic brain injury.
Confirmation of the transgenic mouse design
We developed a transgenic mouse that globally expresses elevated levels of CD39, which has the potential to be used to generate other cell-specific CD39-overexpressing mice, to express elevated levels of CD39 in specific tissues. To accomplish this, a vector containing a floxed stop sequence between the promoter and the transcriptional start sequence of human CD39 was obtained, such that in the presence of the enzyme Cre recombinase43, 44, the stop sequence would be deleted and expression of human CD39 would be driven by a CAG promoter/enhancer sequence. This mouse was developed to aid in future studies of CD39 which center on questions of the role of CD39 in specific tissues, and further characterization of CD39 function across different disease models. We have demonstrated that in our CD39 transgenic mice, human CD39 is detectable in DNA, RNA, and in protein samples. We are able to distinguish between the endogenous and transgene at the RNA level, and furthermore, we can see that this additional expression of CD39 elevates the total CD39 protein across a panel of different tissues.
Having confirmed the expected genotype, protein, and RNA expression for the globally-transgenic mice, we have shown that CD39 overexpression is functional at baseline, as bone marrow-derived macrophages from transgenic mice exhibited more apyrase activity compared to wild type. Our studies have shown that this baseline difference in apyrase activity conferred a protective effect in transgenic mice against inflammation and tissue death after a cerebral ischemic insult. Most intriguingly, it can be seen that mice that overexpress CD39 specifically in their myeloid lineage cells are also protected in terms of infarct size. The mice used in this study were adult mice (mean age 10 weeks old). Future experiments with older mice, female mice, or mice with hyperlipidemia or hypertension could broaden the applicability of our findings.
In the globally CD39 transgenic mice, while the proportions of infiltrating macrophages and neutrophils were not affected, total inflammation was significantly decreased in the first 48 hours after stroke. This difference was confirmed by MRIs showing a far smaller stroke volume, as well as by neurological scoring reflecting fewer neurological deficits in transgenic mice when compared with their wild type counterparts. The time points examined in this study are within the timeframe in which inflammation has been shown to be increasing after MCAO.45, 46
We investigated whether the smaller infarct volumes observed in transgenic mice were accompanied by expression patterns of cytokines previously demonstrated to be important drivers of tissue damage in cerebral ischemia. A multiplex cytokine array was chosen because it allows the simultaneous and highly sensitive measurement of many cytokines from the same samples under the same experimental conditions, thereby giving more consistent and reliable measurements within the sample set47. Measuring many cytokines of interest together, rather than focusing on just a limited few, presents a complete representation of inflammatory mediators within the tissue. Concordant with the anatomic cerebral and functional protection in stroke conferred by transgenic overexpression of CD39, data from a cytokine/chemokine array performed on tissue from mice post-ischemic stroke revealed a less inflammatory post-stroke phenotype in CD39 transgenic mice. A general pattern of cytokine expression manifested. Across classically pro-inflammatory cytokines, levels in transgenic CD39 ischemic hemispheres were uniformly lower than in the wild-type ischemic hemispheres. Contralateral non-ischemic hemispheres, on the other hand, which served as controls, did not appear to differ between the two genotypes.
In sum, these data support a generally anti-inflammatory state in the transgenic human CD39 overexpressing mice we have developed compared to wild type mice, even when challenged with a profound interruption of blood supply to a critical brain region. The mice we have generated do not appear to be phenotypically different from wild type mice in any of the parameters tested, thus supporting our hypothesis that CD39 overexpression can protect against CNS inflammation and tissue death. The role of TNF-α in models of cerebral ischemia remains debated. The onset of cerebral ischemia is marked by an immediate increase in levels of TNF-α within 1–3 hours of injury, followed by a second peak at 24–36 hours48–50, and microglia and infiltrating leukocytes are largely responsible for this response51. Whereas elevated levels of TNF-α in acute stroke have been associated with impaired survival, inhibition of TNF-α in the long term has been shown to impede repair52–54. Elevation of TNF-α has been shown to increase cerebral infarct volumes in a model of cerebral ischemia55, whereas tolerance to ischemic injury via ischemic pre-conditioning is associated with elevated TNF-α expression56. In this current study, we found that elevated TNF-α was associated with greater ischemic damage, and that the overexpression of CD39 suppressed inflammation. This was associated with diminished expression of TNF-α in the ischemic hemisphere of transgenic mouse brains. The decreased TNF-α expression at early ischemic time points in transgenic mice was seen in both direct measurements of the cytokine in ischemic tissue, as well as in staining of coronal brain sections. In these samples, not only did the ischemic hemispheres of transgenic mice contain less TNF-α, but the expression of TNF-α was localized to a much smaller area relative to wild type mice.
