

Abstract
In eukaryotic cells a surveillance mechanism, the S phase checkpoint, detects and responds to insults that challenge chromosomal replication, arresting cell cycle progression and triggering appropriate events to prevent genomic instability. In the budding yeast Saccharomyces cerevisiae, Mec1/ATM/ATR, and its downstream kinase Rad53/Chk2, mediate the response to genotoxic stress. In this study, we place Cip1, a recently identified Cdk1 inhibitor (CKI), under the regulation of Mec1 and Rad53 in response to genotoxic stress. Cip1 accumulates dramatically in a Mec1 and Rad53 dependent manner upon replication stress. This increase requires the activity of MBF, but not the transcriptional activator kinase Dun1. At the protein level, stabilization of replication stress induced Cip1 requires continued de novo protein synthesis. In addition, Cip1 is phosphorylated at an S/TQ motif in a Mec1 dependent manner. Deletion of Cip1 affects proliferation in hydroxyurea containing plates. Significantly, the sensitivity is increased when the dosage of the G1 cyclin CLN2 is increased, compatible to a role of Cip1 as a G1-CDK inhibitor. In all, our results place Cip1 under the S phase checkpoint response to genotoxic stress. Furthermore, Cip1 plays a significant role to preserve viability in response to insults that threaten chromosome replication.
Keywords: YPL014W, Cip1, Cdk1, Cln2, S phase checkpoint, Rad53, MBF, Mec1
Graphical Abstract
We place Cip1, a recently identified Cdk1 inhibitor (CKI), under the regulation of Mec1 and Rad53 in response to genotoxic stress. Cip1 accumulates dramatically in a Mec1 and Rad53 dependent manner upon replication stress. Cip1 is phosphorylated at an S/TQ motif in a Mec1 dependent manner.
Introduction
Cells are under constant pressure from endogenous and exogenous agents that cause DNA damage or interfere with DNA replication, globally termed as genotoxic stress. In response to genotoxic stress, cells activate a crucial surveillance mechanism, S phase checkpoint, which protects ongoing DNA replication and arrests cell cycle progression in order to repair DNA damage.
In budding yeast, replication forks stall upon replication stress, leading to the activation of the central transducer protein kinases Mec1 and Tel1 (Sanchez et al. 1996), the orthologs of human ATM and ATR. Mec1, in turn, activates the effector protein kinases Chk1 and Rad53 (Sanchez et al. 1999), the orthologs of human Chk1 and Chk2, respectively. The checkpoint response includes the stabilization of stalled replisomes, suppression of recombination at arrested replication forks, block of late firing origins of replication (Santocanale & Diffley 1998; Tercero & Diffley 2001), prevents cells entry into mitosis and the segregation of sister chromatids (Cohen-Fix & Koshland 1997), and also activates a transcriptional response that includes the induction of ribonucleotide reductase genes to counteract the stress (Zhou & Elledge 1993).
In human cells, the S phase checkpoint is now regarded as an anti-cancer barrier in early tumorigenesis (Bartkova, Bakkenist, et al. 2005; Bartkova, Horejsí, et al. 2005). Cyclin Dependent Kinase (CDK) activity is under strict checkpoint regulation upon different genotoxic stresses (Hartwell & Weinert 1989; Furnari et al. 1997; Palou et al. 2015). Overexpression of G1 cyclin E or cyclin D causes genomic instability such as increased aneuploidy and gene amplification (Zhou et al. 1996; Spruck et al. 1999). In budding yeast, deregulated G1 cyclin expression also induces genomic instability (Tanaka & Diffley 2002).
We previously reported that budding yeast Cip1 (YPL014W) specifically interacts with G1 cyclins as an inhibitor of cyclin dependent kinase Cdk1 (Ren et al. 2016). Our results now place Cip1 under the S phase checkpoint. Cip1 accumulates in response to genotoxic replication stress in a Mec1 and Rad53 dependent manner. In addition, in response to replication stress Cip1 is phosphorylated at an S/TQ consensus motif in a Mec1 dependent manner. Deletion of Cip1 affects proliferation in hydroxyurea containing plates. Significantly, the sensitivity is increased when the dosage of the G1 cyclin CLN2 is increased, suggesting that Cip1 may play a role as a G1-CDK inhibitor as part of the S phase checkpoint response.
