

Abstract
We describe the parallel synthesis of novel analogs of GW0742, a peroxisome proliferator-activated receptor δ (PPARδ) agonist. For that purpose, modified reaction conditions were applied, such as a solid-phase palladium-catalyzed Suzuki coupling. In addition, tetrazole-based compounds were generated as a bioisostere for carboxylic acid-containing ligand GW0742. The new compounds were investigated for their ability to activate PPARδ mediated transcription and their cross-reactivity with the vitamin D receptor (VDR), another member of the nuclear receptor superfamily. We identified many potent PPARδ agonists that were less toxic than GW0742, where ~65 of the compounds synthesized exhibited partial PPARδ-activity (23-98%) with EC50 values ranging from 0.007 – 18.2 μM. Some ligands, such as compound 32, were more potent inhibitors of VDR-mediated transcription with significantly reduced PPARδ activity than GW0742, however, none of the ligands were completely selective for VDR inhibition over PPARδ activation of transcription.
Keywords: Nuclear Receptor (NR); Vitamin D Receptor (VDR); Peroxisome Proliferation-Activated Receptors α, δ, γ (PPAR α, δ, γ); GW0742; steroid receptor coactivator 2
INTRODUCTION
Nuclear receptors (NRs) are ligand-activated transcription factors representing one of the largest classes of proteins targeted for drug intervention.1 Thus far, 48 identified proteins belong to this superfamily, which is typically subdivided into three classes that share similar protein structure and ligand binding. These classes are steroid receptors, retinoid X receptor (RXR) heterodimers, and orphan receptors.2 Regardless of their class, NRs are modular proteins that contain a DNA-binding domain (DBD) and a ligand-binding domain (LBD). The LBD is a highly structured region that contains the ligand-dependent activation function or AF-2, which acts as the major interface for the recruitment of coactivators or corepressors. In general, the NR-LBD consists of a three-layered α-helical sandwich containing up to 12 helices designated as H1–H12. In the absence of endogenous ligand, corepressor proteins of the SMRT/NCoR family might associate with NR-LBD and block transcription.3 In the presence of ligand, NRs undergo a conformational change that both stabilizes the binding of the ligand and accommodates coactivator proteins such as SRC-1 that bind the hydrophobic cleft formed by rearranged H12.4
The vitamin D receptor (VDR) and peroxisome proliferator-activated receptor δ (PPARδ) form RXR heterodimers. The biological role of PPARδ has only recently been characterized, in part, due to its broad tissue expression and the lack of selective chemical probes to distinguish the pharmacology between the PPAR isoforms (δ, α, γ). To help reveal the biological function of PPARδ, GlaxoSmithKline (GSK) used combinatorial chemistry and structure-based drug design to develop GW0742 and GW501516, both highly selective and potent PPARδ agonists (EC50 = ~0.001 μM).5 Since their discovery, it has been revealed that PPARδ plays a role in lipid, lipoprotein, and glucose homeostasis thus making it a suitable target for hypertension,6 diabetes7, obesity, inflammation8 and cancer9 therapies. Although GW501516 has shown beneficial effects in primate models of metabolic disorders,5b, 10 only PPARα and PPARγ ligands have been approved for human use. Fibrates (PPAR α ligands) and thiazolidinethiones (PPARγ ligands) are prescribed for the treatment of hypercholesterolemia and diabetes mellitus type 2, respectively, but have shown adverse side effects after long-term use possibly due to poor isoform selectivity and off-target effects.11 Due to its complementary role in lipid homeostasis and energy metabolism, a strong interest in developing selective PPARδ ligands without adverse side effects has emerged. VDR, on the other hand, mediates the biological action of the active form of vitamin D, 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3) and regulates calcium homeostasis, immunity, and cellular growth and differentiation.12 Although a lot of research has been directed towards developing new VDR modulators, there still remains a need for potent and VDR selective drugs to treat diseases like osteoporosis, cancer, inflammation and cardiovascular disease that do not cause detrimental side effects such as hypercalcemia.13
High throughput screening (HTS) has quickly evolved over the last decades to become one of the main methods for the identification of lead compounds in drug discovery.14 The advantages of using HTS to identify hit compounds includes the quick retrieval of quality data from large libraries of compounds in a cost-effective manner.15 Previously we reported our collaborative work conducted with the NIH Chemical and Genomics Center (NCGC) to identify VDR ligands that did not possess the secosteroid scaffold of 1,25(OH)2D3 in order to overcome hypercalcemia induced by these endogenous ligands.16 By using a fluorescence polarization (FP) assay employing VDR-LBD and a fluorescently labeled peptide, SRC2-3, we derived GW0742 as a non-secosteroid inhibitor of VDR (IC50 = 27 ± 2.7 μM) out of a library of ~390,000 compounds (PubChem Assay Identifier (AID): 504847). We found GW0742 possessed moderate inhibitory effects for VDR and the androgen receptor (AR) in addition to greatly activating PPARδ. Evaluation of VDR-target gene regulation in LNCaP cells in the presence of GW0742 resulted in negative regulation of CYP24A1, IGFBP-3 and TRPV6 when stimulated with1, 25(OH)2D3. Additionally, GW0742 was capable of inhibiting (IC50 = 37.6 μM) cell differentiation induced by 1,25(OH)2D3 in HL-60 cells, a process governed by VDR gene expression.
Herein, we discuss the medicinal chemistry approach used to optimize GW0742 as a potent VDR antagonist with decreased PPARδ activity. Four major regions of GW0742 were modified as described in Figure 1. The SAR included the replacement of the phenyl ring (pink region) with substituted-aryl or heteroaryl groups, exchanging of the methyl (cyan region) with a hydrogen, substitution of the linker atoms (green region) with oxygen, nitrogen or sulfur, and bioisosteric substitution of the carboxylic acid (blue region).
Figure 1.

Design of GW0742 derivatives.
RESULTS AND DISCUSSION
Synthesis
Over 100 compounds were synthesized based on the GW0742 core scaffold using a parallel chemistry approach that efficiently produced the desired compounds at sufficient yields. Mono-, poly-, and aromatic-substituted GW0742 analogues were synthesized according to reaction Scheme 1. Sodium borohydride was used to reduce ethyl 2-bromo-4-methylthiazole-5-carboxylate to the corresponding primary alcohol 1a. Subsequent reaction with thionyl chloride afforded 1b, which was coupled with 4-hydroxy-3-methylthiophenol in the presence of cesium carbonate to give 1c. Suzuki coupling methodology was applied to enable diversity in this position via different boronic acids and a unique solid supported diphenylphosphine palladium (II) heterogeneous catalyst that could be recovered and used again. The resulting esters were then cleaved with trifluoroacetic acid in CH2Cl2 to afford the final carboxylic acid products (1-78).
Scheme 1.

General synthetic route for mono, poly, and aromatic-substituted ligands.
i) NaBH4, EtOH, R.T., 66%, ii) SOCl2, CH2Cl2, R.T., 86%; iii) a) 4-hydroxy-3-methylthiophenol, Cs2CO3, MeCN, R.T.; b) tert. butyl bromoacetate, Cs2CO3, MeCN, R.T., overall yield = 89%; iv) Boronic acid Na2CO3, SiliaCat® DPP-Pd, DME, 150 °C, MW, 0.5 h, 33-60%; v) TFA, CH2Cl2, R.T., 1h, ~100%.
