

Abstract
Trafficking proteins inside cells is an emerging field with potential utility in basic cell biology and biological therapeutics. A robust and sustainable delivery strategy demands not only a good protection of the cargo, but also for reversibility in conjugation and activity. We report a protein-templated polymer self-assembly strategy for the formation of a sheath around the proteins and then tracelessly releasing them in the cytosol. The demonstrated versatility of the approach suggests that the strategy could be compatible with a wide array of biologics.
Graphical Abstract
Using proteins as a therapeutic is attractive, as this promises to directly address genetic deficiencies and therefore mitigates side-effects that plague many small molecule drugs.1 Potential side-effects from small molecule binders are understandable, as these molecules must be designed to target a specific protein in our complex biological system, highlighted by the nearly 20,000 protein-encoding genes. On the other hand, proteins can directly compensate for a specific deficiency and therefore the drug development is less heuristic. However, realizing the full potential of protein-based therapeutics has been difficult, mainly due to their in vivo instability and immunogenicity. To overcome these issues, approaches to modify protein surfaces have been taken, starting with PEGylation that has been shown to enhance protein circulation lifetimes.2 More recently, strategies that allow for attaching other polymers to proteins have been developed in order to endow these conjugates with stimulus-responsive characteristics or to realize new self-assembled structures.3 These reports provide examples of innovative strategies that allow for enhanced circulation lifetimes and thus have impacted the utility of proteins that function in the extracellular environment.2,4
The next level of challenge involves the ability to develop systems that can handle intracellular proteins, where trafficking the cargo across a cellular membrane is a major hurdle. Two limiting approaches have been taken to address this need, both of which involve non-covalent self-assembly. First involves electrostatic binding of proteins to complementarily charged polymers and nanoparticles.5 The second approach includes encapsulating proteins in water-filled compartments, such as liposomes.6 A limitation of the former approach comprises non-specific fouling of the complex’ surfaces due to electrostatic interactions and the associated toxicities.7 The latter approach has the potential to address the fouling issues, but is fraught with low loading capacities, especially when charge-neutral lipids are used.
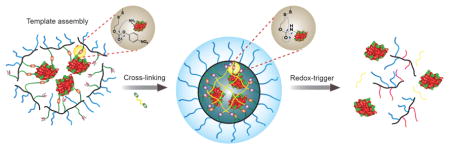
We envisaged that a covalent self-assembly approach, where the protein cargo itself acts as the template for the polymer to self-assemble around it, has the potential to encapsulate proteins with high fidelity and present charge-neutral surface functionalities (Figure 1). The key design hypothesis here is that an initial reaction between the side chain functionalities of a random copolymer and multiple surface-exposed functional groups of a target protein would cause a few polymer chains to organize around the protein. This covalent capture then can act as a template to form a polymer sheath around the protein through a polymer side-chain crosslinking step, as schematically illustrated in Figure 1. We envisaged that the high-fidelity protein encapsulation within this sheath would be aided by: (a) convex surface of globular proteins on which the reactive functional groups are presented, and (b) high-yielding and multivalent reactions are presented; and (b) high-yielding and multivalent reactions between the protein and polymer side chains.
Figure 1.

Schematic representation of the formation of a covalent polymer network using the protein as the template and its traceless and triggered release in a reducing environment.
Cysteine and lysine are two popular handles for conjugating polymers with proteins, because of their nucleophilicity.8 Because of the surface availability in larger number of proteins, lysines are generally preferred. However, this amino acid presents a challenge in that it is more difficult to functionalize them in a form, whereby they can be tracelessly liberated in the intracellular environment. We hypothesized that placing reactive side-chain functionalities in a polymer, with responsive self-immolation characteristics,9 would result in a novel and general system that is capable of encapsulating proteins with high fidelity and tracelessly releasing them upon encountering a target microenvironment.
The structure of the target polymer P1 (Scheme 1), which satisfies all the design requirements, was synthesized by RAFT polymerization. Reaction of an amine with the p-nitrophenylcarbonate (NPC) moiety in P1 will produce the corresponding carbamate, as shown in P2. The polymer is first treated with the protein, where multiple lysine moieties are reacted with the NPC groups in the polymer chains. The remaining NPCs are reacted with a diamine crosslinker to complete the sheath formation around the protein (Figure 1 and Scheme 1). Note that a disulfide moiety is placed at the β-position, relative to the carbamate oxygen, which serves to render the polymer responsive to the more reductive intracellular environment, compared to the extracellular space.8d Reductive cleavage of the disulfide will result in carbamate cleavage to release the original amine. This reaction will cause both the polymer being uncrosslinked and the protein being tracelessly liberated from the polymer.