Future therapies derived from understanding CD39 and its physiologic roles may offer an alternative to currently available thienopyridine anti-platelet therapies, which disable platelet function by binding P2Y12 receptors. This is due to the fact that CD39 phosphohydrolyzes ADP, the substrate of P2Y12 receptors, and so mitigates platelet activation indirectly. Our previously published work has demonstrated that upregulation of endogenous CD39 is possible through the use of PDEIII inhibiting drugs that are clinically used57, 58, which gives further translational relevance to our findings in this paper. Repurposing of existing drugs may in the future be used to drive expression of the endogenous CD39 gene, or delivery of compounds which mimic CD39 phosphohydrolytic activity may be an alternative strategy. In either case, the studies presented here suggest that catalytic cleave of purinergic nucleotides could represent an important therapeutic target in stroke.
Supplementary Material
Clinical Perspective.
What is New?
CD39 is an ectoenzyme with apyrase activity, which cleaves ATP and ADP to form AMP, that is in turn converted to adenosine by CD73.
CD39 is expressed on the surface of myeloid and vascular endothelial cells, where it dissipates high local concentrations of ATP and ADP, which would otherwise serve as potent pro-inflammatory and pro-thrombotic signals, respectively.
In this work, for the first time, we show that the genetic overexpression of CD39 limits the brain infarct volume and neurologic deficits caused by cerebral ischemia.
What are the Clinical Implications?
In our model of permanent middle cerebral artery occlusion, CD39 overexpression reduced edema, infarct volume, and inflammation, with corresponding improvements in neurological outcomes compared to control mice.
Overexpression of CD39 in only myeloid cells also reduced cerebral infarct volume.
Amplification of endogenous CD39 expression or even administration of exogenous circulating CD39 could be of future interest as a therapeutic target, for minimizing ischemic injury caused by cerebral ischemia.
Acknowledgments
We thank the University of Michigan Transgenic Animal Model Core for generating our transgenic founder mice, the University of Michigan DNA Sequencing Core for sequencing the constructs used to generate transgenic mice, and Steven Whitesall of the University of Michigan Physiology Phenotyping Core for performing heart rate and blood pressure measurements.
Sources of Funding:
This work was funded by an American Heart Association Pre-Doctoral fellowship (AEB), the Systems and Integrative Biology Training Grant (T32 GM008322 (AEB)), NIH/NHLBI (National Heart, Lung, and Blood Institute) (R01NS087147 (DJP), R01HL127151 (DJP), T32 HL007853 (NRS, YK, DJP)), NIH (K08HL131993 (YK)), the J. Griswold Ruth and Margery Hopkins Ruth Professorship in Internal Medicine, and the A. Alfred Taubman Medical Research Institute.
Footnotes
Disclosures:
There is nothing to disclose.