Bioisosteric substitution is a common approach in medicinal chemistry to improve potency, selectivity and/or metabolic stability while maintaining similar physical (e.g. size, shape, polarity) and chemical properties (e.g. pKa). In our case, a tetrazole was introduced in place of the carboxylate as depicted in Scheme 2. 2-methyl-4-thiocyanatophenol and 2-bromoacetonitrile were coupled under basic conditions to produce 2a. Reaction with Na2S yielded the free thiol that was sequentially coupled to 1b to yield 2c. The cyano-substituted analogues 2d-2g were formed by Suzuki coupling and subsequently treated with sodium azide to form tetrazoles analogues (79-82) by [2+3] cycloaddition.
Scheme 2.

General synthetic route for bioisostere-substituted analogues.
i) Cs2CO3, MeCN, R.T., 12 h, 42%; ii) Na2S, EtOH•H2O, R.T., 3 h, 91%; iii) 1b, Cs2CO3, MeCN, R.T., 1 h, 75%; iv) Boronic acid, Na2CO3, SiliaCat® DPP-Pd, DME, 150 °C, MW, 0.5 h; 36%-45%; v) NaN3, NH4Cl, DMF, 100°C, 12 h, 35-69%.
The synthesis of linker-modified GW0742 analogues (87, 92 and 93) started with reduction of ethyl 2-bromo-4-methylthiazole-5-carboxylateto obtain 1a.17 This compound was coupled with methyl 3-(4-hydroxyphenyl)propionate under Mitsunobu reaction conditions yielding thiazole ester 3a.18 A microwave-assisted Suzuki reaction allowed for the introduction of the third aromatic ring. Conveniently, the carbon-carbon bond formation and hydrolysis occurred under microwave conditions to form the final carboxylic acids 87, 92 and 93.
Biological Evaluation of GW0742 Analogues
GW0742 analogues were characterized using several biochemical and cell-based assays to determine their activity towards VDR and PPARδ. Cytotoxicity of each compounds was also investigated in HEK293T cells. This included a high-throughput FP assay investigating the interaction between VDR-LBD and coactivator SRC2-3, VDR- and PPARδ-mediated transcription assays anda cytotoxicity assay to differentiate between inhibition of transcription and cytotoxicity.16, 19 We report the results in the following tables: mono-substituted analogues (Table 1), poly-substituted analogues (Table 2), aromatic-substituted analogues (Table 3), tetrazole-substituted analogues (Table 4) and linker-substituted analogues (Table 5).
Table 1.
Evaluation of mono-substituted GW0742 analogues
| Compound | R | PPARδ EC50 (μM) a,b | Toxicity CC50(μM)c |
|---|---|---|---|
| 1 | H | 0.26 ± 0.35 (90 ± 7.5) | >100 |
| 2 | o-CH3 | 0.60 ± 0.17 (66 ± 5.2) | >100 |
| 3 | m-CH3 | 1.62 ± 0.41 (74 ± 4.3) | >80 |
| 4 | p-CH3 | 0.05 ± 0.02 (104 ± 12) | >80 |
| 5 | o-Cl | 0.58 ± 0.20 (71 ± 4.8) | >80 |
| 6 | m-Cl | 0.50 ± 0.16 (74 ± 4.5) | 28.8 ± 5.0 |
| 7 | p-Cl | 0.13 ± 0.04 (66 ± 5.8) | >100 |
| 8 | o-F | 0.26 ± 0.08 (94 ± 5.9) | >80 |
| 9 | m-F | 0.43 ± 0.23 (96 ± 12) | >100 |
| 10 | p- F | 0.16 ± 0.04 (93 ± 3.3) | Non-toxic |
| 11 | o-CF3 | 2.4 ± 0.95 (39 ± 2.8) | >100 |
| 12 | m-CF3 | 1.1 ± 0.6 (79 ± 14) | >50 |
| 13 (GW501516) | p-CF3 | 0.013 ± 0.004 (95 ± 3.3) | >33 |
| 14 | o-OCH3 | 2.3± 0.70 (27 ± 4.2) | >100 |
| 15 | m-OCH3 | 2.36 ± 0.67 (96 ± 7.2) | >80 |
| 16 | m-OCF3 | 0.45 ± 0.15 (40 ± 2.4) | >50 |
| 17 | p-OCF3 | 0.014 ± 0.007 (49 ± 2.2) | >50 |
| 18 | m-CN | 3.2 ± 1.0 (74 ± 5.4) | >80 |
| 19 | p-CN | 0.12 ± 0.42 (126 ± 11) | >80 |
| 20 | p-N(CH3)2 | 0.16 ± 0.02 (140 ± 14) | >50 |
| 21 | p-NHCH3 | 0.57 ± 0.24 (83 ± 7.4) | Non-toxic |
| 22 | p-CO2Et | 0.009 ± 0.002 (123 ± 5.8) | >80 |
| 23 | m-methylsulfinyl | 6.7 ± 3.2 (59 ± 4.9) | Non-toxic |
| 24 | p-methylsulfonamido | >100 | Non-toxic |
| 25 | p-NHCOCH3 | 3.9 ± 2.3 (97 ± 4.7) | >100 |
| 26 | m-CONH(CH2)3N(CH3)2 | 2.4 ± 1.3 (70 ± 7.5) | Non-toxic |
| 27 | p-CONH(CH2)2N(CH3)2 | 2.1 ± 1.4 (65 ± 11) | Non-toxic |
| 28 | p-(4-methylpiperazinyl) methanone | 6.9 ± 5.53 (70 ± 13) | Non-toxic |
| 29 | p-piperazinyl | 16.8 ± 5.7 (37 ± 3.4) | >80 |
| 30 | Morpholino-methanone | 3.8 ± 1.5 (37 ± 2.6) | Non-toxic |
Two-hybrid assay using a CMV-PPARδ-LBD-GAL4-DBD plasmid and a 6xGal4-luc reporter vector. The maximum concentration used for this assay was 100 μM of each compound;
Efficacy in PPARδ assay in respect to full activation with GW0742 are in parentheses;
Cell-TiterGlo (Promega). The maximum concentration used for transcription and toxicity assay was 100 μM of each compound. Data were analyzed using a nonlinear regression with a variable slope (GraphPad Prism).
Table 2.
Evaluation of poly-substituted GW0742 analogues.
