

Abstract
Recent studies suggested that activation of the PRR upregulates profibrotic markers through reactive oxygen species (ROS) formation; however, the exact mechanisms have not been investigated in CD cells. We hypothesized that activation of the PRR increases the expression of profibrotic markers through MAPK-dependent ROS formation in CD cells. Mouse renal CD cell line (M-1) was treated with recombinant prorenin plus ROS or MAPK inhibitors and PRR-shRNA to evaluate their effect on the expression of profibrotic markers. PRR immunostaining revealed plasma membrane and intracellular localization. Recombinant prorenin increases ROS formation (6.0 ± 0.5 vs. 3.9 ± 0.1 nM DCF/μg total protein, P<0.05) and expression of profibrotic markers CTGF (149 ± 12%, P<0.05), α-SMA (160 ± 20%, P<0.05), and PAI-I (153 ± 13%, P<0.05) at 10-8 M. Recombinant prorenin induced phospho ERK 1/2 (p44 and p42) at 10-8 and 10-6 M after 20 min. Prorenin-dependent ROS formation and augmentation of profibrotic factors were blunted by ROS scavengers (trolox, p-coumaric acid, ascorbic acid), the MEK inhibitor PD98059 and PRR transfections with PRR-shRNA. No effects were observed in the presence of antioxidants alone. Prorenin-induced upregulation of collagen I and fibronectin was blunted by ROS scavenging or MEK inhibition independently. PRR-shRNA partially prevented this induction. After 24 h prorenin treatment M-1 cells undergo to epithelial mesenchymal transition phenotype, however MEK inhibitor PD98059 and PRR knockdown prevented this effect. These results suggest that PRR might have a significant role in tubular damage during conditions of high prorenin-renin secretion in the CD.
Keywords: Prorenin receptor, ROS, intrarenal renin-angiotensin system, collecting duct renin, Fibrosis
Introduction
The (pro)renin receptor (PRR) is a 350-amino acid protein with a single transmembrane domain originally described as ATPase H(+)-transporting lysosomal accessory protein 2 and named ATP6AP2 1-4. PRR is expressed in glomerular mesangial cells, podocytes, and intercalated cells in the collecting duct (CD) 2,5,6. PRR binds renin or prorenin, increasing renin catalytic activity and fully activating prorenin 3,4. PRR activation also triggers phosphorylation of mitogen activated protein kinases/extracellular regulated kinases 1/2 (MAPK/ERK1/2) 7-9. Importantly, the actions of PRR have been associated with renal tissue damage 6; indeed, rats overexpressing PRR develop glumeruloesclerosis and hyper-filtration 10,11. PRR-mediated activation of the MAPK/ERK1/2 pathway upregulates cyclooxygenase-2 (COX-2) expression in cardiac tissues 12 and renal cortex 11. More recently, we reported that PRR activation by recombinant prorenin stimulates COX-2 expression independently of angiotensin (ANG) II signaling in renal CD cells 9.
We have demonstrated that chronic ANG II infusions increase CD renin and prorenin synthesis and secretion despite the suppression of renin expression in juxtaglomerular cells 13-17. In addition, we have shown that cultured CD cells undergo to epithelial-mesenchymal transition and have induced expression of fibronectin and collagen I after ANG II treatment 18.
It has been shown that despite the low plasma renin activity in patients with diabetic nephropathy, high plasma prorenin levels are detected 19. Furthermore, this is associated with the occurrence of microvascular complications, such as microalbuminuria and retinopathy 20. Cells exposed to prorenin show upregulation of fibronectin 21. Prorenin also induces cell proliferation in human umbilical artery smooth muscle cells through reactive oxygen species (ROS) generation and ERK1/2 activation 22. In kidney proximal tubular cells the activation of PRR by prorenin increases TGF-β1 and α-smooth muscle actin (α-SMA) 23. Clavreul et al., demonstrated that the induced expression of fibrotic genes in non-differentiated human kidney embryonic (HEK) cells is mediated by a Nox4-dependent mechanism 21 indicating a possible role for ROS as a second messenger in cell damage. Indeed, ROS can activate ERK pathway 24, however the possibility of ROS formation via MAPK/ERK1/2 has not been explored.
High circulating levels of prorenin were described in diabetic patients more than 20 years ago 19. Induced expression of prorenin in the collecting duct has been observed in diabetic animal models 25, which is expected to be higher than plasma. This suggest that PRR activation by tubular prorenin might promote the activation of signaling pathways such as MAPK/ERK 1/2 and ROS, which may contribute to tubular fibrosis. The mechanisms by which PRR could regulate the expression of profibrotic factors and its effects on renal CD cells are not fully understood.
In the present study, we hypothesized that PRR activation by recombinant prorenin enhance the expression of profibrotic factors through MAPK-dependent ROS formation in CD cells. To test this hypothesis, M-1 cells were treated with recombinant prorenin with or without antioxidants or MAPK/ERK1/2 pathway inhibition to evaluate the expression of alpha smooth muscle actin (α-SMA), connecting tissue growth factor (CTGF), plasminogen activator inhibitor-1 (PAI-I), fibronectin, collagen I and the effect on fibroblast-like phenotype.
Results
Band identity of prorenin, renin and immunohistochemical localization of PRR in M-1 cells
Figure 1A shows the identity of Western blot bands detected using anti-ATP6AP2 (Cat. HPA003156, Sigma) in M-1 cells and medullary extracts from mouse kidneys. As shown in the left side of the panel, M-1 shows three main isoforms suggesting the presence of the full length, soluble and truncate forms of the PRR. Figure 1B shows prorenin and renin band identity in M-1 cells using prorenin and renin standards (see methods section for details) and the specific renin antibody (B12, Santa Cruz, CA), demonstrating that prorenin is the main form detected in these cells. To further confirm the presence of PRR in M-1 CD cells we performed immunostaining analysis in sub-confluent cells. Due to the fact that M-1 cells are a mixed population of intercalated and principal cells, we did a further analysis using Anion Exchanger-1 antibody as a marker of intercalated cells and co-localization with PRR. As shown in Figure 1C, some cells express both markers evidencing that they are intercalated cells. Figure 1D and 1E show the presence of PRR (green) in the membrane at different magnifications. Figure 1F shows another co-localization of PRR and NH(3)-specific transporter (Rhcg) a specific marker of A-type intercalated cell. As suggested in previous reports 26 PRR is detected in plasma membrane but also in a perinuclear location and probably associated to the endoplasmic reticulum 27.
Figure 1. PRR is expressed in M-1 collecting duct cells.
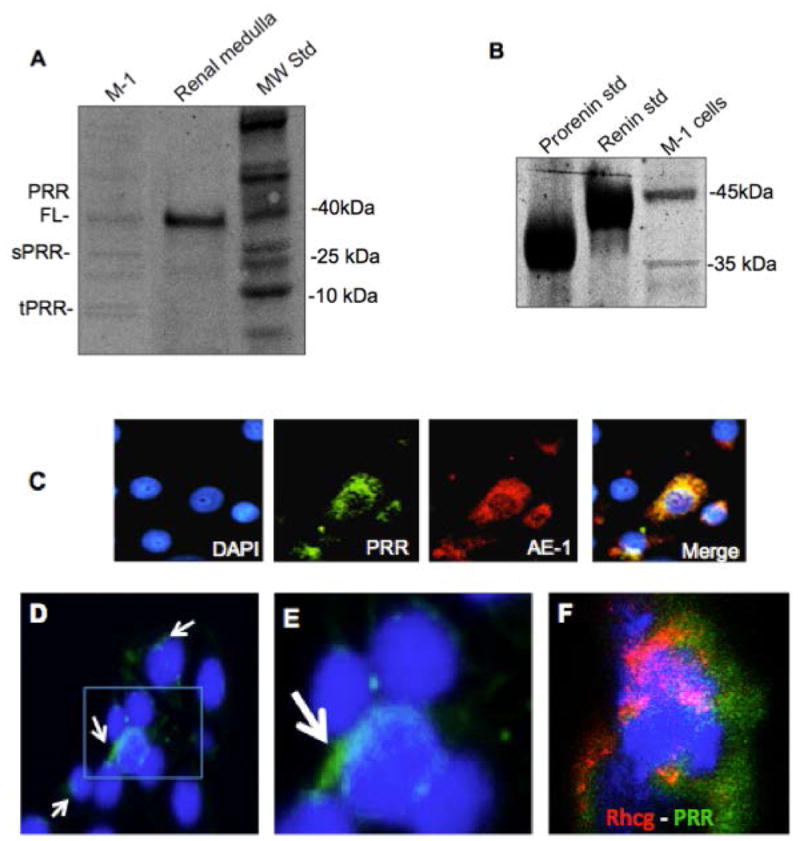
(A). Western blot analysis showing the expression of PRR in M-1 cells and medullary tissues as positive control (see methods section). PRR was detected at the expected size (FL: 38 kDa). We observed a 25 kDa, which probably correspond to the soluble form of the PRR and the truncated form of 8.9 kDa. (B). Protein band identity of renin-detected bands in M-1 cells using recombinant prorenin and renin standards. (C). Immunofluoresence studies of co-localization using anti PRR antibody (green) and anion exchanger 1 (AE-1) as marker of intercalated cells. (D). PRR positive cells (arrows) showing PRR in plasma membrane (magnification image showed in E). (F). Co-localization of PRR (green) and NH(3)-specific transporter (Rhcg) a marker of A-type intercalated cell. Blue color indicated nuclei labeling with DAPI (4′,6-diamidino-2-phenylindole).