Results
Cip1 accumulates upon genotoxic stress
In a proteomic study to identify proteins differentially associated with Cdk1in response to Rad53 activation, we identified 1,078 proteins (Figure S1). 90 proteins were found specifically associated to Cdk1 in wild type cells but not in rad53 mutant cells upon hydroxyurea (HU) treatment (Figure S1, Table S3). Validating the findings, 40 out of 90 proteins were previously known to be directly connected to Cdk1 and/or to replication stress. For example, the second hit, Srl3 is a Cdk1 interacting protein previously identified as a suppressor of the lethality of a rad53 null mutation when overexpressed (Desany et al. 1998). More recently, Srl3 has been shown to be part of the negative control of Start (Yahya et al. 2014).
We recently characterized the most intense, Rad53 dependent Cdk1 interactor, Cip1, as a G1-Cdk1 inhibitor (Ren et al. 2016). Because we found Cip1 associated to Cdk1 in cells under replication stress and in a Rad53 dependent manner, we subsequently wished to explore the role of Cip1 in response to genotoxic stress. We first explored the abundance of Cip1 protein during an unperturbed cell cycle and in cells under replication stress. Cells synchronized in pre-Start G1 were synchronously released into S phase, either in the presence or in the absence of hydroxyurea, a reagent that generates replication stress by depleting the pool of dNTPs. As shown in Figure 1A, when cells enter S phase in the presence of HU, Cip1 accumulates dramatically and the electrophoretic mobility decreases, in a manner compatible with phosphorylation.
Figure 1. Cip1 accumulates upon genotoxic stress.

(A) Cip1-13myc cells (YFL117 strain) were synchronized with α-factor and released into S phase in the presence of HU. The samples were taken at the indicated time points (min). Left panel, fluorescence-activated cell sorting analysis of DNA content. The presence of HU abolishes DNA replication, but not cell cycle entry, as assessed by the progression of the budding indexes. Right panel, TCA whole cell extracts were analyzed by immunoblotting with anti-Myc (Cip1) antibodies. A Ponceau S-stained region of the same membrane used for immunoblotting is shown as a loading control. (B) Wild type cells (YFL110) were released from the G1 arrest, and allowed to progress into S phase in the presence of Noc, or in the presence of HU, MMS, and CPT respectively. The samples were taken at the indicated time points. Left panel, fluorescence-activated cell sorting analysis of DNA content. Right panel, whole cell extracts were analyzed by immunoblotting with anti-Myc (Cip1). A Ponceau S-stained region of the same membrane used for immunoblotting is shown as a loading control.
Significantly, the accumulation of Cip1 occurs not only in response to replication stress, but also in response to DNA damage generated with different standard reagents (Figure 1B), such as methyl methane-sulphonate (MMS), a reagent that generates DNA methylation damage, and camptothecin (CPT), a Topoisomerase I poison that generates single strand breaks on the DNA. In contrast, Cip1 accumulation is not significant when cells were arrested in G2/M in the presence of the spindle depolymerizing drug nocodazole (Noc), and are similar to those seen in an unperturbed cell cycle in cells in mitosis (Figure 1A, YPD, 90 min time point).
The increase of Cip1 abundance under replication stress depends on the S phase checkpoint
The three different types of genotoxic stress shown above to result in an increase of Cip1 levels are known to activate the checkpoint kinase Rad53. We therefore next explored whether the increase in Cip1 abundance depends on Rad53. The experiment described in Figure 1A was repeated, now comparing wild type cells and rad53 null mutant cells. As shown in Figure 2A, the rad53 mutant fails to increase the Cip1 protein levels, even though DNA replication is effectively blocked in both cases (see left panel).
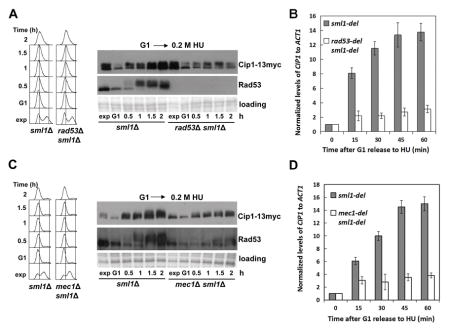
Figure 2. Increase of Cip1 abundance upon replication stress depends on the S phase checkpoint kinases Mec1 and Rad53.