| Compound | R | R1 | R2 | R3 | R4 | PPARδ EC50 (μM)a, b | Toxicity CC50 (μM)d |
|---|---|---|---|---|---|---|---|
| 31 (GW0742) | H | H | CF3 | F | H | 0.0035 ± 0.0003 (97 ± 4.4) | >50 |
| 32 | H | CF3 | H | CF3 | H | 0.99 ± 0.42 (46 ± 5.4) | >50 |
| 33 | CF3 | H | CF3 | H | H | 0.20 ± 0.09 (50 ± 2.8) | >50 |
| 34 | H | H | CF3 | H | Cl | 0.028 ± 0.013 (88 ± 2.8) | >50 |
| 35 | H | H | CF3 | Cl | H | 0.007 ± 0.003 (75 ± 5.4) | >33 |
| 36 | H | H | OCF3 | Cl | H | 0.009 ± 0.004 (69 ± 3.1) | >50 |
| 37 | H | H | OCF3 | F | H | 0.020 ± 0.010 (100 ± 6.3) | >50 |
| 38 | H | OCH3 | OCH3 | H | H | 1.06 ± 0.47 (44 ± 4.6) | >100 |
| 39 | H | F | OCH3 | H | H | 0.074 ± 0.031 (112 ± 5.2) | >100 |
| 40 | H | H | OCH2CF3 | F | H | 0.050 ± 0.028 (91 ± 7.9) | >50 |
| 41 | H | CF3 | F | H | H | 0.49 ± 0.31 (116 ± 14) | >50 |
| 42 | H | H | F | F | F | 0.042 ± 0.009 (122 ± 7.3) | >80 |
| 43 | H | F | F | F | H | 0.046 ± 0.019 (106 ± 19) | >100 |
| 44 | H | H | F | CN | H | 1.07 ± 0.59 (150 ± 24) | >100 |
| 45 | H | H | Cl | H | CF3 | 0.18 ± 0.12 (63 ± 5.3) | >50 |
| 46 | H | H | Cl | Cl | H | 0.692 ± 0.137 (79 ± 4.5) | >50 |
| 47 | H | H | CN | H | Cl | 0.066 ± 0.04 (108 ± 11) | >100 |
| 48 | H | F | H | Cl | H | 0.17 ± 0.05 (62 ± 3.4) | >50 |
| 49 | H | Cl | H | CF3 | H | 0.60 ± 0.28 (23 ± 2.3) | >33 |
| 50 | H | Cl | H | Cl | H | 0.71 ± 0.27 (78 ± 6.0) | >50 |
| 51 | Cl | H | H | H | Cl | 6.6 ± 4.8 (84 ± 11) | >100 |
| 52 | H | H | H | Cl | Cl | 0.028 ± 0.017 (78 ± 7.5) | >100 |
| 53 | H | Cl | H | H | Cl | 1.40 ± 0.74 (75 ± 2.1) | >80 |
| 54 | H | F | Benzyl-morpholine | H | H | 4.3 ± 1.9 (50 ± 4.4) | >100 |
Two-hybrid assay using a CMV-PPARδ-LBD-GAL4-DBD plasmid and a 6xGal4-luc reporter vector. The maximum concentration used for this assay was 100 μM of each compound;
Efficacy in PPARδ assay in respect to full activation with GW0742 are in parentheses;
Cell-TiterGlo (Promega). The maximum concentration used for transcription and toxicity assay was 100 μM of each compound. Data were analyzed using a nonlinear regression with a variable slope (GraphPad Prism).
Table 3.
Evaluation of substituted GW0742 analogues
| Compound | R | PPARδ EC50 (μM) a,b | Toxicity CC50 (μM) c |
|---|---|---|---|
| 55 | 3,5-dimethylisoxazol-4-yl | 0.29 ± 0.12 (103 ± 17) | >100 |
| 56 | 1H-indazol-4-yl | 0.052 ± 0.035 (101 ± 14) | >100 |
| 57 | 4-pyridine | 9.1 ± 3.6 (81 ± 6.4) | >80 |
| 58 | 2-(benzofuran-2-yl) | 0.061 ± 0.028 (97 ± 9.1) | >80 |
| 59 | 2,3-dihydrobenz[1,4]dioxin-6-yl | 0.72 ± 0.23 (127 ± 6.2) | >80 |
| 60 | Naphthalene-1-yl | 1.69 ± 0.34 (36 ± 1.7) | >50 |
| 61 | Benzo[1,3]dioxol-5-yl | 0.26 ± 0.12 (52 ± 3.8) | >50 |
| 62 | Benzo[1,2,5]oxadiazol-5-yl | 0.16 ± 0.04 (35 ± 2.2) | >100 |
| 63 | Furan-2-yl | 6.0 ± 2.7 (96 ± 11) | >100 |
| 64 | Pyridin-3-yl | 8.1 ± 5.6 (87 ± 12) | >100 |
| 65 | Piperazin-1-yl | 0.57 ± 0.39 (46 ± 9.6) | >100 |
| 66 | Pyrimidin-yl | 1.57 ± 0.62 (51 ± 3.8) | Non-toxic |
| 67 | 1H-indazol-6-yl | 3.18 ± 1.80 (122 ± 36) | >100 |
| 68 | Benzothiazol-6-yl | 0.073 ± 0.053 (99 ± 23) | 100 |
| 69 | Isoquinolin-4-yl | 0.35 ± 0.18 (51 ± 6.7) | Non-toxic |
| 70 | 6-fluoropyridin-3-yl | 0.192 ± 0.082 (110 ± 9.5) | Non-toxic |
| 71 | 1-methyl-1H-indazol-6-yl | 0.58 ± 0.34 (99 ± 6.7) | >100 |
| 72 | 6-(trifluoromethyl)pyridin-3-yl | 0.013 ± 0.006 (91 ± 4.9) | >100 |
| 73 | 1-methyl-1H-indol-6-yl | 0.52 ± 0.29 (103 ± 5.0) | >100 |
| 74 | 1H-pyrazol-4-yl | 3.9 ± 1.7 (74 ± 5.7) | Non-toxic |
| 75 | 1H-indazol-5-yl | 1.44 ± 0.66 (64 ± 5.5) | Non-toxic |
| 76 | Benzothiophen-5-yl | 0.14 ± 0.08 (95 ± 12) | >50 |
| 77 | 1H-indol-4-yl | >100 | Non-toxic |
| 78 | 1H-indol-2-yl | 0.035 ± 0.015 (108 ± 5.5) | >50 |
Two-hybrid assay using a CMV-PPARδ-LBD-GAL4-DBD plasmid and a 6xGal4-luc reporter vector. The maximum concentration used for this assay was 100 μM of each compound;
Efficacy in PPARδ assay in respect to full activation with GW0742 are in parentheses;
Cell-TiterGlo (Promega). The maximum concentration used for transcription and toxicity assay was 100 μM of each compound. Data were analyzed using a nonlinear regression with a variable slope (GraphPad Prism).
Table 4.
Evaluation of tetrazole-substituted GW0742 analogues
| Compound | R1 | R2 | R3 | R4 | PPARδ EC50 (μM) a, b | Toxicity CC50 (μM)c | Entry for Corresponding Carboxylic Acid |
|---|---|---|---|---|---|---|---|
| 79 | H | CF3 | F | H | 0.04 ± 0.018 (29 ± 1.5) | >33 | 31 (GW0742) |
| 80 | Cl | H | Cl | H | 0.594 ± 0.435 (29 ± 5.2) | >50 | 53 |
| 81 | H | Cl | H | CF3 | 0.51 ± 0.18 (51 ± 4.8) | >33 | 46 |
| 82 | H | Cl | Cl | H | 0.24 ± 0.11 (26 ± 1.9) | >33 | 49 |
Two-hybrid assay using a CMV-PPARδ-LBD-GAL4-DBD plasmid and a 6xGal4-luc reporter vector. The maximum concentration used for this assay was 100 μM of each compound;
Efficacy in PPARδ assay in respect to full activation with GW0742 are in parentheses;
Cell-TiterGlo (Promega). The maximum concentration used for transcription and toxicity assay was 100 μM of each compound. Data were analyzed using a nonlinear regression with a variable slope (GraphPad Prism).
Table 5.