Scheme 1.

Chemical structures of polymers and the reaction scheme for protein conjugation, crosslinking to generate the nanoassembly and its release in the presence of a reducing agent.
To test our design strategy, we chose cytochrome C (CytC, pI 9.6) as a model protein, because of its distinct cellular readout in the form of apoptotic cell death. After initially reacting CytC with P1, the polymer-protein conjugate was further secured by crosslinking with ethylenediamine (ED) or tetraethyleneoxide-bis-amine (PEG) to afford nanoassemblies NA-CytCED and NA-CytCPEG, respectively. Note that the reaction between the NPC moiety and an amine produces p-nitrophenol as a by-product, the distinct absorption of which is conveniently monitored. Therefore, the protein conjugation step was quantified using the evolution of the absorption spectrum (Figure S5). Encapsulation efficiency and loading capacity were found to be 64–67% and 5–7%, respectively. Dynamic light scattering (DLS) measurements revealed the hydrodynamic diameters of native CytC and the protein-containing nanoassembly to be ~4 nm and ~8–10 nm, respectively (Figure S6). Moreover, zeta potential measurements revealed that the surface of the complex is charge-neutral, suggesting that the complex surface is dominated by the PEG moieties from P1 (see SI). Figure 2a (top) shows a Cryo-EM image of NA-CytCPEG. The average individual particle size is in the 10–30 nm range which is in agreement with DLS data. To obtain a more detailed insight into the protein distribution within the nano-assemblies we employed High Angle Annular Dark Field Microscopy (HAADF) and Energy Dispersive X-Ray Spectroscopy (EDS) at cryo temperatures (see SI for sample preparation). Figure 2a (bottom) shows a HAADF image of an individual nano-assembly. The whole assembly has a diameter of about 20 nm and the bright spots in the 2–3 nm range are caused by the Fe-content of discrete Cyt-C molecules (compare with the empty assemblies in the inset) and EDS analysis confirmed the presence of Fe (see SI).
Figure 2.

(a) Top: Bright Field Cryo-TEM image of NA-CytCPEG showing both individual and small clustered particles. Inset shows a cluster of 3 protein nanoassemblies, Bottom: HAADF image of a NA-CytCPEG nanoassembly. The bright spots with diameters of 2–3 nm are caused by the Fe-content of Cyt C, inset is NA-EmptyPEG particle with no detectable Fe from EDS (~50 nm, see SI); (b) MALDI-MS analysis of the trypsin digest from encapsulated and naked CytC; (c, d) SDS-PAGE of the NA-CytCPEG under non-reducing and reducing conditions (10 mM DTT, 37 °C for 4 h).
An important objective of this work is to use the polymer sheath to protect the protein from protease degradation. To rigorously test for this, we subjected the polymer-protein conjugate to protease digest with trypsin and analyzed the products using MALDI mass spectrometry. While the unprotected CytC afforded characteristic peptide fragment peaks, the conjugate at the same protein concentration did not afford any discernible fragments (Figure 2). These results show that assemblies do indeed protect the protein. The conjugates also seem to be generally stable in serum (Figure S13).
Next, we were interested in testing whether the encapsulated protein can be released in a reducing environment. We first tested this possibility using gel electrophoresis (SDS-PAGE). As anticipated, when the protein is conjugated to the protein, no bands corresponding to the protein was found (Figure 2). When the same SDS-PAGE gel was run under reducing conditions, appearance of protein bands clearly indicated that the encapsulated protein can be released. This is the first indicator, suggesting that the protein release using the reductive self-immolative linker is feasible. We utilized SDS-PAGE experiments to quantify the amount of proteins inside our nanoassemblies. After treating the nanoassemblies with excess dithiothreitol (DTT), the intensity of the protein band in the gel is compared with native proteins of different concentrations to estimate the amount of proteins present inside the nanoassembly (Figure 2).