(A) Accumulation of Cip1 protein upon replication stress is dependent on Rad53. Rad53 positive cells (sml1Δ, YFL101 strain) and rad53Δ null mutant cells (rad53Δ sml1Δ, strain YFL102) were synchronized in pre-Start G1 (α) and released in the presence of 200 mM HU. Samples were taken at the indicated times (min). Left panel, fluorescence-activated cell sorting analysis of DNA content. Right panel, TCA whole cell extracts were analyzed by immunoblotting with anti-Myc (Cip1) or anti-Rad53 (Rad53) antibodies. (B) Accumulation of CIP1 mRNA is dependent on Rad53. The normalized relative amount of CIP1 mRNA to ACT1 control in different strains in the presence of 200 mM HU is shown. (C) Accumulation of Cip1 protein upon replication stress is dependent on Mec1. Mec1 positive cells (sml1Δ, YFL101 strain) and mec1Δ null mutant cells (mec1Δ sml1Δ, strain YZZ2) were synchronized in pre-Start G1 (α) and released in the presence of 200 mM HU. Samples were taken at the indicated times (min). Left panel, fluorescence-activated cell sorting analysis of DNA content. Right panel, TCA whole cell extracts were analyzed by immunoblotting with anti-Myc (Cip1) or anti-Rad53 (Rad53) antibodies. (D) Accumulation of CIP1 mRNA is dependent on Mec1. The normalized relative amount of CIP1 mRNA to ACT1 control in different strains in the presence of 200 mM HU is shown.
The accumulation of Cip1 protein might be achieved through increased transcription or through protein stabilization. The checkpoint kinase Rad53 mediates the transcriptional response to genotoxic stress (Zhou & Elledge 1993; Bastos de Oliveira et al. 2012; Jaehnig et al. 2013). Analysis of the CIP1 mRNA levels shows that CIP1 transcript accumulates in the response to replication stress (Figure 2B). In addition, also the observed protein accumulation in response to replication stress, but not the slower electrophoretic mobility, are dependent on the checkpoint kinase Rad53.
Cip1 upregulation in response to genotoxic stress also depends on the checkpoint central transducer kinase Mec1. We repeated the experiment described in Figure 2A and 2B now comparing a mec1 null mutant with its isogenic MEC1 control. As shown in Figure 2C and 2D, the accumulation of Cip1 mRNA and protein in response to replication stress is also dependent on Mec1. Interestingly, the slower mobility of Cip1 is also affected in mec1 null mutant cells, which suggests a Mec1 dependent modification of Cip1 upon replication stress.
In all, these results indicate that the upregulation of Cip1 upon replication stress depends is part of the S phase checkpoint response mediated by the conserved Mec1 and Rad53 kinases.
Cip1 expression is induced via Nrm1 upon DNA replication stress
Accumulation of CIP1 transcript might result either from mRNA stabilization or from induction of transcription. Two major pathways mediate the transcriptional response triggered by Rad53 in response to genotoxic stress. In one of the pathways, Rad53 activates the downstream kinase Dun1, which subsequently phosphorylates and inactivates the transcriptional repressor Crt1, thereby upregulating the expression of a cluster of genes including those encoding the ribonucleotide reductase subunits RNR2,3,4 (Zhou & Elledge 1993; Huang et al. 1998). Another branch of the transcriptional response mediated by Rad53 depends on the selective reactivation of MBF transcription. When S phase is challenged by genotoxic stress, Rad53 inactivates the Nrm1 repressor that suppresses MBF transcription as cells enter S phase (de Bruin et al. 2008; Travesa et al. 2012).
As shown in Figure 3A, a dun1Δ mutant strain remains proficient to increase the levels of Cip1 in response to replication stress to the same extent as wild type cells. Therefore, increase of Cip1 expression in the presence of HU does not require the Dun1 mediated transcriptional program under Rad53.
Figure 3. Increase of Cip1 abundance upon replication stress depends on MBF transcription.