Antioxidants prevented the prorenin-induced ROS formation and upregulation of profibrotic proteins in M-1 cells
Due to M-1 cells express renin and mostly prorenin (Figure 1B) we performed treatments with different doses of recombinant prorenin (Figure 2A) and renin (Figure 2B) evaluating ROS generation. As shown in Figure 2A, the concentration of DCF (the product of H2DCFDA oxidation) was augmented at prorenin dose of 10-8 M and 10-6 M (6.0 ± 0.5 and 6.3 ± 0.6 vs. 3.9 ± 0.1 nM DCF/μg total protein, P<0.05). Renin caused a modest increase in ROS formation (4.2 ± 0.2 and 4.4 ± 0.3 vs. 3.8 ± 0.2 nM DCF/μg total protein, P=non significant, n=12). Because prorenin is mostly secreted by CD cells 25 and that the affinity of the receptor for renin and prorenin are in the nanomolar range 28 we performed the following experiments in doses of 10-8 M prorenin. Next, we tested the ROS suppression efficiency of the scavenger p-coumaric acid (PCA), which efficiently prevents lipid peroxidation and cytoplasmic ROS formation 29,30. Previous treatment with PCA abolishes the prorenin-induced ROS formation in M-1 cells. Positive controls performed with 200 mM H2O2 greatly increases ROS formation (Figure 2C). We next examined whether prorenin treatment (4 h) induces the expression of profibrotic proteins through ROS formation. As shown in Figure 3A and Figure 3B, prorenin induced the expression of profibrotic markers CTGF (149 ± 12%, P<0.05), α-SMA (160 ± 20%, P<0.05), and PAI-I (153 ± 13%, P<0.05) while PCA prevented this effect. In a similar experiment we evaluated the effect of treatment with two additional antioxidants, trolox C (TLX) a lipophilic antioxidant 31 and ascorbic acid (AA) that acts as a cytoplasmic antioxidant 32. TLX and AA partially but not significantly suppressed the effect of prorenin when measuring protein (Figure 4A and Figure 4B) and mRNA levels (Figure 4C). The results indicate that PRR mediates the upregulation of α-SMA, CTGF and PAI-I via ROS formation at both the cytoplasmic and plasma membrane levels and suggested that p-coumaric acid is more effective at suppressing ROS formation in both compartments. No effects were observed by using antioxidant treatments alone (Figure 4D).
Figure 2. Prorenin induces ROS formation at nanomolar range and ROS scavenger p-coumaric acid prevented this effect.
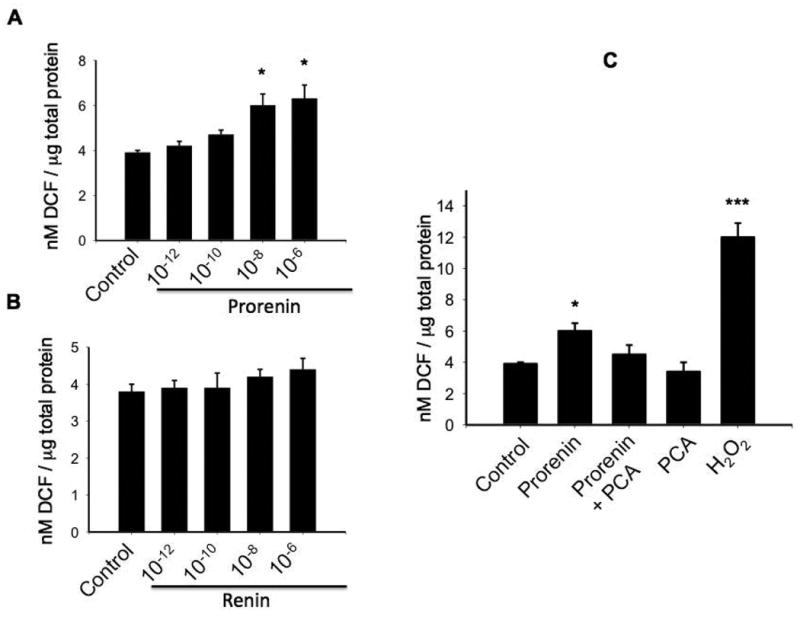
M-1 cells were incubated with prorenin and renin (10-12, 10-10 , 10-8 , 10-6 M). ROS production was measured after 10 min using DCFHDA probe and levels of DCF formed (nM/μg total protein). ROS formation was augmented by prorenin at 10-8 M. Renin increases ROS formation at non-physiological dose (10-5 M). P-coumaric acid (PCA, 10-7 M) added 10 min before prorenin treatment prevented the induction of ROS. As a positive control 200 mM H2O2 was used. *P<0.05 versus control, ***P<0.001 versus control, n=12.
Figure 3. Prorenin-induced upregulation of profibrotic proteins is prevented by p-coumaric acid.
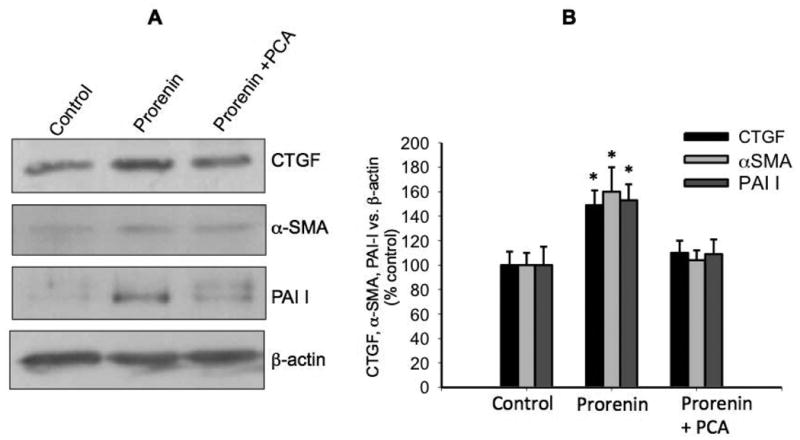
P-coumaric acid (PCA, 10-7 M) was added 10 min before prorenin treatment. Expression of profibrotic proteins CTGF, α-SMA, PAI-I was measured after 6 h. (A). Representative immunoblot of 5 experiments. (B) Quantitative blot analysis of each treatment showing band intensity vs. β-actin as percentage of control. *P<0.05 versus control, n=5.
Figure 4. Prorenin-induced upregulation of profibrotic markers is only partially prevented by lipophilic antioxidant Trolox and cytoplasmic antioxidant ascorbic acid.