References
- 1.Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, Cohen G. Recombinant tissue plasminogen activator for acute ischaemic stroke: An updated systematic review and meta-analysis. Lancet. 2012;379:2364–2372. doi: 10.1016/S0140-6736(12)60738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marler JR, Goldstein LB. Medicine. Stroke–tpa and the clinic. Science. 2003;301:1677. doi: 10.1126/science.1090270. [DOI] [PubMed] [Google Scholar]
- 3.Adams H, Adams R, Del Zoppo G, Goldstein LB. Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update a scientific statement from the stroke council of the american heart association/american stroke association. Stroke. 2005;36:916–923. doi: 10.1161/01.STR.0000163257.66207.2d. [DOI] [PubMed] [Google Scholar]
- 4.Jauch EC, Saver JL, Adams HP, Jr, Bruno A, Connors JJ, Demaerschalk BM, Khatri P, McMullan PW, Jr, Qureshi AI, Rosenfield K, Scott PA, Summers DR, Wang DZ, Wintermark M, Yonas H. Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the american heart association/american stroke association. Stroke. 2013;44:870–947. doi: 10.1161/STR.0b013e318284056a. [DOI] [PubMed] [Google Scholar]
- 5.Herz J, Sabellek P, Lane TE, Gunzer M, Hermann DM, Doeppner TR. Role of neutrophils in exacerbation of brain injury after focal cerebral ischemia in hyperlipidemic mice. Stroke. 2015;46:2916–2925. doi: 10.1161/STROKEAHA.115.010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jalkanen J, Yegutkin GG, Hollmen M, Aalto K, Kiviniemi T, Salomaa V, Jalkanen S, Hakovirta H. Aberrant circulating levels of purinergic signaling markers are associated with several key aspects of peripheral atherosclerosis and thrombosis. Circ Res. 2015;116:1206–1215. doi: 10.1161/CIRCRESAHA.116.305715. [DOI] [PubMed] [Google Scholar]
- 8.Fung CY, Marcus AJ, Broekman MJ, Mahaut-Smith MP. P2x(1) receptor inhibition and soluble cd39 administration as novel approaches to widen the cardiovascular therapeutic window. Trends Cardiovasc Med. 2009;19:1–5. doi: 10.1016/j.tcm.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qawi I, Robson SC. New developments in anti-platelet therapies: Potential use of cd39/vascular atp diphosphohydrolase in thrombotic disorders. Curr Drug Targets. 2000;1:285–296. doi: 10.2174/1389450003349173. [DOI] [PubMed] [Google Scholar]
- 10.Marcus AJ, Broekman MJ, Drosopoulos JH, Pinsky DJ, Islam N, Maliszewsk CR. Inhibition of platelet recruitment by endothelial cell cd39/ecto-adpase: Significance for occlusive vascular diseases. Ital Heart J. 2001;2:824–830. [PubMed] [Google Scholar]
- 11.Kanthi Y, Hyman MC, Liao H, Baek AE, Visovatti SH, Sutton NR, Goonewardena SN, Neral MK, Jo H, Pinsky DJ. Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J Clin Invest. 2015;125:3027–3036. doi: 10.1172/JCI79514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanthi YM, Sutton NR, Pinsky DJ. Cd39: Interface between vascular thrombosis and inflammation. Curr Atheroscler Rep. 2014;16:425. doi: 10.1007/s11883-014-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, Bach FH, Robson SC. Identification and characterization of cd39/vascular atp diphosphohydrolase. J Biol Chem. 1996;271:33116–33122. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- 14.Csoka B, Nemeth ZH, Toro G, Koscso B, Kokai E, Robson SC, Enjyoji K, Rolandelli RH, Erdelyi K, Pacher P, Hasko G. Cd39 improves survival in microbial sepsis by attenuating systemic inflammation. Faseb J. 2015;29:25–36. doi: 10.1096/fj.14-253567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367:2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dwyer KM, Robson SC, Nandurkar HH, Campbell DJ, Gock H, Murray-Segal LJ, Fisicaro N, Mysore TB, Kaczmarek E, Cowan PJ, d’Apice AJ. Thromboregulatory manifestations in human cd39 transgenic mice and the implications for thrombotic disease and transplantation. J Clin Invest. 2004;113:1440–1446. doi: 10.1172/JCI19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 18.Chiang T, Messing RO, Chou WH. Mouse model of middle cerebral artery occlusion. J Vis Exp. 2011;48:2761. doi: 10.3791/2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, Gao Y, Pietras K, Mann K, Yepes M, Strickland DK, Betsholtz C, Eriksson U, Lawrence DA. Activation of pdgf-cc by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–737. