

Abstract
Employment of simple transition metal (TM= Co, Fe, Cu, Pd, Pt, Au)-based photocatalyst (PC) led to the dramatic acceleration of known TM-catalyzed reactions, as well as to the discovery of unprecedented chemical transformations. Compared to the conventional cooperative/dual photocatalysis (type B, Figure 1), this new class of unconventional PCs operates via a single photoexcitation/catalytic cycle, where the TM complex plays a “double duty” role by harvesting light and catalyzing the chemical transformation. Also, these TM photocatalysts participate in the bond-forming/breaking event in the transformation via a substrate-TM interaction, an aspect that is uncommon for conventional photocatalysis (type A, Figure 1). This tutorial review highlights the recent advances in this emerging area.
1. Introduction
Visible light-induced reactions have become a powerful avenue in organic synthesis. This area can be systemized into three main categories: (A) Visible light photocatalysis, which involves photosensitization by a photocatalyst (PC) that enables the transformation to occur either via redox-, atom transfer-, or energy transfer process (Figure 1).1 (B) Cooperative/dual photocatalysis,2 which synergistically combines the first strategy with a transition metal catalysis (TM-cat.). In this case, the PC employed is the sole photo-absorbing species that engages either a redox- or energy transfer process with the traditional TM catalyst in order to facilitate the transformation. Lately, this important fast growing area has been extensively reviewed.2 Typically, for the first two categories, the PCs employed are photoactive organic molecules, or Ir-, Ru-, or Cu-TM complexes bearing conventional bipyridyl and/or phenanthroline ligands.1 Recently, a new area which deviates from the above-mentioned paradigm has emerged (C). In this case, “traditional” transition-metal complexes serving as PCs are employed. These catalysts are TM complexes (Co, Fe, Cu, Pd, Pt, Au) possessing typical phosphine-, amine-, oxime-, or acetylides, ligands that are usually employed in conventional catalytic transformations. Importantly, no exogenous photosensitizer (PC) is required to promote these photocatalytic transformations. Consequently, the mechanism of these methods is distinct from the traditional mode. The bond-forming/breaking process for these visible light-induced TM-catalyzed transformations occurs with direct involvement of the metal center (i.e. substrate-TM interaction), a feature that is not commonly observed for conventional visible light photocatalysis (A). Moreover, these transformations operate via single catalytic cycle, where the TM complex plays “double duty” by harvesting visible light and enabling catalysis. This direction differs from the seemingly alike cooperative/dual photocatalysis (B), where an exogenous photosensitizer (PC) is required to harvest light and to enable a redox or energy transfer process to the performing TM catalyst.
Figure 1.
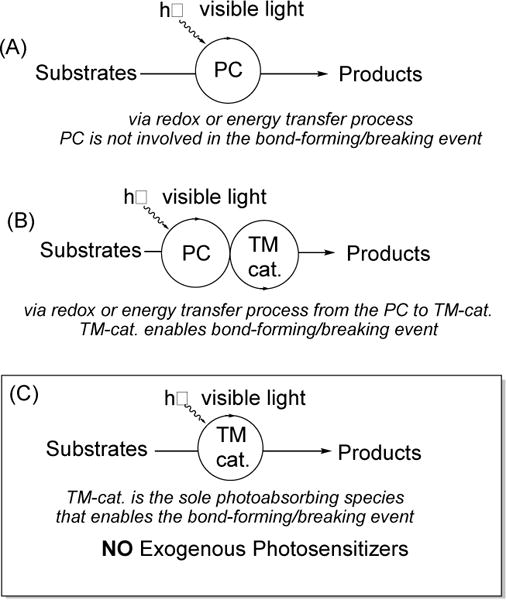
Photocatalytic approaches: a) Traditional photocatalysis. b) Cooperative/dual photocatalysis. c) Visible light-induced transition metal catalysis: focus of this tutorial review.
The aim of this tutorial review is to highlight recent synthetic methodology development employing unconventional TM-photocatalyst under visible light. The synthetic applications and mechanistic aspects of the featured methods are discussed. However, the proposed mechanisms provided throughout this tutorial review only reflect the best hypotheses by the authors. Naturally, due to the limited knowledge on the photochemistry of TM-complexes, some of these mechanisms are underdeveloped. Related methodologies involving TM-catalysis under UV irradiation,3 and photoinduced heterogeneous TM-catalysis/semiconductor photocatalysis4 are not discussed herein.