(A) Mutant dun1Δ cells (YFL122 strain) and wild type cells (YFL100 strain) were synchronized in pre-Start G1 (α) and then released in the presence of 200 mM HU. Samples were taken at the indicated times (min). TCA whole cell extracts were analyzed by immunoblotting with anti-Myc (Cip1) or anti-Rad53 antibodies. (B) Gal-Nrm1ΔN-myc cells (YFL120 strain) and wild type cells (YFL100 strain) were synchronized in pre-Start G1 (α) in YPRaff. The cells were then released from the α-factor arrest into YPGal supplemented with 200 mM HU. Samples were taken at the indicated times (min). TCA whole cell extracts were analyzed by immunoblotting with anti-Myc (Cip1, Nrm1ΔN) or anti-Rad53 (Rad53) antibodies. (C) The normalized relative amount of CIP1 mRNA to ACT1 control in the indicated conditions is shown.
We then checked whether CIP1 is one of the MBF genes reactivated by Rad53 by inactivation of the MBF repressor Nrm1. We took advantage of a dominant allele of Nrm1, Nrm1ΔN, that supersedes Rad53 when overexpressed and keeps MBF expression off (Palou et al. 2010; Bastos de Oliveira et al. 2012). As shown in Figure 3B and 3C, whereas Cip1 expression at both mRNA and protein levels increases in the presence of HU in control cells, the response is abrogated in cells overexpressing Nrm1ΔN, in the continued presence of HU and active Rad53. These observations indicate that MBF transcription is required to maintain the presence of Cip1 in a compromised S phase.
Stabilization of replication stress induced Cip1 requires continued de novo protein synthesis
As mentioned above, Cip1 accumulation might also result from reduced protein turnover. We therefore explored whether Cip1 protein is stabilized in response to replication stress. We analyzed the evolution of Cip1 protein levels when protein synthesis is inhibited with of cycloheximide (CHX) in cells undergoing replication stress in S phase. Wild type cells were synchronized in pre-Start G1 with α-factor, and released into the S phase in the presence of HU. After 1 h the culture was split in two, and CHX was added to one of the cultures.
As shown in Figure 4A, ells are completely unable to accumulate Cip1 when protein synthesis is blocked. Interestingly, the population of Cip1 with lower mobility shows a much more rapid elimination upon inhibition of protein synthesis. Therefore, the excess Cip1 protein accumulated upon replication stress remains highly unstable, indicating that Cip1 abundance is regulated through de novo protein synthesis.
Figure 4. Increase of Cip1 abundance upon replication stress requires continued de novo protein synthesis.

Cip1-13myc cells (YFL100 strain) were synchronized in pre-Start G1 (α) and released in the presence of 200 mM HU for 1h. The culture was then split in two. One half was further incubated under the same conditions (HU) (A), whereas cycloheximide was added to the other half (B), keeping the presence of hydroxyurea (HU + CHX). The samples were collected every half hour, and whole cell extracts were analyzed by immunoblot with anti-Myc antibodies (Cip1). Budding indexes (BI) are shown on top as measures of synchronicity and cell cycle progression to confirm that cells progress into the cell cycle despite the absence of DNA replication or protein synthesis.
Mec1 dependent phosphorylation of Cip1 in response to DNA replication stress
We showed above that in addition to the increase in Cip1 protein levels, the protein displays a slower electrophoretic mobility in response to DNA replication stress (Figure 1A). Despite the increase of Cip1 expression is dependent on the downstream kinase Rad53, the slower mobility appears to depend on Mec1 rather than on Rad53 (Figure 2). A recent high-throughput proteomic study to profile DNA damage-induced phosphorylation in budding yeast suggested that Cip1 is phosphorylated at an SQ motif (Zhou et al. 2016). SQ/TQ is the consensus phosphorylation sequence for the DNA damage response ATM/ATR kinases and their yeast homologs, Tel1/Mec1 (Abraham 2001). Taking these observations into consideration, we asked whether Cip1 is phosphorylated by Mec1 in response to DNA replication stress.
As shown in Figure 5A, Cip1 has three putative Mec1 phosphorylation sites, amino acid residues S18, T42 and S288. We used antibodies that specifically detect phosphorylated S/TQ motifs (anti-pSQ/pTQ antibodies), and analyzed Cip1 from wild type cells and mec1 null cells exposed or not to treatment with HU. Cip1 was immunopurified from native whole cell extracts and western-blotted with anti-pSQ/pTQ antibodies. As seen in Figure 5A, at least one SQ/TQ site of Cip1 is phosphorylated in wild type cells treated with HU (Lane 1), but not in untreated wild type cells (Lane 2). Interestingly, the phosphorylation signal is absent in mec1 null mutants exposed to replication stress (lane 3). These results indicate that Cip1 is phosphorylated at S/TQ motif in a Mec1 dependent upon DNA replication stress, robustly placing Cip1 as part of the S phase checkpoint response. In addition, the Mec1 paralog Tel1 is unable to replace Mec1 in such role.