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrovic-Djergovic D, Hyman MC, Ray JJ, Bouis D, Visovatti SH, Hayasaki T, Pinsky DJ. Tissue-resident ecto-5′ nucleotidase (cd73) regulates leukocyte trafficking in the ischemic brain. J Immunol. 2012;188:2387–2398. doi: 10.4049/jimmunol.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durukan A, Tatlisumak T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87:179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 22.Handa M, Guidotti G. Purification and cloning of a soluble atp-diphosphohydrolase (apyrase) from potato tubers (solanum tuberosum) Biochem Biophys Res Commun. 1996;218:916–923. doi: 10.1006/bbrc.1996.0162. [DOI] [PubMed] [Google Scholar]
- 23.Chadwick BP, Williamson J, Sheer D, Frischauf AM. Cdna cloning and chromosomal mapping of a mouse gene with homology to ntpases. Mamm Genome. 1998;9:162–164. doi: 10.1007/s003359900710. [DOI] [PubMed] [Google Scholar]
- 24.Li T, Hung MS, Wang Y, Mao JH, Tan JL, Jahan K, Roos H, Xu Z, Jablons DM, You L. Transgenic mice for cre-inducible overexpression of the cul4a gene. Genesis. 2011;49:134–141. doi: 10.1002/dvg.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sunmonu NA, Chen L, Li JY. Misexpression of gbx2 throughout the mesencephalon by a conditional gain-of-function transgene leads to deletion of the midbrain and cerebellum in mice. Genesis. 2009;47:667–673. doi: 10.1002/dvg.20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murthy SC, Bhat GP, Thimmappaya B. Adenovirus eiia early promoter: Transcriptional control elements and induction by the viral pre-early eia gene, which appears to be sequence independent. Proc Natl Acad Sci U S A. 1985;82:2230–2234. doi: 10.1073/pnas.82.8.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hata R, Mies G, Wiessner C, Fritze K, Hesselbarth D, Brinker G, Hossmann KA. A reproducible model of middle cerebral artery occlusion in mice: Hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18:367–375. doi: 10.1097/00004647-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 28.North RA. Molecular physiology of p2x receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 29.Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic p2z receptor for extracellular atp identified as a p2x receptor (p2x7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- 30.Khakh BS, North RA. P2x receptors as cell-surface atp sensors in health and disease. Nature. 2006;442:527–532. doi: 10.1038/nature04886. [DOI] [PubMed] [Google Scholar]
- 31.Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D, Investigators E Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 32.van Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, Cairns B, Tumas D, Gerlai R, Williams SP, van Lookeren Campagne M, Ferrara N. Vegf antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. doi: 10.1172/JCI8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nour M, Scalzo F, Liebeskind DS. Ischemia-reperfusion injury in stroke. Interv Neurol. 2013;1:185–199. doi: 10.1159/000353125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan J, Konstas AA, Bateman B, Ortolano GA, Pile-Spellman J. Reperfusion injury following cerebral ischemia: Pathophysiology, mr imaging, and potential therapies. Neuroradiology. 2007;49:93–102. doi: 10.1007/s00234-006-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 36.Antonioli L, Pacher P, Vizi ES, Hasko G. Cd39 and cd73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen HB, Briggs KT, Marino JP, Ravid K, Robson SC, Mosser DM. Tlr stimulation initiates a cd39-based autoregulatory mechanism that limits macrophage inflammatory responses. Blood. 2013;122:1935–1945. doi: 10.1182/blood-2013-04-496216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutton NR, Hayasaki T, Hyman MC, Anyanwu AC, Liao H, Petrovic-Djergovic D, Badri L, Baek AE, Walker N, Fukase K, Kanthi Y, Visovatti SH, Horste EL, Ray JJ, Goonewardena SN, Pinsky DJ. Ectonucleotidase cd39-driven control of postinfarction myocardial repair and rupture. JCI insight. 2017;2:e89504. doi: 10.1172/jci.insight.89504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui M, Ding H, Chen F, Zhao Y, Yang Q, Dong Q. Mdivi-1 protects against ischemic brain injury via elevating extracellular adenosine in a camp/creb-cd39-dependent manner. Mol Neurobiol. 2016;53:240–253. doi: 10.1007/s12035-014-9002-4. [DOI] [PubMed] [Google Scholar]