2. Visible light-induced Co-catalyzed reactions
2.1. C–C bond forming reactions
In 1990, Scheffold and co-workers reported the synthesis of prostaglandin F2α core 1 via cascade intramolecular cyclization of alkyl bromide, followed by intermolecular coupling with ynones (Scheme 1a).5 The reaction was catalyzed by vitamin B12a (2) under photo- and electrochemical conditions. Control studies indicated that all of the reaction components were necessary to promote the reaction. It was surmised that the alkyl Co(III) intermediate 3, formed via SET with alkyl-Br and 2, undergoes photoinduced homolysis to generate the transient alkyl radical species and the persistent radical/TM Co(II) complex 4 (Scheme 1b). The former undergoes typical reductive radical type reactions, whereas the latter is reduced by a one-electron electrochemical process to produce the active Co(I) complex 2.
Scheme 1.

a) Scheffold’s photoinduced/electrochemical Co-catalyzed reductive cascade reaction. b) Catalytic cycle.
Cobaloximes complexes (5) have been rigorously studied for decades.6 Photophysical analysis of these complexes reveals that they are capable of harvesting visible light efficiently. However, employment of these complexes by photocatalytic means for synthesis was not exploited until 2011, when Carreira and co-workers reported the first visible light-induced, Co-catalyzed alkyl Heck reaction of 6→7; en route to (+)-daphmanidin E (Scheme 2).7 In their follow-up report, the authors reported the full scope and mechanistic details of their novel photoinduced intramolecular alkyl Heck reaction (Scheme 3).8 Exo-trig cyclization of systems containing sensitive functionalities (8a–d), such as amides, aldehydes, and heterocycles, reacted efficiently. Notably, substrates possessing usually reactive aryl-halides did not affect the outcome of the reaction (8d). The novelty of their methodology was showcased by the efficient formation of 8e, an important intermediate for the synthesis of (±)-samin. The authors proposed that the operative mechanism (Scheme 4) starts with the release of ISnPh3 and formation of Co(III) 10 via irradiation of complex 5 in the presence of alkyl iodide substrate 9. Intermediate 10 undergoes 5-exo-trig cyclization to produce 11, followed by elimination to furnish the Heck product 12 and the hydridocobalt species. Deprotonation of the latter turns over the catalytic system by producing anionic Co(I) complex, which then undergoes a chain process with the starting alkyl halide (9). Although, no studies were conducted to test the intermediacy of radicals, it likely that the reaction proceeds via a hybrid Co-radical pathway due the literature precedents on UV-induced homolysis of alkyl Co(III) species.3a
Scheme 2.
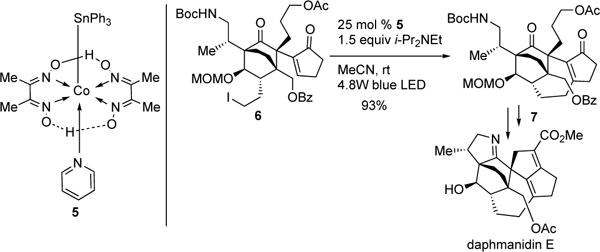
Carreira’s photoinduced Co-catalyzed Heck reaction.
Scheme 3.
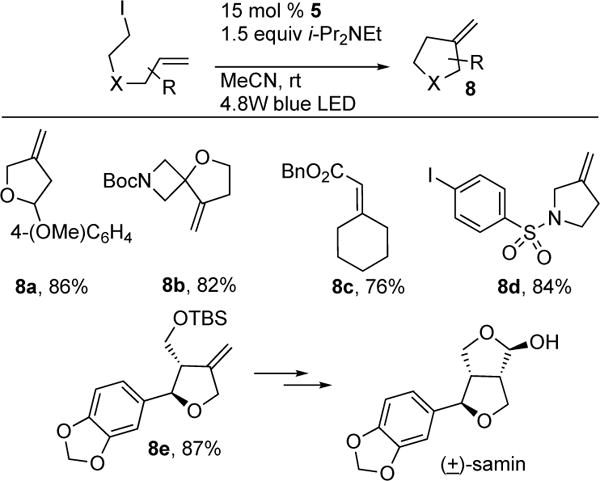
Scope of Carreira’s photoinduced Co-catalyzed intramolecular Heck reaction of alkyl halides.
Scheme 4.