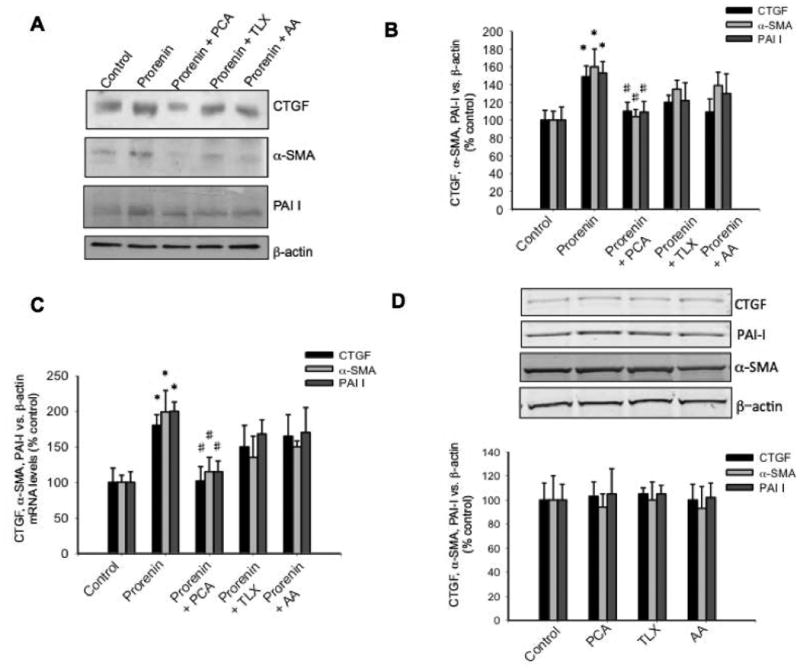
Similarly to previous experiments, the cells were incubated with prorenin at 10-8 M and the expression of profibrotic proteins CTGF, α-SMA, PAI-I measured after 6 h. P-coumaric acid (PCA, 10-7 M), trolox (TLX, 10-7 M) or ascorbic acid (AA, 10-5 M) were added 10 min before prorenin treatment. (A). Representative immunoblot of 5 experiments. (B) Quantitative blot analysis of each treatment showing band intensity vs. β-actin as percentage of control. (C). Real-time quantitative PCR of CTGF, α-SMA, PAI-I measured after 6 h post treatments. *P<0.05 versus control, #P<0.05 versus prorenin-treated group, n=5. (D). Controls of the effect of antioxidant treatment alone on protein expression after 6 h treatment. No effects were observed on the expression of CTGF, α-SMA or PAI-I.
Prorenin treatment increases ERK 1/2 phosphorylation in M-1 cells
To confirm that prorenin treatment increases phosphorylation of ERK 1/2 in M-1 collecting duct cells, we performed a western blot analysis in cell lysates from M-1 cells treated for 20 min with recombinant prorenin at 10-10, 10-8 and 10-6 M. As shown in Figure 5, prorenin significantly increases P-ERK1/2 vs. total ERK 1/2 ratio (in percentage of control) at 10-8 M (185 ± 15 vs. 100 ± 10, P<0.05) and 10-6 M (195 ± 23 vs. 100 ± 10, P<0.05).
Figure 5. Prorenin induces ERK 1/2 phosphorylation in M-1 cells.
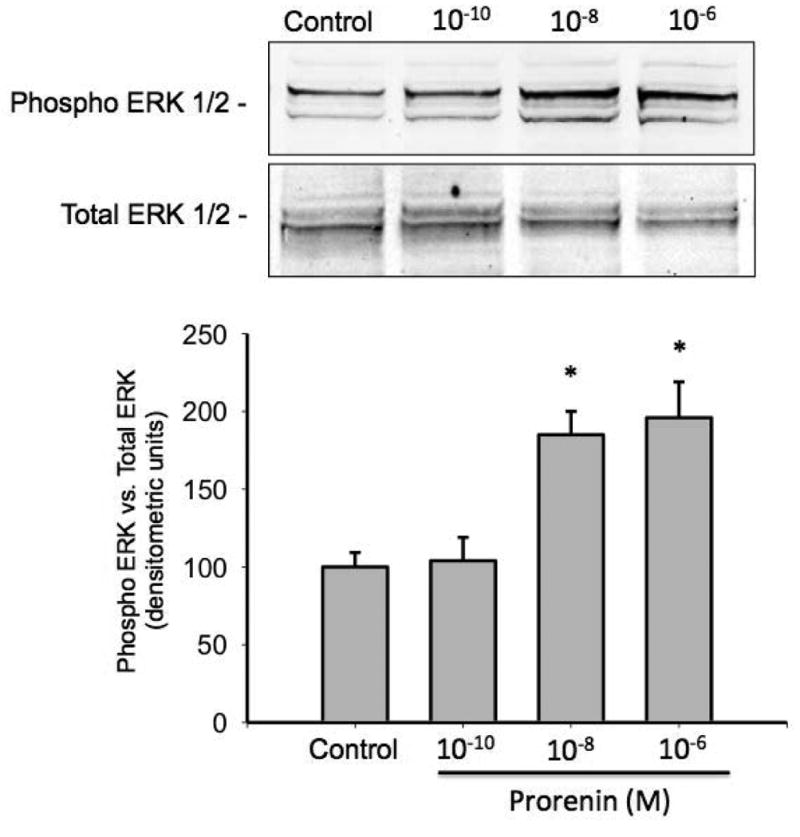
Cultured M-1 cells were incubated with prorenin at 10-10, 10-8 and 10-6 M and phosphor-ERK 1/2 and total ERK 1/2 measured after 20 min. Phospho ERK 1/2 (p44 and p42 intensity) was significantly augmented as compared to controls at 10-8 and 10-6 M . *P<0.05 versus control, n=5.
MEK inhibition suppresses ROS formation and upregulation of profibrotic markers in M-1 cells
We next evaluated MEK inhibition on prorenin-induced ROS formation and upregulation of profibrotic factors. Figure 6A shows the effect of prorenin and prorenin plus MEK inhibitor PD98059 treatment (4 h) on ROS formation. MEK inhibition blunted the augmentation of ROS mediated by prorenin (control: 3.91 ± 0.12; prorenin: 6.2 ± 0.5, P<0.05; prorenin + PD98059: 4.6 ± 0.6, nM DCF/μg total protein) indicating that ROS formation depends on MAPK/ERK activation. We next evaluated the behavior of ROS formation every 20 s after rapid addition of prorenin or prorenin + PD98059 to the M-1 cells. As shown in Figure 6B MEK inhibition blunted the augmentation of ROS after prorenin treatment, this effect was observed even after 600 sec. Finally we evaluated the effect of MEK inhibition on prorenin-induced profibrotic factors. As observed above, prorenin induced the expression of CTGF, α -SMA and PAI-I (149 ± 12%; 160 ± 20%; 153 ± 13, P<0.05) while MEK inhibition blunted this response (Figure 7A and Figure 7B). No significant effects were observed with PD98059 alone (data not shown).
Figure 6. MEK inhibition prevented the prorenin-induced ROS formation.
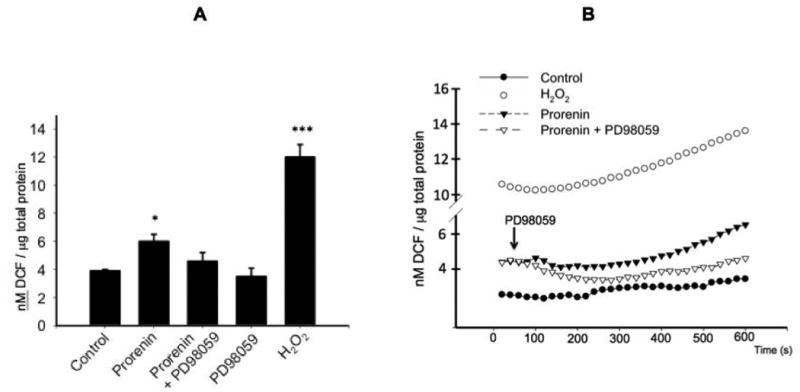
M-1 cells were incubated with prorenin at 10-8 M and ROS measured after 10 min (A) or every 15 s during 600 s (B). ROS formation was measured using DCFHDA probe and levels of DCF formed (nM/μg total protein). MEK inhibitor PD98059 (30 μM, white triangles) was added 20 s after prorenin treatment (black triangles). Controls are indicated in black circles. As a positive control 200 mM H2O2 was used (white circles). ROS formation was normalized by total protein. *P<0.05 versus control, ***P<0.001 versus control, n=12.