Figure 5. Mec1 dependent phosphorylation of Cip1 in response to replication stress.

(A) Upper panel, a schematic diagram of Cip1 showing the three SQ/TQ motif sites present. Lower panel, Mec1 positive cells (sml1Δ, YFL101 strain) and mec1Δ null mutant cells (mec1Δ sml1Δ, strain YZZ2) were treated with or without 0.2 M HU and subjected to Cip1-13myc immunopurification followed by immunoblotting with anti-Myc (Cip1) or anti-pS/TQ antibodies. (B) GAL-Cip1 (strain YFL97), GAL-Cip1-3AQ (strain YZZ9) or wild-type control cells (strain YFL3) were synchronized in pre-Start G1 (α). Galactose was added while keeping the presence of α-factor for 30 min. Cells were then released from the α-factor while keeping the presence of galactose in the medium. Samples were taken at the indicated points (min) and the budding indexes (BI) were measured. (C) Cultures of GAL-Cip1 (strain YFL97), GAL-Cip1-3AQ (strain YZZ9) or wild-type control cells (strain YFL3) were 10-fold serial spotted on YPD or YPGal plates. (D) Whole cell extracts analyzed by immunoblot with anti-Myc (Cip1) antibodies show that the proteins are being overexpressed equally in the indicated strains. A coomassie blue-stained region of the same membrane used for immunoblot is shown as a loading control.
We previously showed that cells over-expressing Cip1 upon release from the α-factor arrest significantly delay cell budding, an event triggered by Cln1/2–Cdk1 (Ren et al. 2016). To explore whether Cip1 phosphorylation by Mec1 affects Cdk1 activity, we generated a strain that over-expressing non-phosphorylatable mutant of Cip1 (Gal-Cip1-3AQ) and checked budding index in the indicated strains released from the α-factor arrest. As shown in Figure 5B, budding defects in cells over-expressing Cip1 is robustly rescued when Cip1 S/TQ sites were mutated to non-phosphor AQ. The arrested cell cycle progression of cells overexpressing Cip1 is significantly rescued by nearly 10-fold in cells with phosphor-mutated Cip1, as quantified by 10-fold serial dilution assay shown in Figure 5C. The overexpression extends of both wild type and non-phosphorylatable Cip1-3AQ is identical as controlled in Figure 5D.
CIP1 mutants are sensitive to replication stress when the dosage of the G1 cyclin CLN2 is increased
Our results above placed Cip1 under the regulation of Mec1 and Rad53 kinases in response to replication stress. We therefore examined the result of Cip1 ablation in cells exposed to replication stress. As shown in Figure 6, proliferation of the cip1 null mutant is decreased in the presence of hydroxyurea compared to wild type cells. The effect is more evident at 0.2 M HU and when the two circles seeded with the highest cell densities are compared between the two strains.
Figure 6. CIP1 deletion results in sensitivity to replication stress when the dosage of the G1 cyclin CLN2 is increased.

WT cells and cip1Δ mutants transformed with pRS425 or pRS425-CLN2 vectors were 5-fold serial spotted on SD-Leu− with or without the HU at the indicated concentrations.
Because we previously identified Cip1 as a putative G1-CKI (Ren et al. 2016), we asked whether the role of Cip1 in the response to replication stress had to do with control of the G1 cyclin-Cdk1 activity present in S phase (Wittenberg et al. 1990). We took advantage of a 2-micron viral origin driven plasmid (pRS425-CLN2) to create strains carrying multiple copies of the CLN2 gene under its own promoter. As shown in Figure 6, wild type cells carrying the pRS425-CLN2 gene survive normally to replication stress. In deep contrast, upregulation of the G1 cyclin in cells lacking Cip1 results in a dramatic sensitivity to DNA replication stress.