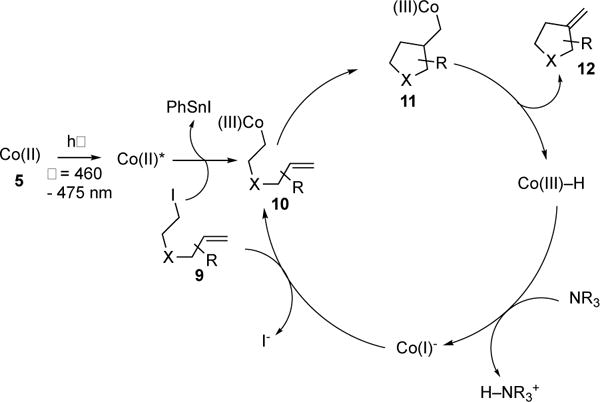
Mechanism of Carreira’s photoinduced Co-catalyzed intramolecular Heck reaction of alkyl halides.
In 2013, Martin and Carreria reported the first intermolecular Co-catalyzed Heck reaction of alkyl iodides under visible light (Scheme 5).9 Their report focuses on the photochemical synthesis of important allylic trifluoromethanes (13) from iodomethyltrifluoromethane and styrenes. Although highly innovative, this method required high catalyst loading to drive the reaction to completion. In addition, an interesting aspect of this work is the development and design of a self-made flow reactor photobox that enabled an eighty-seven-fold increase in productivity. Notably, it was found that their design significantly accelerated the rate of the reaction (30 mins) compared to the batch process (24 hrs).
Scheme 5.
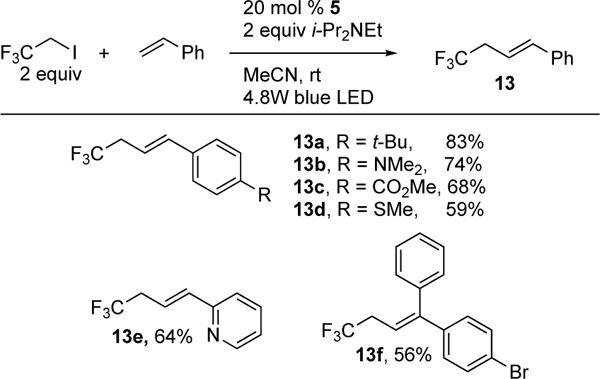
Carreira’s photoinduced Co-catalyzed intermolecular Heck reaction of iodomethyltrifluoromethanes with styrenes.
3. Visible light-induced Fe-catalyzed reactions
3.1. Hydrosilylation reactions
Visible light-induced transformations involving Fe-catalyst are very rare. Most photoinducing Fe-catalyzed methods tend to employ UV-irradiation for ligand dissociation of CO to form an open-coordination site at the metal center, thus allowing other transformations to occur.3a Nonetheless, in 2011, Darcel and Sortais reported a visible light-induced hydrosilylative reduction of ketones and aldehydes using Fe(II) catalyst 14 (Scheme 6).10 It was found that the addition of thermal energy can greatly accelerate the reaction and help reducing both the catalyst load, as well as the equivalence of Si–H substrate (15a vs 15b; 16a vs 16b). Control studies indicated that visible light is required to enable the transformation, even at high temperatures. Thus, indicating that visible light is necessary for formation of the active Fe-catalyst. Over the past few years, the authors have greatly expanded their approach and many methods were developed on the reduction of esters, amides, and imines via hydrosilylation with a diverse set of Fe-catalysis under visible light irradiation.
Scheme 6.

Darcel and Sortais’ photocatalytic hydrosilylation of ketones and aldehydes using Fe(II) catalyst.
4. Visible light-induced Cu-catalyzed reactions
4.1. C–C coupling reactions
In 2012, Hwang and co-workers reported a visible light-induced Cu-catalyzed, “Pd-free,” Sonogashira reaction (Scheme 7).11 Though, Sonogashira reactions of this type have been previously reported, they either contained traces of Pd or employed high reaction temperatures. Also, initial attempts for a UV-induced-metal free Sonogashira reaction was reportedly met with some challenges due to photolytic decomposition and undesired rearrangements. Hwang’s approach involves low loading of cheap CuCl catalyst and operates under visible light with no exogenous photosensitizers. This combination provided a great acceleration effect; the Sonogashira reaction proceeded efficiently at room temperature in the absence of excess ligand, base, or traces of palladium. Aryl iodides and bromides reacted well; producing their respective Sonogashira adducts 17a and 17b in good yields. The scope of the terminal alkynes was quite broad, as terminal alkynes bearing alkyl-Cl (17c), tertiary alcohol (17d), and 4- fluoroarene (17e) were tolerated, but trimethylsilylacetylene (17f) was less efficient. The authors then applied this concept toward a domino Sonogashira reaction sequence involving aryl halides with pendent o-nucleophiles 18, which resulted in synthetically valuable benzofurans (19a) via the 5-exo-dig cyclization, and a mixture of E/Z -benzofuranones/benzopyrrolones (19b/19c) via 5-exo-dig cyclization.