The protein encapsulation and release process would be a futile exercise, if the methodology does not preserve the structure and function of the protein upon release. To this end, the secondary structure of the released protein was examined by circular dichroism (CD) spectroscopy, the spectrum of which was found to be identical to that of the native CytC (Figure 3). This suggests that the conjugation and release processes did not alter the secondary structure of the protein. We also claim that the strategy leads to a traceless release of the protein in reductive environment. To test this hypothesis, the released protein was analyzed by mass spectrometry. The m/z for the released protein matched the native CytC, suggesting that there are no remnants of the polymer after the protein’s reductive release (Figure S21).
Figure 3.

Structure and function of released CytC from the NA-CytCPEG, evaluated by (a) CD spectroscopy; and (b–c) ABTS activity assay: b-assay kinetics, c-% activity of samples treated with DTT.
Next, we investigated whether the function of the protein is maintained by quantifying the released protein’s activity using an ABTS assay (based on 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (Figure 3b–c, see SI).10a While the nanoassembly did not exhibit any enzymatic activity, the activity recovery was nearly quantitative when the assembly was treated with 10 mM DTT (compare the activities of native CytC and released CytC in the presence of 10 mM DTT in Figure 3). This activity recovery was also found to be dependent on the concentration of DTT and thus the extent of protein release. These results show that the polymer shell can act to ‘turn-off’ the protein function, until it is released in its target environment. Both structure and function recoveries were found to be independent of the crosslinker length in ED and PEG linkers, as shown in Figure 3.
The ultimate goal of the proposed research is to utilize this polymer coating to traffic the protein across the cellular membrane and release it in the cytosol. It is the higher redox potential of the cytosol that is being targeted for selective release. To track the location of the protein under confocal laser scanning microscopy (CLSM), CytC was labeled with rhodamine B and the cell nucleus was stained with hoechst 33342. After 4 h incubation, well-distributed red fluorescence from labeled proteins was observed (Figure 4a), while negligible fluorescence was observed from cells that were treated with an identical concentration of naked proteins (Figure S23). These results suggest that the polymer conjugate has better access to the cells, compared to the native protein itself. The time course of cellular internalization process was also monitored under CLSM for this conjugate (see Figure S26 and video in SI). Since the most likely pathway for uptake is endocytosis, we were next interested in evaluating whether the proteins are stuck in the endosome or they escaped the endosome to get into the cytosol. For a cytosolic delivery, the latter is desired. Accordingly, the endosomes were labeled with lysotracker green. The data clearly indicate that the nanoassemblies enter the cells through the endosomes (see co-localization of lysotracker green and rhodamine-B label after 4 h incubation (Figure 4b)), but the proteins escape the endosomes over time as seen by the dominant red color in the cell in the 24 h image (Figure 4c).
Figure 4.

HeLa cells treated with NA-CytCPEG conjugates to study cellular uptake: (a) 4 h post-incubation; (b–c) endosomal co-localization and escape at 4 h and 24 h, respectively; (green: lysotracker; red: rhodamine B; blue: hoechst); (d) cell viability after 72 h (dosage represents amount of NA-Protein conjugate, see SI); (e) detection of activated caspase-3/7 after 72 h using the Cy5-tagged assay reagent; scale bar: (a) 50 μm, (b, c, e) 10 μm.
To evaluate whether the delivered CytC is active, we evaluated the apoptotic cell death in response to the protein delivery. CytC is known to induce apoptosis through interaction with apoptotic protease activating factor 1 (Apaf-1) in cytosol and activation of pro-caspase-9, which in turn initiates pro-caspase-3 leading to activation of caspase dependent apoptotic pathways.10b,c The dose-dependent decrease in cell viability of the nanoassembly (Figure 4d), combined with the lack of toxicity for the corresponding concentration of the free nanoassembly or naked CytC, indicate that the cytosolically delivered CytC is causing apoptosis. Furthermore, the mechanism of action of CytC allows the direct interrogation whether the caspase-dependent pathway is activated in the cytosol. A fluorimetric immunoassay that causes a caspase product to be intercalated the DNA in the nucleus was utilized to asses this possibility. The co-localization of the caspase-processes, cy5-tagged reagent (red) and the nuclear stain (blue) confirmed the apoptotic nuclei in the cells (Figure 4e and SI). Control experiments show that the nanoassembly without the CytC does not cause the activation of the cy5-tagged reagent.