Evaluation of linker-substituted GW0742 analogues.
| Compound | R1 | R2 | Y | R | X | PPARδ EC50 (μM) a,b | Toxicity CC50 (μM)c |
|---|---|---|---|---|---|---|---|
| 83 | H | CF3 | S | H | CH2 | 0.039 ± 0.022 (87 ± 4.3) | >100 |
| 84 | F | CF3 | S | H | -- | 0.018 ± 0.010 (70 ± 3.7) | >100 |
| 85 | F | CF3 | S | H | O | 0.026 ± 0.008 (84 ± 5.1) | >100 |
| 86 | F | CF3 | O | CH3 | O | 0.02 ± 0.02 (73 ± 11) | >100 |
| 87 | F | CF3 | O | H | CH2 | 0.032 ± 0.021 (77 ± 5.0) | >100 |
| 88 | F | CF3 | O | H | S | 0.27 ± 0.023 (75 ± 5.0) | >100 |
| 89 | F | CF3 | N | CH3 | O | 0.18 ± 0.12 (61 ± 5.0) | >100 |
| 90 | F | CF3 | N | H | O | 0.16 ± 0.045 (78 ± 6.0) | >80 |
| 91 | F | CF3 | N | H | CH2 | 0.058 ± 0.021 (95 ± 7.0) | >80 |
| 92 | CH2OH | H | O | H | CH2 | 18.2 ± 2.8 (47 ± 1.8) | Non-toxic |
| 93 | H | CF3 | O | H | CH2 | 0.70 ± 0.11 (111 ± 6.5) | >100 |
Two-hybrid assay using a CMV-PPARδ-LBD-GAL4-DBD plasmid and a 6xGal4-luc reporter vector. The maximum concentration used for this assay was 100 μM of each compound;
Efficacy in PPARδ assay in respect to full activation with GW0742 are in parentheses;
Cell-TiterGlo (Promega). The maximum concentration used for transcription and toxicity assay was 100 μM of each compound. Data were analyzed using a nonlinear regression with a variable slope (GraphPad Prism).
The mono-substituted GW0742 analogues exhibited a wide range of potencies (EC50 = 0.009 – <100 μM) with respect to PPARδ activation in cells (Table 1).

Three mono-substituted compounds modulated transcription with high potency. They were 13 (p-CF3), 17 (p-OCF3), and 22 (p-CO2Et) with EC50 values lower than 15 nM. Compound 13, also known as GW501516, was confirmed as a potent PPARδ agonist.5a The other two compounds have not been reported but possess similar electron donating properties and significant hydrophobicity. Notably, the positioning of substituents around the phenyl ring affected the ligand’s ability to activate PPARδ mediated transcription. A para positioned substituent, in most cases, resulted in a more potent PPARδ agonist than compounds that bear the same group in the ortho or meta position. This relationship was observed with methyl (2-4), trifluoromethyl (11-13), trifluoromethoxy (16-17) and cyano (18-19) substituents. However, ligands with halide substituents, like Cl (5-7) and F (8-10), showed no significant activity difference between ortho, meta, or para positioning possibly due to their atomic size or change in orientation. Compounds with para-positioned fluorine and chlorine substituents had similar but relatively high EC50 values of ~0.14 μM (compounds 10 and 7). The least active PPARδ agonist was polar sulfonamide 24. As evidenced by the crystal structure of PPARδ complexed with GW0742 (PDB: 3TKM), the binding pocket accommodates the pink region of Figure 1 of the ligand which is relatively large and hydrophobicdueto residues V245, V305, V312, L317, and I328 (Figure 2).5a, 20 Therefore, depending on its position within the LBP, it is expected that extra hydrogen bonding capability is not favorable in this region of the ligand. Compound 29 bearing a piperazine was another example where polarity reduced activity towards PPARδ with an EC50= 16.8 ± 5.7 μM.
Figure 2.

Computer docking of 32 (blue) in the ligand binding pocket of PPARδ with GW0742 (green) (PDB: 3TKM). Major nonpolar and hydrogen bonding interactions have been labeled.
Interestingly, while many ligands exhibited partial agonistic activity towards PPARδ, we did not observe a trend between structure and efficacy. This is significant because some nuclear receptor partial agonists possess superior pharmacodynamics due to reduced side effects and moderate activation.21 Finally, most compounds exhibited CC50 values, determined in the presence of HEK293 kidney cells, of 80 μM and higher. An exception was compound 6 (m-Cl) with an CC50 of 28.8 μM.
Poly-substituted ligands found in Table 2 were also capable of activating PPARδ mediated transcription (EC50 = 0.0035 – 6.6 μM).

The least active compound in our series was compound 51 (o,o’-Cl) with an EC50 value of greater than 5 μM, indicating that two substituents occupying both ortho positions of the phenyl ring is not favorable for PPARδ activation. Interestingly, by moving just a chloride to the R3 position (compound 52) activity of this ligand is greatly increased by 230-fold when compared to 51. The positioning of groups like CF3, Cl, F and OCF3 on phenyl ring positions gave some insight about the PPARδ ligand binding pocket. For example, by switching the p-CF3 and m-F substituents on GW0742 to make compound 41 (m-CF3, p-F) a 140-fold decrease in ligand potency was observed. When m-F of GW0742 is exchanged for a m-Cl (compound 35), no significant change in ligand potency was observed. When comparing compound 35 (p-CF3, m-Cl) to compound 34 (p-CF3, o-Cl), only a small decrease in PPARδ activation was observed (4-fold change). When p-CF3 of GW0742 was replaced with p-OCF3 regardless of a Cl or F in the meta position (compounds 36 and 37, respectively), PPARδ activation was observed at low nanomolar concentrations. With respect to all fluorine substituents, it appeared that two fluorine substituents were better than one regardless of their positioning. The same trend was observed for chlorine substituents. The toxicity of poly-substituted GW0742 analogues was, in general, more pronounced than their mono-substituted counterparts, however none of them exhibited toxicity below 50 μM.
Aromatic substituents were also coupled to the C-2 position of the thiazole ring and their biological activity is summarized in Table 3.
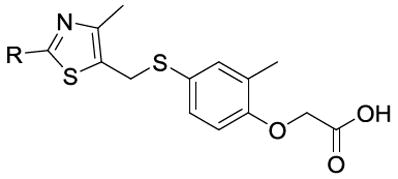
Five compounds activated PPARδ with EC50 values less than 75 nM (56, 58, 68, 72 and 78). Of these, all but one had a bicyclic aromatic ring structure. This result confirmed earlier observations that the LBD of PPARδ is spacious enough to accommodate such ligands, possibly through a unique orientation unlike GW0742. It is worth noting that when compared to VDR, PPARδ has a larger Y-shaped ligand binding pocket which can make contact with the ligand in three different regions, thus possibly explaining the accommodation for large ring systems.20 From Table 1 we concluded that non-substituted phenyl ring structures such as compound 1 made poor PPARδ agonists. Similarly, substitution of the phenyl ring with a pyridine, as in compound 64, resulted in a poor binding. However, when the pyridine ring contained a trifluoromethyl group like compound 72, a 620-fold increase in transcriptional activation was observed when compared to 64. The positioning of heterocyclic rings may be important considering the effect compounds 77 and 78 have on PPARδ activation. Both have an indole ring attached to C-2 of the thiazole but the 1H-indol-4-yl group (77) was nearly 3,000 times less potent than the 1H-indol-2-yl substituent (78).
Bioisoteric substitution of the carboxylate group with a tetrazole ring was used for several analogues to determine its effect on PPARδ-mediated transcription (Table 4).