Figure 7. MEK inhibition prevented prorenin-induced upregulation of CTGF, α-SMA, PAI-I.
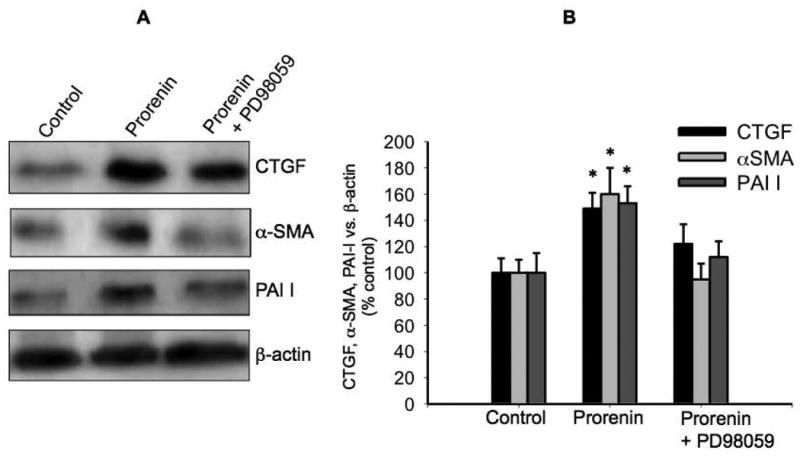
To evaluate if MEK inhibition prevents the upregulation of profibrotic factors, the M- cells were incubated with prorenin at 10-8 M or prorenin plus the MEK inhibitor PD98059. The expression of profibrotic proteins CTGF, α-SMA, PAI-I was evaluated after 6 h. PD98059 (30 μM) was added 10 min before prorenin treatment. (A). Representative immunoblot. (B) Quantitative blot analysis of each treatment showing band intensity vs. β-actin as percentage of control. *P<0.05 versus control, n=5.
PRR mediates the upregulation of α-SMA, CTGF and PAI-I
To knockdown the expression of PRR, M-1 cells were transfected with GFP-shRNA-PRR (Figure 8A). In our hands, we obtained near 43% reduction in full length plus soluble forms of PRR protein (Figure 8B) as compared to non-transfected cells (43 ± 5% vs. 100 ± 9%, P<0.05). No effects on PRR expression were observed by using scrambled shRNA. No effects were observed in cell viability after transfections (Figure 8C). Using this strategy we were able to partially prevents prorenin-induced ROS generation in M-1 cells transfected with shRNA-PRR (scrambled transfected control: 3.9 ± 0.2; prorenin: 6.3 ± 0.4; PRR-shRNA: 5.0 ± 0.5 nM DCF/μg total protein, Figure 8D) and the induction of profibrotic markers CTGF (125 ± 12%, P=0.039 vs. control: 100 ± 6%), α-SMA (113 ± 8% vs. control: 100 ± 9% P=non significant vs. control), and PAI-I (123 ± 11%, control: 100 ± 9% P=non significant vs. control, Figure 8E).
Figure 8. PRR mediates the upregulation of α-SMA, CTGF and PAI-I.
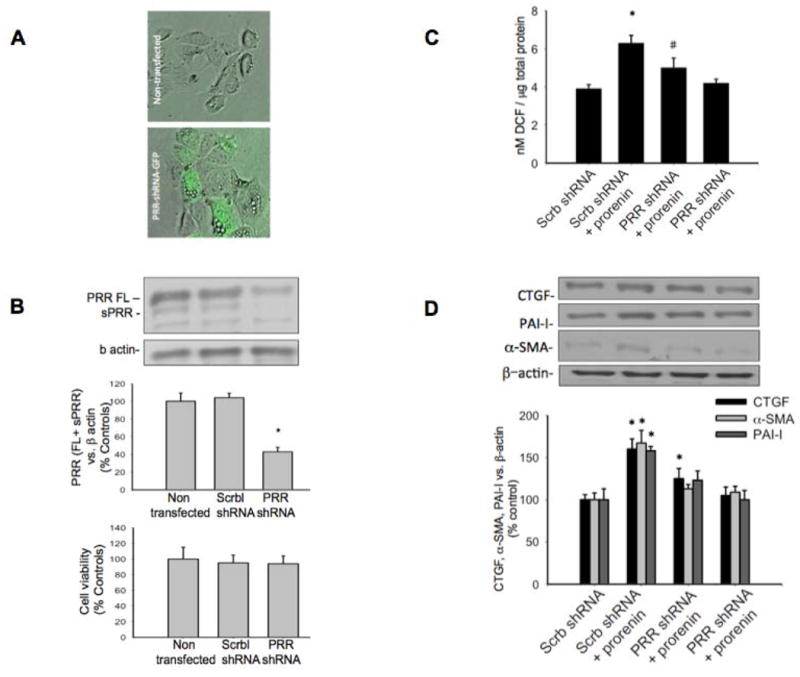
Knockdown of PRR expression. M-1 cells were transfected with GFP-shRNA-PRR (A). In our hands, we obtained near 43% reduction in full length plus soluble forms of PRR protein (B) as compared to non-transfected cells (43 ± 5% vs. 100 ± 9%, P<0.05). No effects on PRR expression were observed by using scrambled shRNA. No effects were observed in cell viability after transfections (C). PRR knockdown partially prevents prorenin-induced ROS generation (D) and the induction of profibrotic markers CTGF, α-SMA, and PAI-I (E). *P<0.05 versus control, #P<0.05 PRR-shRNA plus prorenin versus Scrb-shRNA plus prorenin, n=5 for protein analysis, n=12 for ROS measurements.
Prorenin-mediated upregulation of profibrotic factors Collagen I, fibronectin and induced fibroblast-like phenotype is mediated by PRR activation through ERK 1/2-dependent ROS formation
Because augmented expression of collagen I and fibronectin is associated with epithelial mesenchymal transition 33,34, we evaluated the expression of both markers after 16 h of prorenin treatment with or without ROS inhibition, PD98059 or PRR knockdown. Prorenin induced collagen I (170 ± 20%, P<0.05 vs. control) and fibronectin (198 ± 40%, P<0.05 vs. control), protein expression were effectively prevented by PCA (98 ± 23% and 100 ± 20%, P=non significant vs. control) and MEK inhibitor PD98059 (102 ± 11% and 109 ± 20%, P=non significant vs. control). Transfections with shRNA-PRR were able to partially reduce prorenin-induced collagen I and fibronectin protein expression (143 ± 26% and 134 ± 23%, respectively, P<0.05 vs. Prorenin group). Figure 9A shows a summary of these results in a representative Western blot (n=6). We next evaluated whether prorenin treatment induces epithelial-mesenchymal transition. M-1 cells were treated with prorenin (10-8 M). After 24 h, cells were fixed in methanol and staining with β-actin to visualize the cell-shape using immunofluorescence. Figure 9B shows representative microscopic fields that suggest that prorenin treatment alters the phenotype of epithelial cells with some cells having extensive projections. Despite the significant increase in the percentage of fibroblast-like phenotype (Figure 9C) mediated by prorenin treatment (58 ± 10% vs. 10 ± 23%, p<0.05), MEK inhibitor PD98059, PCA prevented this effect (15 ± 3% and 13 ± 5% respectively P=non significant versus control group), while PRR-shRNA partially reduced the effect (34 ± 8% P<0.05 versus control group).
Figure 9. Prorenin induces the upregulation of collagen I and fibronectin and myofibroblast-like phenotype through PRR activation and MAK/ERK dependent ROS formation.