The true testament to the versatility of this strategy is the applicability to a broad range of proteins. To test this, we used two other proteins, viz. lysozyme (Lys) and ribonuclease A (RNaseA). Both proteins were found to be successfully conjugated with the polymer P1 (see SI for characterizations). SDS-PAGE, CD, and activity studies also show that the encapsulated protein can be released with high fidelity under reducing conditions with high retention in both structure and function (Figure 5a–b and SI). It is interesting to note that the overall kinetics of proteins release was observed to have the order: CytC<RNaseA<Lys (see SI). The release kinetics seems to correlate with the number of surface exposed lysines in each protein (CytC: 19, RNaseA: 10, Lys: 611). This is understandable, since the higher number of anchoring points requires more sites for the reducing agent to process during release. Finally, the intracellular delivery and activity of these proteins were also evaluated (see Figure 5 and SI). While lysozyme is expected to be innocuous to cells, RNaseA with access to cytosolic RNA can initiate cell death.12 Although both proteins were taken up by the cells as assessed by CLSM studies, the lysosome-containing nanoassembly did not induce any cell death, whereas the RNaseA-bearing nanoassembly had a profound effect on cell viability (Figure 5). The proteins studied so far are basic (pI >8.5) with a MW of 12–15 kDa. To further test this system, we also investigated the encapsulation and release properties of eGFP with higher MW and lower pI (MW ~27 kDa, pI 5.5). SDS-PAGE and cellular uptake studies show that this protein, too, can be successfully encapsulated and delivered (see SI).
Figure 5.

SDS-PAGE showing protein release (reducing conditions), cellular uptake (4 h) and viability (72 h) in HeLa cells for (a) NA-Lys and (b) NA-RNaseA conjugates, scale bar: 50 μm. TOC graphic
In summary, we have developed a versatile strategy for the encapsulation of proteins and their tracelss release in response to a specific trigger. The encapsulation is templated by the lysine handles in the protein itself, which are then used to wrap the protein with a polymer sheath in a secondary crosslinking step. The versatility of the approach is highlighted by the fact that: (i) it utilizes a functional handle that is abundantly available on the surface of >85% the globular proteins, which renders the strategy broadly applicable; (ii) the target protein is encapsulated with high fidelity, i.e. high loading capacity; (iii) the cargo is protected from degradation by proteases; (iv) the protein activity is masked in the encapsulated state; (iv) the polymer sheath is removed tracelessly with high efficiency in response to a target intracellular environment; (v) the native structure and function are retained upon release; (vi) the protein can be delivered with high fidelity into the cytosol; and (vii) activity of the protein is regained in the cytosol. Thus, we believe that this simple and general strategy will serve to produce a potent protein therapeutic delivery platform for a broad range of proteins.
Supplementary Material
Acknowledgments
We thank Army Research Office (W911NF-15-1-0568) for support, NIH (T32GM008515) for CBI-training grant to KD, Kishore Raghupathi for helpful discussions, Francesca Anson for eGFP, and James Chambers for help in CLSM.
Footnotes
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website. Synthesis and characterization details (PDF) and cellular uptake video (MP4).