Discussion
In this study, we showed that Cip1 is upregulated by the S phase checkpoint in response to genotoxic stress. CIP1 expression is induced in response in a Rad53 dependent manner. Rad53/Chk2 is the checkpoint effector kinase responsible for the transcriptional response to genotoxic stress (Zhou & Elledge 1993; Bastos de Oliveira et al. 2012; Jaehnig et al. 2013). As a result, Cip1 protein levels accumulate, despite the lower mobility pool of the protein remains highly unstable. In addition, Cip1 is phosphorylated at an SQ/TQ motif in a Mec1 dependent manner. Mec1/ATM/ATR is the central transducer kinase of the S phase and DNA damage checkpoint (Sanchez et al. 1996; McGowan & Russell 2004).
The fact that Cip1 is upregulated by the S phase checkpoint prompted us to explore the function of Cip1 in response to DNA replication stress. Whereas Cip1 null mutants are as viable as wild type cells under normal conditions, cip1 mutant cells are clearly sensitive to replication stress. Because we previously identified Cip1 as a putative G1-CKI (Ren et al. 2016), the simplest explanation is that checkpoint mediated accumulation of Cip1 is required to downregulate G1 cyclin-Cdk1 activity. A prediction derived from such hypothesis is that increased G1 cyclin-Cdk1 activity should enhance the hydroxyurea sensitivity phenotype in cip1 null cells. Indeed, cip1 mutants carrying multiple copies of the CLN2 gene are unable to cope with replication stress, whereas CIP1+ cells carrying the same multicopy plasmid respond normally to the stress.
Replication stress is sensed in S phase. Despite G1 cyclins start declining by the G1-S transition, their presence lingers well into S phase (Wittenberg et al. 1990), where G1 cyclin-Cdk1 activity has been proposed to be continuously required for proper polar growth (McCusker et al. 2007). We showed that Cln2 levels in cells under replication stress in S phase parallel those of an unperturbed S phase. Based on our results, residual G1 cyclin-Cdk1 activity during genotoxic stress may be deleterious.
Several possibilities may account for the observed loss of viability in cip1 mutants. One, deregulated G1 cyclin-Cdk1 may result in inadequate polarization of the cell. Persistent G1 cyclin-Cdk1 activity has been shown to result in excessive polar growth (Lew & Reed 1993; Barral et al. 1995). Because the S phase checkpoint keeps mitotic cyclin-Cdk1 inactive in response to genotoxic stress (Palou et al. 2015), the polar-to-isotropic growth switch is expectably delayed. Under those conditions Cip1 may be required to fine-tune the Cln1,2-Cdk1 activity to maintain the adequate polarity during the cell cycle arrest.
A second possibility is that unrestrained G1 cyclin-Cdk1 activity during genotoxic stress results in genomic instability. G1 cyclin overexpression has been reported to cause genomic instability in human cells (Zhou et al. 1996; Spruck et al. 1999) and in budding yeast (Tanaka & Diffley 2002). The observed sensitivity of cip1 mutants to replication stress might be the result of cells being unable to preserve genomic integrity under such conditions. The dramatic loss of viability observed in cip1 null cells may indeed underscore a significant degree of genomic instability.
Finally, the strong and lasting accumulation of Cip1 protein, past the presence of Cln2, is also compatible with Cip1 playing a role other than as a G1-CKI. Work is currently going on in our lab to distinguish between the different possible possibilities, and determine what is the role of Cip1 to prevent loss of viability in the presence of genotoxic stress.
In all, our results place Cip1 under the DNA damage response. Significantly, Cip1 is required for survival in the presence of replication stress. The loss of viability is aggravated when the dosage of Cln2 cyclin is increased, which strengthens the proposed role of Cip1 as a G1 cyclin-Cdk1 inhibitor (G1-CKI).
Experimental procedures
Strains, Constructs, Culture Media, Cell Synchronization, DNA Content Analysis and Inhibition of Protein Synthesis
The strains used in this study are listed in Table S1. All strains were derived from Saccharomyces cerevisiae W303-1a (Thomas & Rothstein 1989). Yeast cells used in this study were grown at 30°C in either YPD medium or Synthetic Dextrose (SD) medium supplemented with the required essential nutrients. For the hydroxyurea sensitivity tests, SD medium plates were chosen and incubated at 30°C, to minimize the effect of the Trp auxotrophy reported for the limiting tryptophan concentration in rich medium plates and at lower temperatures (Godin et al. 2016). Cell cycle synchronization with the pheromone α-factor, and DNA content analysis by propidium iodide Fluorescent Activated Cell Sorting (FACS) were done as previously described (Palou et al. 2015; Ren et al. 2016). Where indicated, protein synthesis was inhibited by adding 100 μg/ml cycloheximide (CHX) to the medium as previously described (Palou et al. 2010).