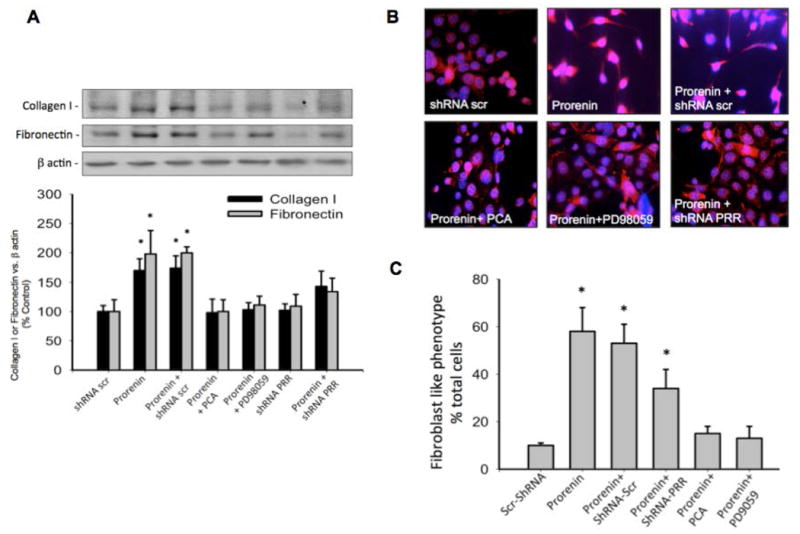
(A). Collagen I and fibronectin were evaluated after 16 h of prorenin treatment in M-1 cells previously transfected with PRR-shRNA or pretreated with p-coumaric acid or MEK inhibitor PD98059 (10 min). Prorenin-induced upregulation of collagen I and fibronectin was blunted by ROS scavenging or MEK inhibition independently. PRR-shRNA partially prevented this induction (B). M-1 cells were treated with prorenin and morphology analyzed after 24 h using β-actin immunostaining. As shown in representative microscopic fields, M-1 cells undergo to epithelial mesenchymal transition phenotype after prorenin treatment as judged by morphology, however MEK inhibitor PD98059 and PRR knockdown prevented this effect. (C) Cell counting of fibroblast like phenotype (cell extensions) versus total cell number in each field (n=10). *P<0.05 versus scrambled shRNA-transfected cells.
Discussion
The present findings demonstrate that PRR activation by recombinant prorenin increases the mRNA and protein levels of profibrotic factors CTGF, α-SMA, PAI-I and induced an early expression of Fibronectin and collagen I in mouse renal CD cell line M-1. Additionally we have shown that long treatment with prorenin induced a fibroblast-like phenotype suggesting epithelial-mesenchymal transition events in M-1 CD cells. We showed that this effect depends on ROS formation through the activation of MAPK pathway since ROS scavengers; MEK inhibition and PRR knockdown prevented the upregulation of profibrotic proteins and fibroblast-like phenotype. These observations suggested that PRR is implicated in the development of fibrosis in the distal nephron through a direct activation of MAPK pathway.
Elucidating pathways involved in the activation of PRR has brought new perspectives about its possible role in renal tissue damage. PRR is abundantly expressed in mesangial cells, podocytes, and intercalated type A cells of collecting ducts 2,9,35,36. PRR was initially described as an associated protein with the V-ATPase (vacuolar H+-ATPase), however, its ability to bind renin and prorenin with an affinity in the nanomolar range and its capacity to activate MAPK/ERK1/2 are particularly important facts that might help to understand its role in the development of renal damage and diabetic nephropathy 37,38. We have detected the expression of PRR in M-1 at the expected size (38 kDa) we also observed a 25 kDa, which probably correspond to the soluble form of the PRR (sPRR) and the truncated form of 8.9 kDa. As shown in Figure 1, M-1 cells express mostly prorenin at basal conditions. This agrees with our previous reports showing that prorenin is the most prominent form in intracellular and extracellular media 39. It is also possible that higher basal levels of secreted prorenin by these cells may add further effects to the added recombinant prorenin in each treatment. High prorenin levels can be observed in pathological conditions. Despite the low levels of plasma renin in patients with diabetic nephropathy, the high levels of plasma prorenin found in these patients are associated with the occurrence of microvascular complications, microalbuminuria and retinopathy 19,20. Furthermore, normal concentrations of plasma prorenin are usually 10 times higher than renin and became even higher in diabetic patients 40. Thus, it is possible that augmented plasma prorenin may interact with CD PRR as it has already been confirmed in animal models of diabetic disease 25. Furthermore, PRR expression is augmented in podocytes and tubular segments when diabetic nephropathy is present. Additionally, we have shown that not only PRR but also prorenin and renin expression is augmented in the CD of hypertensive animals 14,15,17,41.
Direct association of PRR and kidney damage has been reported. In 2006, Kaneshihiro et al., demonstrated that the overexpression of PRR leads to augmentation of cycloocygenase-2 (COX-2) 11 suggesting a possible role in inflammation. In diabetic transgenic mice expressing COX-2 selectively in podocytes developed progressive albuminuria accompanied with high PRR expression, while COX-2 inhibitor prevented the upregulation of PRR suggesting that he PRR may act as a mediator for increased susceptibility to injury 42. More importantly it has been shown that blockade of prorenin binding to the PRR prevent diabetic nephropathy 43.
Nguyen and associates originally reported the presence of PRR predominantly in glomerular mesangial cells and in vascular smooth muscle cells of renal arteries 44. Later, Advani et al., showed the predominant expression of the PRR at the apex of acid-secreting cells in the CD 2. The same group demonstrated that in collecting Madin-Darby canine kidney cells, MAPK/ERK1/2 pathway is induced by either renin or prorenin 2 (2). In a previous study in non-differentiated human embryonic (HEK) cells that possessed a functional PRR. Clavreul et al., reported that prorenin promotes fibrotic gene expression through a Nox4-dependent mechanism, suggesting a direct role of ROS in fibrosis 21. Despite this evidence, there are no specific reports about the specific role of PRR-dependent activation of MAPK/ERK 1/2 on ROS formation and fibrosis in differentiated CD cells. In 2008, Yogi et al., showed that renal redox-sensitive signaling was attenuated by Nox1 knockout in ANG II-dependent chronic hypertension 45. On the other hand, Clavreul et al., suggested that fibrosis and ERK signaling in kidneys of hypertensive animals are mediated through a Nox4-dependent increase of ROS 21. Here we have shown for the first time that ROS formation depends on MAPK/ERK 1/2 activation, since MEK inhibitor PD98059 was able to prevent ROS formation and profibrotic gene induction in CD cells. Our group has reported previous evidence showing the activation of MAPK/ERK 1/2 pathway by PRR in CD cells 9. We also demonstrated that PRR activation by nanomolar concentrations of recombinant prorenin was able to induce COX-2 expression in CD intercalated cells in primary cultures 9 as observed in glomeruli 10,11. Additionally we demonstrated that PRR is expressed in M-1 CD cells specifically in AE-1 expressing cells, confirming the expression of PRR only in intercalated cells (Figure 1D) and particularly in an intracellular perinuclear location, with only a minor portion on the cell surface. 27
Cumulated evidence indicates that in hypertensive models of ANG II-dependent hypertension there is an increased expression of prorenin and renin in the CD 13-17. We have previously shown that renal PRR is also increased, and that there is a time dependent pattern of expression of PRR and COX-2 during chronic ANG II infusion 41. Then, it is possible that increased prorenin synthesis and secretion by principal cell may interact with PRR in the neighbor-intercalated cell in the CD to promote activation of MAP/ERK 1/2 cascade independently of ANG II as we previously show 9. Although divergence of angiotensinogen mRNA transcription and angiotensinogen protein synthesis and metabolism has been reported in different segments of the normal rat proximal tubules 46, it is also possible that gene expression of angiotensinogen might be present in other nephron segments, suggesting that an ANG II-dependent pathway cannot be avoided.