In general, increased toxicity and decreased potency was observed for tetrazole analogues when compared to their carboxylate counterparts. The tetrazole substitution of GW0742, resulting in compound 79, had an 11-fold decrease in potency and a two-fold increase of toxicity. Although similar in acidity, the spacial ability of this particular tetrazole to form hydrogen bond interactions is different from a carboxylic acid. It is known that both oxygens on the carboxylate group make hydrogen bonds with key residues like His413 (helix 10/11), Try437 (helix 12), His287 (helix 7) of PPARδ LBP and are implicated with maintaining the locked conformation of helix 12 (Figure 2).20 Thus, the inability of the tetrazole to form all necessary hydrogen bonds might be responsible for low efficacy of these compounds ranging between 26-51%.
The replacement of the sulfur and oxygen linkers of GW0742 allowed structure-activity relationship investigations into the central part of PPARδ agonists (Table 5).

In addition, the role of the ortho methyl group (R) was investigated in terms of favorable PPARδ binding. Compound 85 is structurally similar to GW0742 except for the lack of methyl group in the R position: it was 7 times less potent than GW0742 and surprisingly, behaved as a partial agonist (84% in comparison with GW0742). A large decrease in potency (~200-fold) was observed for compound 93 when Y was replaced with a smaller oxygen atom and X with a CH2 in the absence of a fluoride and methyl group in positions R1 and R, respectively. When compared to compound 87, bearing a fluorine in the R1 position, the activity difference was less, suggesting an additional hydrophobic interactions between 87 and PPARδ LBP. This behavior was not observed for Y = nitrogen when comparing 89 and 90. The presence or absence of an R positioned methyl had little effect on activation of transcription thus producing agonists that were ~50 times less potent than GW0742. However, CH2 in place of oxygen (91) improved PPARδ binding when compared to 89 and 90. The least potent compound was 92, bearing a m-CH2OH substituent on the phenyl ring with linkers similar to that of 93. This compound was approximately 5,000 times less potent than GW0742 confirming that hydrogen bond donor and acceptor moieties in the pink region of Figure 1 of ligands are not favorable for PPARδ binding.
In addition to GW0742 and GW501516, compounds 7, 10, 32, 46, and 83 were patented by GSK.22 I was reported that most of the GW0742-like compounds showed ~50% activation of hPPARδ at concentrations of 10 nM or less in a β-galactosidase transactivation assay using CV-1 cells. Other than compounds 32 and 46, this correlates well with what we have observed in our PPARδ-mediated transcription assay. Also, these compounds were reported being at least 10-fold selective for hPPARδ over hPPARα and PPARγ.
The SAR study of GW0742 analogues has given us some insight into the structural requirements for these highly potent PPARδ ligands. In addition, structural elements have been identified that significantly reduce the ability of ligands to activate PPARδ-mediated transcription. The in vitro data was further supported by molecular modeling using a crystal structure of GW0742 bound to PPARδ LBD (PDB: 3TKM). Using the Schrödinger software to perform standard precision grid-based ligand docking with energetics (Glide) studies, we were able to show that 32 aligns favorably with GW0742. However, occupation of the highly hydrophobic region on the left side of the pocket (Figure 2) differs significantly. It is evident that the addition of two bulky trifluoromethyl groups in both meta positions orients the pink region of Figure 1 of the molecule downwards therefore causing a 283-fold decrease in PPARδ activity (GW0742, EC50 = 0.0034 μM and 32, EC50 = 0.99 μM).
Having identified several PPARδ ligands with EC50 values greater than 1 μM, we next set out to test their ability to bind to VDR. All “poor” PPARδ ligands were investigated with respect to their ability to inhibit the interaction between VDR and coactivator peptide SRC2-3 and their ability to inhibit VDR-mediated transcription in cells. The results are summarized in Table 6.
Table 6.
Evaluation of “poor” PPARδ ligands (EC50< 1μM) in respect to VDR binding.
| Entry | VDR-SRC2-3 interaction IC50 (μM)a | VDR transcription IC50 (μM)b | PPARδ EC50 (μM) a,b |
|---|---|---|---|
| 25 | 40.11 ± 9.26 | inactive | 3.9 ± 2.3 (97 ± 4.7) |
| 11 | 32.27 ± 5.28 | 31.5 ± 9.1 | 2.4 ± 0.95 (39 ± 2.8) |
| 14 | 45.18 ± 5.65 | 31.4 ± 8.1 | 2.3± 0.70 (27 ± 4.2) |
| 18 | >100 | 25.1 ± 10.0 | 3.2 ± 1.0 (74 ± 5.4) |
| 23 | >100 | >100 | 6.7 ± 3.2 (59 ± 4.9) |
| 26 | 30.28 ± 4.5 | inactive | 2.4 ± 1.3 (70 ± 7.5) |
| 27 | 52.03 ± 12.79 | >100 | 2.1 ± 1.4 (65 ± 11) |
| 32 | 9.088 ± 0.982 | 15.0 ± 4.7 | 0.99 ± 0.42 (46 ± 5.4) |
| 38 | >100 | >50 | 1.06 ± 0.47 (44 ± 4.6) |
| 44 | 53.53 ± 15.95 | 23.7 ± 4.7 | 1.07 ± 0.59 (150 ± 24) |
| 51 | 40.28 ± 8.07 | 33.0 ± 7.2 | 6.6 ± 4.8 (84 ± 11) |
| 53 | 14.47 ±2.21 | >50 | 1.40 ± 0.74 (75 ± 2.1) |
| 57 | 43.88 ± 8.54 | inactive | 9.1 ± 3.6 (81 ± 6.4) |
| 63 | 20.19 ± 1.89 | 26.3 ± 0 8.3 | 1.69 ± 0.34 (36 ± 1.7) |
| 30 | >100 | >100 | 3.8 ± 1.5 (37 ± 2.6) |
| 54 | 68.79 ± 13.04 | >50 | 4.3 ± 1.9 (50 ± 4.4) |
| 63 | >100 | >50 | 6.0 ± 2.7 (96 ± 11) |
| 64 | >100 | >100 | 8.1 ± 5.6 (87 ± 12) |
| 66 | >100 | inactive | 1.57 ± 0.62 (51 ± 3.8) |
| 67 | 57.78 ± 8.69 | inactive | 3.18 ± 1.80 (122 ± 36) |
| 74 | >100 | inactive | 3.9 ± 1.7 (74 ± 5.7) |
| 75 | 51.14 ± 5.55 | inactive | 1.44 ± 0.66 (64 ± 5.5) |
| 77 | 28.93 ± 4.69 | inactive | >100 |
| 3 | 47.45 ± 6.61 | 38.2 ± 8.6 | 1.62 ± 0.41 (74 ± 4.3) |
| 12 | 24.5 ± 2.27 | 19.0 ± 6.01 | 1.1 ± 0.6 (79 ± 14) |
| 15 | 48.82 ± 6.56 | 26.3 ± 6.9 | 2.36 ± 0.67 (96 ± 7.2) |
| 28 | >100 | 24.5 ± 6.1 | 6.9 ± 5.53 (70 ± 13) |
| 29 | >100 | inactive | 16.8 ± 5.7 (37 ± 3.4) |
| 92 | inactive | inactive | 18.2 ± 2.8 (47 ± 1.8) |
VDR-LBD concentration used was 0.1μM. Inhibition of VDR-SRC2-3 interaction in the presence of LG190178 (0.75 μM). The maximum concentration used for this assay was 300 μM of each compound;
Transcription assay using a CMV-VDR plasmid and a luciferase reporter plasmid under control of a 24-hydroxylase promoter with GW0742 analogues. The maximum concentration used for this assay was 100 μM of each compound. Data were analyzed using a nonlinear regression with a variable slope (GraphPad Prism).