Scheme 7.

Scope of Hwang’s photoinduced Cu-catalyzed, “Pd-free,” Sonogashira reaction.
The authors performed various photophysical mechanistic studies. Analysis of the absorbance spectrum of Cu-acetylide 20 (Scheme 8) revealed a λmax at 476 nm, which is in agreement of a photoinduced ligand to metal charge transfer (LMCT). It was found that the addition of 0.1% of Cu(I)Cl to the stoichiometric reaction of 20 with Ar-I greatly accelerated the reaction, which presumes residual copper π-coordination of Cu-acetylide intermediates. The proposed mechanism starts with a based-assisted formation of acetylide 20. Then irradiation of the latter results in excited complex 21, which undergoes a concerted aryl transfer with the aryl halide substrate (via complex 22) to generate the Sonogashira adduct 17 and regenerate the catalyst. However, based on the known propensity of 20 to undergo a facile intersystem crossing (ISC) to furnish excited triplet 21, Jacobi von Wangelin proposed an alternative mechanism12 where intermediate 21 can undergo a single electron transfer (SET) process with aryl halides resulting in Cu(II)X species and aryl radical 23, followed by recombination to generate Cu(III) complex 24. Subsequent reductive elimination of the latter produces 17. Hwang’s initial work did not provide enough evidence to rule out the latter pathway, though, his later studies supported the triplet nature of 21 (vide infra).
Scheme 8.
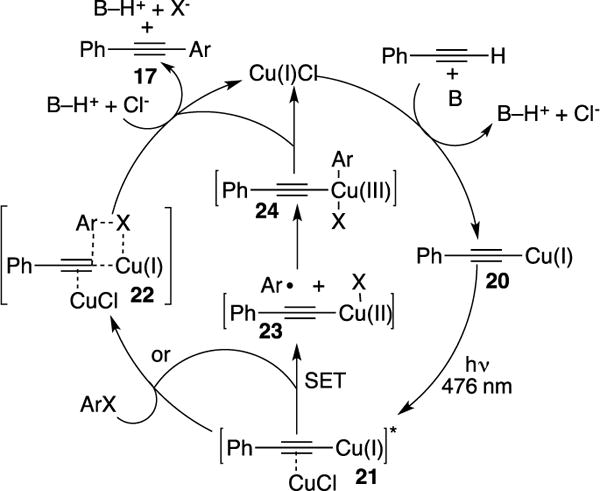
Catalytic-cycle of the Hwang’s photoinduced Cu-catalyzed, “Pd-free,” Sonogashira reaction.
Recently, the same group employed a similar strategy toward the oxidative synthesis of unsymmetrical conjugated diynes, Scheme 9a.13 Compared to the well-established protocols for the synthesis of diynes via classical Glaser-Hay and/or Cadiot-Chodkiewicz coupling, Hwang’s approach obviates the need for high reaction temperature, pre-functionalized substrates, and employment of excess base, ligands, and costly oxidants. More importantly, Hwang’s photoinduced approach allows for efficient coupling of electron deficient alkynes (27c-d), which overcomes the inherent limitations of the aforementioned protocols. The reaction appeared to be very general in scope, as halogen-containing-(27b) and alkyl alkynes (27e-f) were well tolerated. It should be noted, however, that alkyne homo-coupling products were obtained as minor products (25-25, ~11% and 26-26, ~48%). The use of non-polar solvents, such as toluene, led to lower reaction efficiency indicating a solvatochromic effect; whereas the employment of Cu(II) sources resulted in no reaction. The green metrics14 for synthesis of 27d were studied and resulted in a E-factor of 11.7 with 93% atom-economy, which is superior to the green metrics of Lei’s previously reported approach (E-factor= 27 with 56% atom-economy).15 Control studies indicated that all reaction components: light, the Cu-catalyst, and O2, are necessary to promote the reaction. In addition, EPR studies, using superoxide quencher DMPO, indicated the formation of a superoxide radical anion during the course of the reaction. The proposed mechanism presumes coordination of the Cu(I) catalyst with the electron rich alkyne to produce complex 20, followed by π-coordination of the electron-deficient alkyne which affords intermediate 28 (Scheme 9b). Notably, the author’s mechanism favors a π-π coordination of 20 with the alkyne ligands (complex 28) over the possible π-coordination of 21 with residual copper (Scheme 8), as supported by the results of isothermal titration calorimetry (ITC) experiments. Irradiation of 28 generates the excited complex 29 via the LMCT at λmax=479 nm, which subsequently undergoes SET with O2 to furnish Cu(II) complex 30 and the superoxide radical anion. Then, hydrogen atom abstraction of the superoxide radical anion with 30 furnishes radical alkyne complex 31. The latter produces the conjugated diyne 27 and regenerates the catalyst via a direct ligand cross-coupling (31→27). Alternatively, radical complex 31 can collapse to form high valent Cu(III) species 32, where a facile reductive elimination yields the same products.3b–c,12 The authors recently reported a follow up report on the homo-coupling of alkynes.16
Scheme 9.