Although overexpression of PRR has not resulted in a significant cardiovascular injury in rats and mice 45,47; high synthesis of intratubular prorenin and renin might reach enough amounts to activate plasma membrane PRR in the CD. Then, prorenin-induced activation of profibrotic synthesis at supra physiological dose might be better prevented by blocking prorenin-PRR binding as suggested before 48 or through specific deletion of PRR, however, all conditional knockout studies of PRR published to date have shown that this protein is crucial for the development and survival of cells 30,49. Our studies showed that specific transient knockdown of PRR in CD M-1 cells did not show a decrease in cell viability, although more accurate studies should be performed. On the other hand, a study from Sequeira-Lopez et al, showed that deletion of renin in the tubular compartment did not result in any discernible morphological or physiological changes in kidney CD duct cells. 50
CD cells have the predisposition to epithelial-mesenchymal transition 33,34. We have shown that ANG II treatment stimulates fibronectin and collagen I, both markers of fibrosis, via β-catenin pathway in M-1 CD cells 18. Here we have shown that prorenin treatment at nanomlar concentrations induces ROS formation and enhance the expression of CTGF, α-SMA, PAI-I, Fibronectin and collagen I, all markers of fibrosis (Figure 2 and Figure 3). Renin treatment at very high doses caused a slight but non significant increase in ROS formation, probably due to enhanced effect caused by preexisting prorenin secretion as observed in Figure 1B. Importantly, we demonstrated that antioxidant actions in membrane and cytoplasmic compartments have different effects. Using a lipophilic antioxidant (trolox C) or soluble ascorbic acid (AA) separately, we were not able to obtain the suppression observed with p-coumaric acid, which quenches the singlet oxygen in solution and also reacts with lipid peroxides in the membrane lipids 51. The water and lipid solubility of p-coumaric acid, together with its substantial efficiency to scavenge peroxides and to inhibit lipid peroxidation may provide a strategy for prevention of tubular injury as reported in several studies showing its protective effects against nephrotoxicity 52, cardiac damage 53.
In summary, the present results suggest that activation of PRR by nanomolar concentrations of prorenin induces fibrosis through generation of oxidative stress, which is dependent on MAPK/ERK 1/2 activation. It is possible that activation of MAPK/ERK pathway could enhance the activity of NOX-4, which is the main NOX isoform in CD cells 54,55.
Material and Methods
Cell line culture and treatments
M-1 cortical collecting duct cells (American Type Culture Collection; CRL-2038) which maintain the phenotype of CD cell 56 were grown in DMEM-F12 media supplemented with 10% FBS, 5 μM dexamethasone, 1× insulin-transferrin-selenium, and 100 U/ml penicillin/streptomycin in a humid atmosphere of 5% CO2-95% room air at 37°C. Cells were then treated with recombinant prorenin (Cayman Ann Arbor, MI) at nanomolar concentrations 57. Experiments with dose responses and time dependent effects are indicated in the results section.
Measurement of reactive oxygen species in M-1 cells
M-1 cells (200,000 per well) were seeded in 96-well plates. Groups were pretreated with p-coumaric acid 10-7 M (PCA, Sigma), trolox ((±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid, Sigma, USA) at 10-7 M, ascorbic acid 10-5 M (AA, Sigma, USA) or MEK inhibitor PD98059 (30 μM) in each case, measurements were taken 10 minutes post treatment. All groups were treated with probe carboxy-2′, 7′-dichloro-dihydro-fluorescein diacetate (DCFHDA, Sigma, USA) at 0.1 mM for 10 min. Cell culture media was removed and cells were treated with 10-8 M of recombinant prorenin during 10 min. Fluorescence measurements of DCF (the product of H2DCFDA oxidation: excitation 495 nm, emission 529nm) were performed on a plate reader (Appliskan, thermo, USA). To normalize results, total protein from each well was quantified by the bicinchoninic acid (BCA) method. A positive control was conducted using 200 mM H2O2. Kinetic for active ROS formation: ROS formation was measured at 20 seconds intervals using the same approach mentioned above. For experiments using the MEK inhibitor, cells were treated after 20 s of prorenin treatment (Figure 5).
Measurements of fibronectin, collagen I by Quantitative real-time RT-PCR (qRT-PCR)
qRT-PCR was performed using the TaqMan PCR system. Total RNA (20 ng) was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA). The following primers and probes were used to amplify the genes:
CTGF: 5- CAAAGCAGCTGCAAATACCA-3 (sense), 5-ggccaaatgtgtcttccagt-3 (antisense) and 5-6-FAM-GGAGTGGGTGTGTGACGAG (BQH1a-6FAM)-3 (fluorogenic probe); α-SMA: 5-CTGACAGAGGCACCACTGAA-3 (sense), 5-TGTGCTGGACTCTGGAGATG-3 (antisense), and 5-6-FAM-GCTGTCCCTCTATGCCTCTG (BQH1a-6FAM)-3 (fluorogenic probe); PAI-I: 5- AGTCTTTCCGACCAAGAGCA -3 (sense), 5- ATCACTTGCCCCATGAAGAG -3 (antisense) and 5-6-FAM- ATCGAGGTAAACGAGAGCG (BQH1a-6FAM)-3 (fluorogenic probe). Data were normalized against β-actin mRNA levels using primer and probe sequences as previously described 18.
Protein expression analysis
Forty micrograms of total protein were used for Western blot analysis. Protein expression levels were quantified after immunoblotting using a 1:1,000 dilution of the following specific antibodies: anti-ATP6AP2 (Cat. HPA003156, Sigma); connecting tissue growth factor (CTGF; Cat. No. sc-25440, Santa Cruz Biotechnology), α-smooth muscle actin (α -SMA; Cat. no. sc-53142, Santa Cruz Biotechnology), PAI-I (Cat. no. SC-8979, Santa Cruz Biotechnology), anti-ERK1 (phospho T202 + Y204) antibody (ab24157, Abcam Cambridge, UK) and anti-ERK1 + ERK2 antibody [ERK-7D8] (ab54230, Abcam Cambridge, UK). Primary antibodies were followed by incubation with either donkey anti-rabbit or anti-mouse IgG IRDye 800 CW (Li-cor Biosciences, Lincoln, NE) at a 1:30,000 dilution. Densitometric analyses were performed by normalization against β-actin (Cat. no. ab8227, Abcam Cambridge, UK). Recombinant mouse prorenin (AnaSpec, Fremont, CA) and renin (Lee Biosolutions, St. Louis, MO) were used as standards. Medullary enriched kidney tissues were used as positive controls for PRR.
Immunofluorescence in M-1 cells
Sub-confluent M-1 cells (50– 60%) cultured in chamber slides (Nalge Nunc, Rochester, NY) were fixed in cold methanol for 20 min, blocked with PBS-Tween (0.1%) plus BSA (3%) for 1 h, and stained with anti-ATP6AP2 (Cat. HPA003156, Sigma); anti-anion exchanger type 1 antibody at 1:500 dilution (AE1, Cat. # AE11-A; Alpha Diagnostic Intl; San Antonio, TX); collagen I (Cat. no. sc-8784; Santa Cruz Biotechnology, Santa Cruz, CA), fibronectin (Cat. no. sc-8422; Santa Cruz Biotechnology, Santa Cruz, CA) or anti- β-actin antibody (Cat. no. ab8227, Abcam Cambridge, UK) at 1:50 dilutions and detected with secondary antibody Alexa Fluor 488 (or 594 for AE-1 co-localization experiments) conjugated to anti-rabbit IgG (Invitrogen, Carlsbad, CA) at 1:500 dilutions. Samples were counterstained with 4,6-diamidino-2-phenylindole (Invitrogen, Carlsbad, CA). Negative controls were obtained by omission of the specific primary antibody. Measurements of fluorescence intensity was performed with NIS Elements ® software (Nikon). Fibroblast like phenotype was performed by cell counting (flat, spindle-shaped cells with multiple projections) versus total cell number in each field (n=10).
PRR knockdown of PRR
Silencing of PRR expression was performed using SureSilencing™ shRNA plasmid for Mouse Atp6ap2 (Cat. KM26726, SABioscience, Qiagen, Valencia, CA) according to Manufaturer's instructions. Sub-confluent cells (60%) were transfected 36 h prior treatments. Green fluorescent protein (GFP) positive cells (Figure 8A) confirmed effectiveness of transfections, which reached a rate 72 ± 5% (n=10). Effectiveness of PRR silencing was also verified by band intensity as compared to non-transfected cells and cells transfected with scramble-shRNA. PRR protein expression (full length plus soluble form bands) was reduced in 43 ± 7% (n=12). (Figure 8B). Cell viability after PRR knockdown (48 h) was assessed by AlamarBlue® Cell Viability Reagent (ThermoFisher DAL 1025) (Figure 8C).