Site-directed DNA mutagenesis
The site-directed DNA mutagenesis for generation of CIP1-3AQ mutants was carried out using a modified method based on Single-Primer Reactions IN Parallel (SPRINP) method (Edelheit et al. 2009; Zeng et al. 2017). Cip1-3AQ refers to the mutation of the three S/TQ sites of Cip1 to Alanine (S18A, T42Q, S228A).
Western Blotting and Antibodies
Whole cell extracts for western blotting were prepared by glass beads beating in trichloroacetic acid (TCA), then resolved by SDS-PAGE as previously described (Palou et al. 2015; Ren et al. 2016). The primary antibodies used in this study were anti-Rad53 (Santa cruz sc-6749), anti-Myc (9E10, monoclonal mouse hybridoma supernatant), and anti-pS/TQ (Cell Signaling Technologies 2851).
RNA Extraction and qRT-PCR
Around 109 cells were harvested for each sample and washed once with pre-chilled diethy pyrocarbonate (DEPC)-treated water. Cell pellets were snap-frozen in liquid nitrogen and stored at −80°C until RNA extraction. Total RNA was obtained using Trizol reagent (Invitrogen) as recommended by the manufacturer. RNA was then reverse transcribed using PrimeScriptTM RT kit (Takara, RR014A). The cDNA library was used as template in real-time PCR using Takara SYBR Premix Ex-Taq (Tli RNaseH Plus) kit (Takara, RR420A). The primers for CIP1 and the control ACT1 used in this study are listed in Table S2.
Immunoprecipitation
For immunoprecipitation, around 109 cells treated with 0.2 M HU for 2 hours were collected and extracted by glass beads beating in lysis buffer (50 mM Tris-HCl pH 7.8, 175 mM NaCl, 1 mM EDTA, 0.1% (v/v) Nonidet-P40) supplemented with protease inhibitors (1 mM AEBSF, 0.15 mM aprotinin, 1 mM leupeptin, 1 mM pepstatin) and phosphatase inhibitors (0.5 mM sodium pyrophosphate, 2 mM NaF, 2 mM β-glycerophosphate). 20 μl of anti-myc HA-7 agarose matrix (Sigma A2095, mouse monoclonal purified IgG) were added to extracts to immunopurify Cip1-13myc. The samples were gently rotated for 1 h at 4°C. The beads were then washed four times with cold lysis buffer. Finally, the proteins were released by boiling the beads in Laemmli sample buffer. Enriched proteins were resolved by SDS-PAGE and checked by western blotting. For mass spectroscopy analysis of Cdk1 interacting proteins, proteins from the immunoprecipitate were released from the beads by mixing with 50 μl of room temperature 8M urea in 100 mM Tris pH 8.0 for 5 min.
Serial dilution assay
Yeast cells were grown overnight in YPD at 30°C to mid-log phase. Serial 10-fold or 5-fold dilutions from the cultures were spotted on indicated plates and incubated at 30°C for 3 days. For cells containing 2-micron based plasmids pRS425, leucine minus SD medium was used instead of YPD.
Supplementary Material
Acknowledgments
This work was supported by a Starting Grant from Hebei Agricultural University, China (ZD201622) to FZ; by National College Students’ Science and Technology Innovation Project to ZZ from Hebei Education Department (201710086014) and Hebei Agricultural University (2017014); by the Ministry of Science and Competitiveness of Spain (MINECO), and by the European Regional Development Fund (FEDER), Grants BFU2011-28007 (MINECO) and BFU2015-68493-P (MINECO/FEDER) to DG; and grant NIH GM089778 from the National Institutes of Health to JW.