- 40.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Kim M, Song JH, Lee HT. Endogenous a1 adenosine receptors protect against hepatic ischemia reperfusion injury in mice. Liver Transpl. 2008;14:845–854. doi: 10.1002/lt.21432. [DOI] [PubMed] [Google Scholar]
- 42.Hyman MC, Petrovic-Djergovic D, Visovatti SH, Liao H, Yanamadala S, Bouis D, Su EJ, Lawrence DA, Broekman MJ, Marcus AJ, Pinsky DJ. Self-regulation of inflammatory cell trafficking in mice by the leukocyte surface apyrase cd39. J Clin Invest. 2009;119:1136–1149. doi: 10.1172/JCI36433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ansermot N, Albayrak O, Schlapfer J, Crettol S, Croquette-Krokar M, Bourquin M, Deglon JJ, Faouzi M, Scherbaum N, Eap CB. Substitution of (r,s)-methadone by (r)-methadone: Impact on qtc interval. Arch Intern Med. 2010;170:529–536. doi: 10.1001/archinternmed.2010.26. [DOI] [PubMed] [Google Scholar]
- 44.Ding H, Wu X, Nagy A. Mice with cre recombinase activatable pdgf-c expression. Genesis. 2002;32:181–183. doi: 10.1002/gene.10065. [DOI] [PubMed] [Google Scholar]
- 45.Zhang RL, Chopp M, Chen H, Garcia JH. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2h) middle cerebral artery occlusion in the rat. J Neurol Sci. 1994;125:3–10. doi: 10.1016/0022-510x(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 46.Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (wistar rat) Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- 47.Leng SX, McElhaney JE, Walston JD, Xie D, Fedarko NS, Kuchel GA. Elisa and multiplex technologies for cytokine measurement in inflammation and aging research. J Gerontol A Biol Sci Med Sci. 2008;63:879–884. doi: 10.1093/gerona/63.8.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: Modulatory effects of hypothermia. J neuroinflammation. 2010;7:74. doi: 10.1186/1742-2094-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 50.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 51.Dziewulska D, Mossakowski MJ. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin Neuropathol. 2003;22:35–40. [PubMed] [Google Scholar]
- 52.Heldmann U, Thored P, Claasen JH, Arvidsson A, Kokaia Z, Lindvall O. Tnf-alpha antibody infusion impairs survival of stroke-generated neuroblasts in adult rat brain. Exp Neurol. 2005;196:204–208. doi: 10.1016/j.expneurol.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Feuerstein GZ, Xu L, Wang H, Schumacher WA, Ogletree ML, Taub R, Duan JJ, Decicco CP, Liu RQ. Inhibition of tumor necrosis factor-alpha-converting enzyme by a selective antagonist protects brain from focal ischemic injury in rats. Mol Pharmacol. 2004;65:890–896. doi: 10.1124/mol.65.4.890. [DOI] [PubMed] [Google Scholar]
- 54.Pan W, Kastin AJ. Tumor necrosis factor and stroke: Role of the blood-brain barrier. Progress in neurobiology. 2007;83:363–374. doi: 10.1016/j.pneurobio.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pettigrew LC, Kindy MS, Scheff S, Springer JE, Kryscio RJ, Li Y, Grass DS. Focal cerebral ischemia in the tnfalpha-transgenic rat. J Neuroinflammation. 2008;5:47. doi: 10.1186/1742-2094-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pradillo JM, Romera C, Hurtado O, Cardenas A, Moro MA, Leza JC, Davalos A, Castillo J, Lorenzo P, Lizasoain I. Tnfr1 upregulation mediates tolerance after brain ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:193–203. doi: 10.1038/sj.jcbfm.9600019. [DOI] [PubMed] [Google Scholar]
- 57.Baek AE, Kanthi Y, Sutton NR, Liao H, Pinsky DJ. Regulation of ecto-apyrase cd39 (entpd1) expression by phosphodiesterase iii (pde3) Faseb J. 2013;27:4419–4428. doi: 10.1096/fj.13-234625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao H, Hyman MC, Baek AE, Fukase K, Pinsky DJ. Camp/creb-mediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (cd39) expression. J Biol Chem. 2010;285:14791–14805. doi: 10.1074/jbc.M110.116905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.