Statistics analysis
Each experiment consisted of five independent observations (each well represented an independent observation). Experiments were performed in at least three different cell passages with n=5 per treatment. Cells were used until passages 10–12. Differences between groups were assessed by one-way ANOVA followed by Tukey's test using the GraphPad Prism software v 5.0 (GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant.
Acknowledgments
We thank the Imaging and Molecular Cores of the Tulane Renal Hypertension Center of Excellence
Funding: A.A.G. received funds from FONDECYT 11121217, Chile. M.C.P. received funds from the National Institutes of Health (NIH) through the Institutional Developmental Award Program of the National Center for Research Resources (P20RR-017659), HL26371, American Heart Association (AHA; 09BGIA2280440), and Eunice Kennedy Shriver National Institute of Child Health and Human Development (K12HD043451).
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Ludwig J, Kerscher S, Brandt U, et al. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem. 1998;273:10939–47. doi: 10.1074/jbc.273.18.10939. [DOI] [PubMed] [Google Scholar]
- 2.Advani A, Kelly DJ, Cox AJ, et al. The (Pro) Renin Receptor Site-Specific and Functional Linkage to the Vacuolar H(+)-ATPase in the Kidney. Hypertension. 2009;54:261–U129. doi: 10.1161/HYPERTENSIONAHA.109.128645. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen G, Delarue F, Berrou J, Rondeau E, Sraer JD. Specific receptor binding of renin on human mesangial cells in culture increases plasminogen activator inhibitor-1 antigen. Kidney Int. 1996;50:1897–903. doi: 10.1038/ki.1996.511. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen G, Burckle CA, Sraer JD. Renin/prorenin-receptor biochemistry and functional significance. Curr Hypertens Rep. 2004;6:129–32. doi: 10.1007/s11906-004-0088-3. [DOI] [PubMed] [Google Scholar]
- 5.Ichihara A, Kaneshiro FY, Takemitsu T, Sakodo M, Itoh H. The (Pro)Renin receptor and the kidney. Semin Nephrol. 2007;27:524–8. doi: 10.1016/j.semnephrol.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Ichihara A, Sakoda M, Kurauchi-Mito A, Nishiyama A, Itoh H. Involvement of receptor-bound prorenin in development of nephropathy in diabetic db/db mice. J Am Soc Hypertens. 2008;2:332–40. doi: 10.1016/j.jash.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Feldt S, Batenburg WW, Mazak I, et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension. 2008;51:682–8. doi: 10.1161/HYPERTENSIONAHA.107.101444. [DOI] [PubMed] [Google Scholar]
- 8.Muller DN, Klanke B, Feldt S, et al. (Pro) renin receptor peptide inhibitor “handle-region” peptide does not affect hypertensive nephrosclerosis in Goldblatt rats. Hypertension. 2008;51:676–81. doi: 10.1161/HYPERTENSIONAHA.107.101493. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez AA, Luffman C, Bourgeois CRT, Vio CP, Prieto MC. Angiotensin II-Independent Upregulation of Cyclooxygenase-2 by Activation of the (Pro) Renin Receptor in Rat Renal Inner Medullary Cells. Hypertension. 2013;61:443. doi: 10.1161/HYPERTENSIONAHA.112.196303. -+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaneshiro Y, Ichihara A, Sakoda M, et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol. 2007;18:1789–95. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 11.Kaneshiro Y, Ichihara A, Takemitsu T, et al. Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int. 2006;70:641–6. doi: 10.1038/sj.ki.5001627. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez-Barbero A, Dorado F, Velasco S, Pandiella A, Banas B, Lopez-Novoa JM. TGF-beta 1 induces COX-2 expression and PGE(2) synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney Int. 2006;70:901–9. doi: 10.1038/sj.ki.5001626. [DOI] [PubMed] [Google Scholar]
- 13.Prieto-Carrasquero MC, Botros FT, Pagan J, et al. Collecting duct renin is upregulated in both kidneys of 2-kidney, 1-clip Goldblatt hypertensive rats. Hypertension. 2008;51:1590–6. doi: 10.1161/HYPERTENSIONAHA.108.110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, et al. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension. 2004;44:223–9. doi: 10.1161/01.HYP.0000135678.20725.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prieto-Carrasquero MC, Kobori H, Ozawa Y, Guttierrez A, Seth D, Navar LG. AT(1) receptor- mediated enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Am J Physiol-Renal. 2005;289:F632–F7. doi: 10.1152/ajprenal.00462.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Gonzalez AA, McCormack M, et al. Increased renin excretion is associated with augmented urinary angiotensin II levels in chronic angiotensin II-infused hypertensive rats. Am J Physiol-Renal. 2011;301:F1195–F201. doi: 10.1152/ajprenal.00339.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC. Angiotensin II Stimulates Renin in Inner Medullary Collecting Duct Cells via Protein Kinase C and Independent of Epithelial Sodium Channel and Mineralocorticoid Receptor Activity. Hypertension. 2011;57:594–9. doi: 10.1161/HYPERTENSIONAHA.110.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuevas CA, Gonzalez AA, Inestrosa NC, Vio CP, Prieto MC. Angiotensin II increases fibronectin and collagen I through the beta-catenin-dependent signaling in mouse collecting duct cells. Am J Physiol-Renal. 2015;308:F358–F65. doi: 10.1152/ajprenal.00429.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franken AAM, Derkx FHM, Blankestijn PJ, et al. Plasma Prorenin as an Early Marker of Microvascular Disease in Patients with Diabetes-Mellitus. Diabetes Metab. 1992;18:137–43. [PubMed] [Google Scholar]
- 20.Chiarelli F, Pomilio M, De Luca FA, Vecchiet J, Verrotti A. Plasma prorenin levels may predict persistent microalbuminuria in children with diabetes. Pediatr Nephrol. 2001;16:116–20. doi: 10.1007/s004670000514. [DOI] [PubMed] [Google Scholar]
- 21.Clavreul N, Sansilvestri-Morel P, Magard D, Verbeuren TJ, Rupin A. (Pro)renin promotes fibrosis gene expression in HEK cells through a Nox4-dependent mechanism. Am J Physiol-Renal. 2011;300:F1310–F8. doi: 10.1152/ajprenal.00119.2010. [DOI] [PubMed] [Google Scholar]
- 22.Liu FY, Liu XY, Zhang LJ, Cheng YP, Jiang YN. Binding of prorenin to (pro)renin receptor induces the proliferation of human umbilical artery smooth muscle cells via ROS generation and ERK1/2 activation. J Renin-Angio-Aldo S. 2014;15:99–108. doi: 10.1177/1470320314525215. [DOI] [PubMed] [Google Scholar]
- 23.Yisireyili M, Saito S, Abudureyimu S, et al. Indoxyl Sulfate-Induced Activation of (Pro)renin Receptor Promotes Cell Proliferation and Tissue Factor Expression in Vascular Smooth Muscle Cells. Plos One. 2014;9 doi: 10.1371/journal.pone.0109268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorin Y, Ricono JM, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Nox4 mediates angiotensin II-induced activation of Akt/protein kinase B in mesangial cells. Am J Physiol-Renal. 2003;285:F219–F29. doi: 10.1152/ajprenal.00414.2002. [DOI] [PubMed] [Google Scholar]
- 25.Kang JJ, Toma I, Sipos A, Meer EJ, Vargas SL, Peti-Peterdi J. The collecting duct is the major source of prorenin in diabetes. Hypertension. 2008;51:1597–604. doi: 10.1161/HYPERTENSIONAHA.107.107268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schefe JH, Neumann C, Goebel M, et al. Prorenin engages the (pro) renin receptor like renin and both ligand activities are unopposed by aliskiren. J Hypertens. 2008;26:1787–U9. doi: 10.1097/HJH.0b013e3283060f2e. [DOI] [PubMed] [Google Scholar]