References
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Barral Y, Jentsch S, Mann C. G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes Dev. 1995;9:399–409. doi: 10.1101/gad.9.4.399. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Bakkenist CJ, Rajpert-De Meyts E, Skakkebaek NE, Sehested M, Lukas J, Kastan MB, Bartek J. ATM activation in normal human tissues and testicular cancer. Cell Cycle. 2005;4:838–845. doi: 10.4161/cc.4.6.1742. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Ørntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Harris MR, Brazauskas P, de Bruin RAM, Smolka MB. Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 2012;31:1798–1810. doi: 10.1038/emboj.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci USA. 1997;94:14361–14366. doi: 10.1073/pnas.94.26.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RAM, Kalashnikova TI, Aslanian A, Wohlschlegel J, Chahwan C, Yates JR, Russell P, Wittenberg C. DNA replication checkpoint promotes G1-S transcription by inactivating the MBF repressor Nrm1. Proc Natl Acad Sci USA. 2008;105:11230–11235. doi: 10.1073/pnas.0801106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–2970. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelheit O, Hanukoglu A, Hanukoglu I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 2009;9:61. doi: 10.1186/1472-6750-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- Godin SK, Lee AG, Baird JM, Herken BW, Bernstein KA. Tryptophan biosynthesis is important for resistance to replicative stress in Saccharomyces cerevisiae. Yeast. 2016;33:183–189. doi: 10.1002/yea.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- Jaehnig EJJ, Kuo D, Hombauer H, Ideker TGG, Kolodner RDD. Checkpoint Kinases Regulate a Global Network of Transcription Factors in Response to DNA Damage. Cell Rep. 2013;4:174–188. doi: 10.1016/j.celrep.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker D, Denison C, Anderson S, Egelhofer TA, Yates JR, Gygi SP, Kellogg DR. Cdk1 coordinates cell-surface growth with the cell cycle. Nat Cell Biol. 2007;9:506–515. doi: 10.1038/ncb1568. [DOI] [PubMed] [Google Scholar]
- McGowan CH, Russell P. The DNA damage response: sensing and signaling. Curr Opin Cell Biol. 2004;16:629–633. doi: 10.1016/j.ceb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Palou G, Palou R, Guerra-Moreno A, Duch A, Travesa A, Quintana DG. Cyclin regulation by the s phase checkpoint. J Biol Chem. 2010;285:26431–26440. doi: 10.1074/jbc.M110.138669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palou G, Palou R, Zeng F, Vashisht AA, Wohlschlegel JA, Quintana DG. Three Different Pathways Prevent Chromosome Segregation in the Presence of DNA Damage or Replication Stress in Budding Yeast. PLoS Genet. 2015;11:e1005468. doi: 10.1371/journal.pgen.1005468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren P, Malik A, Zeng F. Identification of YPL014W (Cip1) as a novel negative regulator of cyclin-dependent kinase in Saccharomyces cerevisiae. Genes Cells. 2016;21:543–552. doi: 10.1111/gtc.12361. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Diffley JFX. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002;16:2639–2649. doi: 10.1101/gad.1011002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- Travesa A, Kuo D, de Bruin RAM, Kalashnikova TI, Guaderrama M, Thai K, Aslanian A, Smolka MB, Yates JR, Ideker T, Wittenberg C. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012;31:1811–1822. doi: 10.1038/emboj.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg C, Sugimoto K, Reed SI. G1-specific cyclins of S. cerevisiae: cell cycle periodicity, regulation by mating pheromone, and association with the p34CDC28 protein kinase. Cell. 1990;62:225–237. doi: 10.1016/0092-8674(90)90361-h. [DOI] [PubMed] [Google Scholar]
- Yahya G, Parisi E, Flores A, Gallego C, Aldea M. A whi7-anchored loop controls the g1 cdk-cyclin complex at start. Mol Cell. 2014;53:115–126. doi: 10.1016/j.molcel.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Zeng F, Hao Z, Li P, Meng Y, Dong J, Lin Y. A restriction-free method for gene reconstitution using two single-primer PCRs in parallel to generate compatible cohesive ends. BMC Biotechnol. 2017;17:32. doi: 10.1186/s12896-017-0346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Elia AEH, Naylor ML, Dephoure N, Ballif BA, Goel G, Xu Q, Ng A, Chou DM, Xavier RJ, Gygi SP, Elledge SJ. Profiling DNA damage-induced phosphorylation in budding yeast reveals diverse signaling networks. Proc Natl Acad Sci USA. 2016;113:3667–3675. doi: 10.1073/pnas.1602827113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Jiang W, Weghorst CM, Weinstein IB. Overexpression of cyclin D1 enhances gene amplification. Cancer Res. 1996;56:36–39. [PubMed] [Google Scholar]
- Zhou Z, Elledge SJ. DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell. 1993;75:1119–1127. doi: 10.1016/0092-8674(93)90321-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.