Among the ligands investigated only 32, bearing a CF3 group in the meta position instead of the para position like GW0742, exhibited an IC50 value below 10 μM in the VDR FP-based assay. Many of the other ligands were not active. The activity of 32 was confirmed in cells with an IC50 of 15 μM for VDR-mediated transcription. Notably, most ligands including GW0742 in light of the FP-based assay and the VDR-mediated transcription assay, showed full antagonistic activity when compared to the DMSO control (Figures S2 and S3). Compounds whose IC50 values were greater than 100 μM appeared to be trending towards full antagonistic activity although higher concentrations would need to be tested to confirm this. Unfortunately, none of the synthesized ligands exhibited high affinity for VDR. Interestingly, other identified VDR ligands that bear a carboxylic acid function such as lithocholic acid23 or calcitroic acid24 exhibited binding constants toward VDR in the same range (1-10 μM); thus the carboxylic acid function of these molecules might be inferior to the two hydroxyl groups of 1,25(OH)2D3 to the bind VDR.25 On the other hand, 1,25(OH)2D3 interacts with histidine residues via it’s tertiary alcohol. The only ligand bearing a hydroxyl function among the ligands generated is compound 92 that interacted with neither PPARδ nor VDR. Thus, the current investigation illustrates that the change of substituents introduced here to design new VDR ligands based on the GW0742 scaffold is not sufficient. In the future, we will change the length of this scaffold by introducing smaller ring structures and linkers in addition to other carboxylic acid bioisosteres. However, it is encouraging that the simple removal of a para substituent and the addition of meta substituents on the phenyl ring (pink region, Figure 1), as in 32, does greatly reduce PPARδ activity. Thus, an introduction of a 3,4-bis(hydroxymethyl)phenyl group as shown for VDR ligands CD442026 might be advantageous to improve VDR binding and reduce PPARδ activity. On the other hand, we did identify many new potent PPARδ agonists and partial agonists during this study, however, further studies are needed to determine the PPAR subtype selectivity of these ligands. This will be essential to develop new drug candidates for lowering density lipoprotein therapeutics based on PPARδ binding.
EXPERIMENTAL PROCEDURES
Chemistry
General Information for Compounds 1-86 and 88-91
All reagents and solvents used in this study were obtained from commercial suppliers and used without further purification. Preparative purification was performed on a Waters semi-preparative HPLC system using a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. A gradient of 10% to 50% acetonitrile in water over 8 minutes (each containing 0.1% trifluoroacetic acid) was used as a mobile phase during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). Method t1: A 7 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50°C. Method t2: A 3 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 minute run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 micron, 3 × 100 mm) was used at a temperature of 50° C. Method t3: Analysis was performed on an Agilent 1290 Infinity Series HPLC. UHPLC Long Gradient Equivalent 4% to 100% acetonitrile (0.05% trifluoroacetic acid) in water over 3.5 minutes run time of 4 minutes with a flow rate of 0.8 mL/min. > 95 % Purity was determined using an Agilent Diode Array Detector for both Method t1, Method t2 and Method t3. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometer. Chemical shifts are reported in ppm with DMSO at 2.49 ppm as internal standard. High resolution mass spectrometry was recorded on Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02). The synthesis of these compounds is described in more detail in the Supporting Information.
General Information for Compounds 87, 92-93
All starting reagents were purchased from Sigma-Aldrich. Anhydrous solvents were purchased in sure-seal bottles and handled under dry conditions using syringe technique. All glassware was dried overnight at 100°C before use. Thin layer chromatography was performed on pre-coated silica gel 60 F254 plates (Fisher Scientific). Microwave reactions were performed using a CEM Discover SP instrument. Synthesized compounds were purified by normal phase flash chromatography (SPI Biotage, silica gel 230-400 mesh) and concentrated under vacuum. All pure compounds were stored as solids at -20°C. Molecular masses and >95 % purities were determined with a Shimadzu 2020 LC-MS (single quadrupole) instrument. NMR spectra were recorded on a Bruker 500 MHz or 300 MHz instrument with samples diluted in either CDCl3 or DMSO-D6. Chemical shifts are reported in δ (parts per million: ppm) by reference to the hydrogenated residues of the deuterated solvent as an internal standard (CDCl3: δ= 7.24 ppm (1HNMR) and δ= 77 ppm (13CNMR) and DMSO-D6: δ= 2.50 ppm (1HNMR) and δ= 39.5 ppm (13CNMR). The synthesis of these compounds is described in more detail in the Supporting Information.
Biology
Reagents and Instrumentation
LG190178 (a VDR agonist) was synthesized following a published procedure27 and used as a positive control. 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (a VDR-coactivator inhibitor) was used as a second positive control and was synthesized using a previously published method.28 1,25(OH)2D3 was purchased from Endotherm. The VDR-LBDmt DNA was provided by D. Moras29 and cloned into pMAL-z2X vector (New England Biolabs). For a details on the expression and purification of VDR-LBD, see ref30. AlexaFluor 647 -Labeled SRC2-3 was synthesized by coupling SRC2-3 (CKKKENALLRYLLDKDDTKD) with cysteine-reactive AlexaFluor 647 probe followed by reverse phase HPLC purification using a C18 column. VDR-CMV and CYP24A1-luciferase reporter plasmids were provide by D.D. Bikle.
VDR Binding Determined by a Fluorescence Polarization (FP) Assay
Was conducted in 384-well black polystyrene microplates (Corning, #3573). The assay solution contained buffer (25mM PIPES, 50mM NaCl (Fisher), and 0.01% NP-40, at pH 6.75), 0.1 μM VDR-LBD and 7.5 nM Alexa Fluor 647-labeled SRC2-3. For competitive inhibition, 0.75 μM LG190178 was also added to the solution. 10 mM stock solutions of synthesized compounds made in DMSO were serially diluted (1:3), and four 14 μL aliquots of each compound concentration was transferred to opaque 384-well polypropylene plates for storage. Small volume transfers were performed by a Tecan Freedom EVO liquid handling system using a 50H hydrophobic coated pin tool that carried 100 nL (V&P Scientific). To obtain a maximum concentration of 300 μM, 600 nl of each compound concentration was transferred into the 20 μL assay solution. Agonistic binding was determined in the absence of LG190178 while competitive inhibition binding was determine in the presence of LG190187. Fluorescence polarization was detected after 30 minutes at an emission/excitation wavelength of 635/685 nm (Alexa Fluor 647). LG190178 (Ki = 150 nM) and DMSO were used as positive and negative controls in the agonistic binding assay, respectively. In the competitive inhibition binding assay 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one was the positive control while DMSO was used as a negative control. Controls were measured within each plate to determine the z’ factor, which assed the quality of the assay, and enable data normalization. Three independent experiments were carried out in quadruplicate, and data was analyzed using nonlinear regression with a variable slope (GraphPrism). Data reported in the Supporting Information.