References
- 1.Leader B, Baca QJ, Golan DE. Nat Rev Drug Discovery. 2008;7:21. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.(a) Cobo I, Li M, Sumerlin BS, Perrier S. Nat Mater. 2014;14:143. doi: 10.1038/nmat4106. [DOI] [PubMed] [Google Scholar]; (b) Gu Z, Biswas A, Zhao M, Tang Y. Chem Soc Rev. 2011;40:3638–3655. doi: 10.1039/c0cs00227e. [DOI] [PubMed] [Google Scholar]; (c) Abuchowski A, van Es T, Palczuk NC, Davis FF. J Biol Chem. 1977;252:3578. [PubMed] [Google Scholar]
- 3.(a) Stayton PS, Shimoboji T, Long C, Chilkoti A, Ghen G, Harris JM, Hoffman AS. Nature. 1995;378:472. doi: 10.1038/378472a0. [DOI] [PubMed] [Google Scholar]; (b) Lu Y, Sun W, Gu Z. J Controlled Rel. 2014;194:1. doi: 10.1016/j.jconrel.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hannink JM, Cornelissen JJLM, Farrera JA, Foubert P, De Schryver FC, Sommerdijk NAJM, Nolte RJM. Angew Chem, Int Ed. 2001;40:4732. [PubMed] [Google Scholar]; (d) Moatsou D, Li J, Ranji A, Pitto-Barry A, Ntai I, Jewett MC, O’Reilly RK. Bioconjugate Chem. 2015;26:1890. doi: 10.1021/acs.bioconjchem.5b00264. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alconcel SNS, Baas AS, Maynard HD. Polym Chem. 2011;2:1442. [Google Scholar]
- 5.(a) Ray M, Tang R, Jiang Z, Rotello VM. Bioconjugate Chem. 2015;26:1004. doi: 10.1021/acs.bioconjchem.5b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee Y, Ishii T, Cabral H, Kim HJ, Seo JH, Nishiyama N, Oshima H, Osada K, Kataoka K. Angew Chem Int Ed. 2009;48:5309. doi: 10.1002/anie.200900064. [DOI] [PubMed] [Google Scholar]; (c) González-Toro DC, Ryu JH, Chacko RT, Zhuang J, Thayumanavan S. J Am Chem Soc. 2012;134:6964. doi: 10.1021/ja3019143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swaminathan J, Ehrhardt C. Expert Opin Drug Deliv. 2012;9:1489. doi: 10.1517/17425247.2012.735658. [DOI] [PubMed] [Google Scholar]
- 7.Lv H, Zhang S, Wang B, Cui S, Yan J. J Controlled Release. 2006;114:100. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 8.(a) Stenzel MH. ACS Macro Lett. 2013;2:14. doi: 10.1021/mz3005814. [DOI] [PubMed] [Google Scholar]; (b) Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]; (c) Chen JP, Yang HJ, Hoffman AS. Biomaterials. 1990;11:625. doi: 10.1016/0142-9612(90)90019-m. [DOI] [PubMed] [Google Scholar]; (d) Ventura J, Eron SJ, González-Toro DC, Raghupathi K, Wang F, Hardy JA, Thayumanavan S. Biomacromolecules. 2015;16:3161. doi: 10.1021/acs.biomac.5b00779. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Vazquez-Dorbatt V, Tolstyka ZP, Maynard HD. Macromolecules. 2009;42:7650. doi: 10.1021/ma9013803. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wiss KT, Krishna OD, Roth PJ, Kiick KL, Theato PA. Macromolecules. 2009;42:3860. [Google Scholar]; (g) Li H, Bapat AP, Li M, Sumerlin BS. Polym Chem. 2011;2:323. [Google Scholar]; (h) Danial M, Tran CM, Jolliffe KA, Perrier S. J Am Chem Soc. 2014;136:8018. doi: 10.1021/ja5024699. [DOI] [PubMed] [Google Scholar]
- 9.Riber CF, Smith AAA, Zelikin AN. Adv Healthcare Mater. 2015;4:1887. doi: 10.1002/adhm.201500344. [DOI] [PubMed] [Google Scholar]
- 10.(a) Childs RE, Bardsley WG. Biochem J. 1975;145:93. doi: 10.1042/bj1450093. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Santra S, Kaittanis C, Perez JM. Mol Pharmaceutics. 2010;7:1209. doi: 10.1021/mp100043h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morales-Cruz M, Figueroa CM, Gonzalez-Robles T, Delgado Y, Molina A, Méndez J, Morales M, Griebenow K. J Nanobiotechnol. 2014;12:33. doi: 10.1186/s12951-014-0033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Prasanna Murthy SN, Lukas TJ, Jardetzky TS, Lorand L. Biochemistry. 2009;48:2654. doi: 10.1021/bi802323z. [DOI] [PubMed] [Google Scholar]; (b) Bosshard HR, Zurrer M. J Biol Chem. 1980;255:6694. [PubMed] [Google Scholar]
- 12.(a) Wang M, Sun S, Neufeld CI, Perez-Ramirez B, Xu Q. Angew Chem Int Ed. 2014;53:13444. doi: 10.1002/anie.201407234. [DOI] [PubMed] [Google Scholar]; (b) Ellis GA, Palte MJ, Raines RT. J Am Chem Soc. 2012;134:3631. doi: 10.1021/ja210719s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.