- 27.Schefe JH, Menk M, Reinemund J, et al. A novel signal transduction cascade involving direct physical interaction of the renin/prorenin receptor with the transcription factor PLZF. J Hypertens. 2007;25:S392–S. doi: 10.1161/01.RES.0000251700.00994.0d. [DOI] [PubMed] [Google Scholar]
- 28.Batenburg WW, Krop M, Garrelds IM, et al. Prorenin is the endogenous agonist of the (pro)renin receptor: Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. Hypertension. 2007;50:E76–E. doi: 10.1097/HJH.0b013e3282f05bae. [DOI] [PubMed] [Google Scholar]
- 29.Rosendahl A, Niemann G, Lange S, et al. Increased expression of (pro)renin receptor does not cause hypertension or cardiac and renal fibrosis in mice. Lab Invest. 2014;94:863–72. doi: 10.1038/labinvest.2014.83. [DOI] [PubMed] [Google Scholar]
- 30.Oshima Y, Kinouchi K, Ichihara A, et al. Prorenin Receptor Is Essential for Normal Podocyte Structure and Function. J Am Soc Nephrol. 2011;22:2203–12. doi: 10.1681/ASN.2011020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubinstein JD, Lesnefsky EJ, Byler RM, Fennessey PV, Horwitz LD. Trolox-C, a Lipid-Soluble Membrane Protective Agent, Attenuates Myocardial Injury from Ischemia and Reperfusion. Free Radical Bio Med. 1992;13:627–34. doi: 10.1016/0891-5849(92)90037-h. [DOI] [PubMed] [Google Scholar]
- 32.Padayatty SJ, Levine M. Vitamin C: the known and the unknown and Goldilocks. Oral Dis. 2016;22:463–93. doi: 10.1111/odi.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivanova L, Butt MJ, Matsell DG. Mesenchymal transition in kidney collecting duct epithelial cells. Am J Physiol-Renal. 2008;294:F1238–F48. doi: 10.1152/ajprenal.00326.2007. [DOI] [PubMed] [Google Scholar]
- 34.Butt MJ, Tarantal AF, Jimenez DF, Matsell DG. Collecting duct epithelial-mesenchymal transition in fetal urinary tract obstruction. Kidney Int. 2007;72:936–44. doi: 10.1038/sj.ki.5002457. [DOI] [PubMed] [Google Scholar]
- 35.Ichihara A, Sakoda M, Kurauchi-Mito A, Kaneshiro Y, Itoh H. Involvement of (pro)renin receptor in the glomerular filtration barrier. J Mol Med. 2008;86:629–35. doi: 10.1007/s00109-008-0327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen G, Muller DN. The Biology of the (Pro)Renin Receptor. J Am Soc Nephrol. 2010;21:18–23. doi: 10.1681/ASN.2009030300. [DOI] [PubMed] [Google Scholar]
- 37.Satofuka S, Ichihara A, Nagai N, et al. (Pro)renin Receptor-Mediated Signal Transduction and Tissue Renin-Angiotensin System Contribute to Diabetes-Induced Retinal Inflammation. Diabetes. 2009;58:1625–33. doi: 10.2337/db08-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang JQ, Matavelli LC, Siragy HM. Renal (pro)renin receptor contributes to development of diabetic kidney disease through transforming growth factor-beta 1-connective tissue growth factor signalling cascade. Clin Exp Pharmacol P. 2011;38:215–21. doi: 10.1111/j.1440-1681.2011.05486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez AA, Cifuentes-Araneda F, Ibaceta-Gonzalez C, et al. Vasopressin/V2 receptor stimulates renin synthesis in the collecting duct. Am J Physiol-Renal. 2016;310:F284–F93. doi: 10.1152/ajprenal.00360.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deinum J, Ronn B, Mathiesen E, Derkx FHM, Hop WCJ, Schalekamp MADH. Increase in serum prorenin precedes onset of microalbuminuria in patients with insulin-dependent diabetes mellitus (vol 42, pg 1006, 1999) Diabetologia. 1999;42:1444. doi: 10.1007/s001250051260. [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez AA, Green T, Luffman C, Bourgeois CRT, Navar LG, Prieto MC. Renal medullary cyclooxygenase-2 and (pro)renin receptor expression during angiotensin II-dependent hypertension. Am J Physiol-Renal. 2014;307:F962–F70. doi: 10.1152/ajprenal.00267.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng HF, Fan XF, Moeckel GW, Harris RC. Podocyte COX-2 Exacerbates Diabetic Nephropathy by Increasing Podocyte (Pro)renin Receptor Expression. J Am Soc Nephrol. 2011;22:1240–51. doi: 10.1681/ASN.2010111149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ichihara A, Suzuki F, Nakagawa T, et al. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol. 2006;17:1950–61. doi: 10.1681/ASN.2006010029. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen G, Delarue F, Rondeau E, Sraer JD. Characterization of a Specific Receptor for Renin on Human Mesangial Cells in Culture. J Am Soc Nephrol. 1995;6:805. [Google Scholar]
- 45.Yogi A, Mercure C, Touyz J, et al. Renal redox-sensitive signaling, but not blood pressure, is attenuated by Nox1 knockout in angiotensin II-dependent chronic hypertension. Hypertension. 2008;51:500–6. doi: 10.1161/HYPERTENSIONAHA.107.103192. [DOI] [PubMed] [Google Scholar]
- 46.Kamiyama M, Farragut KM, Garner MK, Navar LG, Kobori H. Divergent localization of angiotensinogen mRNA and protein in proximal tubule segments of normal rat kidney. J Hypertens. 2012;30:2365–72. doi: 10.1097/HJH.0b013e3283598eed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mahmud H, Candido WM, van Genne L, et al. Cardiac Function and Architecture Are Maintained in a Model of Cardiorestricted Overexpression of the Prorenin-Renin Receptor. Plos One. 2014;9 doi: 10.1371/journal.pone.0089929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ichihara A, Hayashi M, Kaneshiro Y, et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest. 2004;114:1128–35. doi: 10.1172/JCI21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kinouchi K, Ichihara A, Sano M, et al. ATP6AP2/(pro)renin Receptor is Essential for the Functions of Organella in Murine Cardiomyocytes. Hypertension. 2010;56:E132–E. [Google Scholar]
- 50.Sequeira-Lopez MLS, Nagalakshmi VK, Li MH, Sigmund CD, Gomez RA. Vascular versus tubular renin: role in kidney development. Am J Physiol-Reg I. 2015;309:R650–R7. doi: 10.1152/ajpregu.00313.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zang LY, Cosma G, Gardner H, Shi XL, Castranova V, Vallyathan V. Effect of antioxidant protection by p-coumaric acid on low-density lipoprotein cholesterol oxidation. Am J Physiol-Cell Ph. 2000;279:C954–C60. doi: 10.1152/ajpcell.2000.279.4.C954. [DOI] [PubMed] [Google Scholar]
- 52.Navaneethan D, Rasool M. p-Coumaric acid, a common dietary polyphenol, protects cadmium chloride-induced nephrotoxicity in rats. Renal Failure. 2014;36:244–51. doi: 10.3109/0886022X.2013.835268. [DOI] [PubMed] [Google Scholar]
- 53.Roy AJ, Prince PSM. Preventive effects of p-coumaric acid on lysosomal dysfunction and myocardial infarct size in experimentally induced myocardial infarction. Eur J Pharmacol. 2013;699:33–9. doi: 10.1016/j.ejphar.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 54.Babilonia E, Wei Y, Sterling H, Kaminski P, Wolin M, Wang WH. Superoxide anions are involved in mediating the effect of low K intake on c-Src expression and renal K secretion in the cortical collecting duct. J Biol Chem. 2005;280:10790–6. doi: 10.1074/jbc.M414610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schreck C, O'connor PM. NAD(P)H oxidase and renal epithelial ion transport. Am J Physiol-Reg I. 2011;300:R1023–R9. doi: 10.1152/ajpregu.00618.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoos BA, Narayfejestoth A, Carretero OA, Ito S, Fejestoth G. Characterization of a Mouse Cortical Collecting Duct Cell-Line. Kidney Int. 1991;39:1168–75. doi: 10.1038/ki.1991.148. [DOI] [PubMed] [Google Scholar]
- 57.Nabi AHMN, Biswas KB, Nakagawa T, Ichihara A, Inagami T, Suzuki F. Prorenin has high affinity multiple binding sites for (pro)renin receptor. Bba-Proteins Proteom. 2009;1794:1838–47. doi: 10.1016/j.bbapap.2009.08.024. [DOI] [PubMed] [Google Scholar]