VDR- and PPARδ-Mediated Transcription Assays
Human embryonic kidney (HEK) 293T cells were purchased (ATCC) and cultured in 75 cm2 flasks (CellStar) coated in matrigel (BD Bioscience, #354234). Cells were grown in DMEM/High Glucose (Hyclone, #SH3024301) media to which non-essential amino acids (Hyclone, #SH30238.01), 10 mM HEPES (Hyclone, #SH302237.01), 5 × 106 units of penicillin and streptomycin (Hyclone, #SV30010), and 10% of heat inactivated fetal bovine serum (Gibco, #10082147) were added. For the assay, cells at 70-80% confluency were transfected by lipid-based methods where 2 mL of untreated DMEM/High Glucose media (without additives) containing: 1) transcription assay:0.7 μg of VDR-CMV plasmid, 16 μg of a CYP24A1-luciferase reporter gene, Lipofectamine™ LTX (75 μL, Life Technologies, #15338020), and PLUS™ reagent (25 μL) or 2) 2- Hybrid Assay:1.5 μg of CMV-PPARδ-LBD-GAL4-DBD plasmid, 16 μg of a 6xGal4-luciferase reporter gene, Lipofectamine™ LTX (75 μL) and PLUS™ reagent (25 μL) was added to the flask. After 16 hours of incubation at 37°C with 5% CO2, the cells were harvested with 3mL of 0.05% Trypsin (Hyclone, #SH3023601), added to 10mL of the assay buffer, DMEM/High Modified buffer without phenol red (Hyclone, #SH30284.01) containing all the above mentioned additives plus 10 mM sodium pyruvate and 2% percent charcoal treated FBS (Invitrogen, #12676-011) instead of HI FBS, and spun down for 2 minutes at 1000 rpm. The media was removed and cells were re-suspended in the assay buffer, DMEM/High Modified buffer without phenol red. Prior to adding cells to sterile white, optical bottom 384-well plates (NUNC, #142762), plates were treated with 20 μL per well of a 0.25% Matrigel solution. To each well, 20 μL of cells were added to yield a final concentration of 15,000 cells per well. After 4 hours, plated cells were treated with 200 nL of small molecules (1:3 serial dilution, with maximum final concentration at 100 μM) and controls (VDR: DMSO and 10 nM 1,25(OH)2D3[EC50 = 1 nM] and PPARδ: DMSO and 30 nMGW0742 [PPARδ EC50 = 3.5 nM]) which were added using a Tecan Freedom EVO liquid handling system with a 50H hydrophobic coated pin tool. In the competitive inhibition assay, 1,25(OH)2D3 (10 nM) was also added to the assay wells containing small molecule. After 16 hours of incubation at 37°C with 5% CO2, 20μL of Bright-Glo™ Luciferase Assay Kit (Promega, Madison, WI) was added to each well and luminescence was read. Controls were measured within each plate to determine the z’ factor and to enable data normalization. At least two independent experiments were performed in quadruplicate and data was analyzed using nonlinear regression with variable slope (GraphPrism).
Cytotoxicity Assay
Hek293T cells were plated according to the transcription assay protocol and 200 nL of serially diluted (1:3 in DMSO) small molecules (final maximum concentration at 100 μM) were transferred using a Tecan Freedom EVO liquid handling system with a 50H hydrophobic coated pin tool. The controls for the cytotoxicity assay used were 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (100 μM in DMSO, positive) and DMSO (negative). After 18 hours, cell viability was evaluated by adding 20 μL of Cell Titer-Glo™ Luminescence Assay Kit (Promega, Madison, WI) to each well and reading luminescence on a Tecan Infinite M1000 plate reader. Controls were measured within each plate to determine the z’ factor and to enable data normalization. At least two independent experiments were performed in quadruplicate and data was analyzed using nonlinear regression with variable slope (GraphPrism).
Supplementary Material
Scheme 3.

General synthetic route for linker-substituted GW0742 analogues.
i) PPh3, DIAD, CH2Cl2, methyl 3-(4-hydroxyphenyl) propionate, R.T., 2 h, 30%; ii) PdCl2(PPh3)2, Na2CO3·H2O, DME/H2O/EtOH, 160 °C, 10minutes, MW, 36%-45%.
Acknowledgments
Funding Sources
This work was supported by the University of Wisconsin-Milwaukee (UWM) (L.A.A.), the UWM Research Growth Initiative (RGI Grant) (L.A.A.), National Institutes of Health Grant R03DA031090 (L.A.A.), the UWM Research Foundation (Catalyst grant), the Lynde and Harry Bradley Foundation (L.A.A.), the Richard and Ethel Herzfeld Foundation (L.A.A.), and the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) (G.R., A.S., A.J., A.Y., and D.M.)
ABBREVIATIONS
- NR
Nuclear Receptor
- VDR
Vitamin D Receptor
- PPARδ, γ, α
Peroxisome Proliferator-Activated Receptor δ, γ, α
- SAR
structure-activity relationship
- DBD
DNA-Binding Domain
- LBD
Ligand-Binding Domain
- 125(OH)2D3
1α, 25-dihydroxyvitamind D3
- HTS
High Throughput Screening
- FP
Fluorescence Polarization
- AR
Androgen Receptor
Footnotes
Supporting Information.
The supporting information is available free of charge on the ACS publication website at DOI: It includes detailed descriptions of the chemical synthesis and compound characterization as well as VDR activities for all compounds.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Overington JP, Al-Lazkiani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Bain DL, Heneghan AF, Connaghan-Jones KD, Miura MT. Nuclear Receptor Structure: Implications for Function. Annu Rev Physiol. 2007;69:201–220. doi: 10.1146/annurev.physiol.69.031905.160308. [DOI] [PubMed] [Google Scholar]
- 3.Watson PJ, Fairall L, Schwabe JWR. Nuclear hormone receptor co-repressors: Structure and function. Mol Cell Endocrinol. 2012;348-135(2-3):440–449. doi: 10.1016/j.mce.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rochel N, Moras D. Structural Basis for Ligand Activity in VDR. In: Feldman D, Pike JW, Adams JS, editors. Vitamin D. 3. II. Elsevier Inc; 2011. pp. 171–191. [Google Scholar]
- 5.(a) Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, Sierra ML, LeGrumelec C, Xu HE, Montana VG, Lambert MH, Willson TM, Oliver William RJ, Sternbach DD. Novel Selective Small Molecule Agonists for Peroxisome Proliferator-Activated Receptor δ (PPARδ)- Synthesis and Biological Activity. Bioroganic & Medicinal Chemistry Letters. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]; (b) Oliver WilliamRJ, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. A seletive peroxisome proliferator-activated receptor δ agonist promotes reverse cholesterol transport. Proc Natl Acad Sci. 2001;98(9):5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrington LS, Moreno L, Reed A, Wort SJ, Desvergne B, Garland C, Zhao L, Mitchell JA. The PPARβ/δ Agonist GW0742 Relaxes Pulmonary Vessels and Limits Right Heart Hypertrophy in Rats with Hypoxia-Induced Pulmonary Hypertension. Plos ONE. 2010;5(3):381–385. doi: 10.1371/journal.pone.0009526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quintela AM, Jimenez R, Gomez-Guzman M, Zarzuelo MJ, Galindo P, Sanchez M, Vargas F, Cogolludo A, Tamargo J, Perez-Vizcaino F, Duarte J. Activation of Peroxisome Proliferator-Activated Receptor-β/-δPPARβ/δ) Prevents Endothelial Dysfunction in Type 1 Diebetic Rats. Free Radical Biology and Medicine. 2012;53:730–741. doi: 10.1016/j.freeradbiomed.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 8.Zingarelli B, Piraino G, Hake PW, O’Connor M, Denenberg A, Fan H, Cook JA. Peroxisome Proliferator-Activated Receptor δ Regulates Inflammation via NF-κB Signaling in Polymicrobial Sepsis. The American journal of pathology. 2010;177(4):1834–1847. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nature Medicine. 2004;10:481–483. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]; (b) Wang X, Wang G, Shi Y, Sun L, Gorczynski R, Li Y-J, Xu Z, Spaner DE. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis. 2016;5:1–11. doi: 10.1038/oncsis.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GEO. The peroxisome proliferator-activated receptor B/δ agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. J Mol Endocrinol. 2003;17(12):2477–2493. doi: 10.1210/me.2003-0151. [DOI] [PubMed] [Google Scholar]
- 11.(a) Arner P. The adipocyte in insulin resistance: kety molecules and the impact of the thiazolidinediones. TRENDS in Endocrinology and Metabolism. 2003;14(3):137–145. doi: 10.1016/s1043-2760(03)00024-9. [DOI] [PubMed] [Google Scholar]; (b) Kahn CR, Chen L, Cohen SE. Unraveling the mechanism of action of thiazolidinediones. The Journal of Clinical Investigations. 2000;106(11):1221–1228. doi: 10.1172/JCI11705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, Kravitz BG, Yu D, Heise MA, Aftring RP, Viberti G. Rosiglitazone-associated fractures in type 2 diabetes: an analysis from a diabetes outcome progression trial (ADOPT) Diabetes Care. 2008;31(5):845–851. doi: 10.2337/dc07-2270. [DOI] [PubMed] [Google Scholar]; (d) Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton EH, Winter ML, Porte D, Semenkovich CF, Smith S, Young LH, Kahn R. Thiazolidinedion use, fluid retention, and congestive heart failureL a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care. 2004;27(1):256–263. doi: 10.2337/diacare.27.1.256. [DOI] [PubMed] [Google Scholar]
- 12.Jurutka PW, Whitefield GK, Hsieh J-C, Thompson PD, Haussler CA, Haussler MR. Molecular Nature of the Vitamin D Receptor and its Role in Regulation of Gene Expression. Reviews in Endocrine & Metabollic Disorders. 2001;2:203–216. doi: 10.1023/a:1010062929140. [DOI] [PubMed] [Google Scholar]
- 13.Takada I, Makishima M. Therapeutic application of vitamin D receptor ligands: an updated patent review. Expert Opin Drug Discov. 2015;25(12):1373–1383. doi: 10.1517/13543776.2015.1093113. [DOI] [PubMed] [Google Scholar]
- 14.(a) Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DVS, Hertzber RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam GS. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discovery. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]; (b) Macarron R. Critical Review of the Role of HTS in Drug Discovery. Drug Discovery Today. 2006;11(7/8) doi: 10.1016/j.drudis.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Pinne M, Raucy JL. Advantages of cell-based high-volume screening assays to assess nuclear receptor activation during drug discovery. Expert Opin Drug Discov. 2014;9(6):669–686. doi: 10.1517/17460441.2014.913019. [DOI] [PubMed] [Google Scholar]
- 16.Nandhikonda P, Yasgar A, Baranowski AM, Sidhu PS, McCallum MM, Pawlak AJ, Teske K, Feleke B, Yuan NY, Kevin C, Bikle DD, Ayers SD, Webb P, Rai G, Simeonov A, Jadhav A, Maloney D, Arnold LA. GW0742 interacts weakly with multiple nuclear receptors, including the vitamin D receptor. Biochemistry. 2013;52:4193–203. doi: 10.1021/bi400321p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keil S, Urmann M, Bernardelli P, Glein M, Wendler W, Chandross K. Phenyl-1,2,4-oxadiazolone derivatives, processes for their preparation and their use and pharmaceuticals. 2007 [Google Scholar]
- 18.Moon H-S, Yoo M-H, Kim S-H, Lim J-I. Novel Phenylpropionic Acid Derivatives as Peroxisome Proliferator-Activated Gamma Receptor Modulators. Method of the Same, and Pharmaceutical Composition Comprising the Same. 2008 [Google Scholar]
- 19.Teske K, Nandhikonda P, Bogart JW, Feleke B, Sidhu P, Yuan N, Preston J, Goy R, Arnold L. Modulation of Transcription Mediated by the Vitamin D Receptor and the Peroxisome Proliferator-Activated Receptor delta in the Presence of GW0742 Analogs. Biomolecular Research & Therapeutics. 2014;3(1) doi: 10.4172/2167-7956.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batista FAH, Trivella DBB, Bernardes A, Gratieri J, Oliveira PSL, Figueira ACM, Webb P, Polikarpov I. Structural Insights into Human Peroxisome Proliferator Activated Receptor Delta (PPAR-Delta) Selective Ligand Binding. PLoS One. 2012;7(5):1–7. doi: 10.1371/journal.pone.0033643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kojetin DJ, Burris TP. Small molecule modulation of nuclear receptor conformational dynamics: implications for function and drug discovery. Mol Pharmacol. 2013;83(1):1–8. doi: 10.1124/mol.112.079285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Chao EY-H, Haffner CD, Millard Hurst Lambert I, Maloney PR, Sierra ML, Sternbach DD, Sznaidman ML, Willson TM. Thiazole and oxazole derivatives and their pharmaceutial use. 2001 Jan 4; [Google Scholar]; (b) Buchan KW. Medicaments. WO 02/2843 A2. 2002 Apr 11;
- 23.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 24.Teske KA, Bogart JW, Sanchez LM, Yu OB, Preston JV, Cook JM, Silvaggi NR, Bikle DD, Arnold LA. Synthesis and evaluation of vitamin D receptor-mediated activities of cholesterol and vitamin D metabolites. Eur J Med Chem. 2016;109:238–46. doi: 10.1016/j.ejmech.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masuno H, Ikura T, Morizono D, Orita I, Yamada S, Shimizu M, Ito N. Crystal structures of complexes of vitamin D receptor ligand-binding domain with lithocholic acid derivatives. Journal of lipid research. 2013;54(8):2206–13. doi: 10.1194/jlr.M038307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perakyla M, Malinen M, Herzig KH, Carlberg C. Gene regulatory potential of nonsteroidal vitamin D receptor ligands. Molecular endocrinology. 2005;19(8):2060–73. doi: 10.1210/me.2004-0417. [DOI] [PubMed] [Google Scholar]
- 27.Hakamata W, Sato Y, Okuda H, Honzawa S, Saito N, Kishimoto S, Yamashita A, Sugiura T, Kittaska A, Kurihara M. (2,S,1’R)-Analogue of LG190178 is a major active isomer. Bioorg Med Chem Lett. 2008;18:120–123. doi: 10.1016/j.bmcl.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Nandhikonda P, Lynt WZ, McCallum MM, Ara T, Baranowski AM, Yuan NY, Pearson D, Bikle DD, Guy RK, Arnold LA. Discovery of the first irreversible small molecule inhibitors of the interaction between the vitamin D receptor and coactivators. J Med Chem. 2012;55(10):4640–51. doi: 10.1021/jm300460c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The Crystal Structure of the Nuclear Receptor for Vitamin D Bound to Its Natural Ligand. Mol Cell. 2000;5:173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 30.Teichert A, Arnold LA, Otieno S, Oda Y, Augustinaite I, Geistlinger TR, Kriwacki RW, Guy RK, Bikle DD. Quantification of the vitamin D receptor-coregulator interaction. Biochemistry. 2009;48(7):1454–61. doi: 10.1021/bi801874n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.