a) Scope of Hwang’s photoinduced synthesis of unsymmetrical conjugated diynes. b) Catalytic-cycle.
4.2. C–N coupling reactions
In 2015, Hwang and co-workers reported synthesis of α-ketoamides 33 via oxidative coupling of aryl amines and terminal alkynes (Scheme 10a).17 Compared to the Jiao’s state-of-art approach18 which requires excess base, co-catalyst, and heat, Hwang’s photoinduced approach operates under ambient conditions (room temperature) and does not require ligands or excess base. Due to the employment of milder conditions, Hwang’s method evidently has greater functional group tolerance, which overcomes the limitations reported by Jiao. The novelty of Hwang’s method was highlighted by the synthesis of epoxide hydrolase inhibitor 34 (Scheme 10b), where 86% isolated yield was obtained on a preperative scale, which is by far more efficient than the established multi-step approach (5% total isolated yield) reported by Aavula and co-workers.19 The E-factor14 for Hwang’s aforementioned synthesis was found to be 47.7 with 95% atom-economy.
Scheme 10.
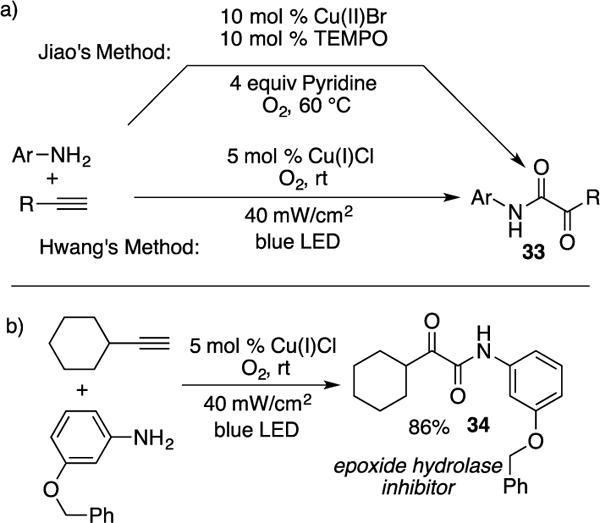
(a) Comparison of the Jiao and Hwang’s method for the synthesis of α-ketoamides (b) Hwang’s synthesis of epoxide hydrolase inhibitor.
It was proposed that once intermediate 20 is formed, it undergoes excitation with blue light to produce complex 35 (Scheme 11). Photophysical studies revealed that complex 35 has triplet character based on its longer lifetime, 10.47 μs, compared to a much shorter lived singlet Cu(I) species (<1 μs). The following SET of 35 with molecular oxygen generates Cu(II) complex 36 and the superoxide radical anion. Next, addition of the aniline radical species, formed via HAT with O2, generates Cu(III) complex 37. A subsequent reductive elimination of the latter produces ynamide 38, which reacts with molecular oxygen to form complex 39. Then, complex 39 readily undergoes isomerization to yield intermediate 40 followed by a subsequent intramolecular attack to generate the active catalytic species and 41. The latter undergoes rapid ring opening to form the α-ketoamide product 33. It should be noted that the authors previously implemented a similar strategy toward synthesis of quinoxalines involving Cu-photocatalysis.20
Scheme 11.
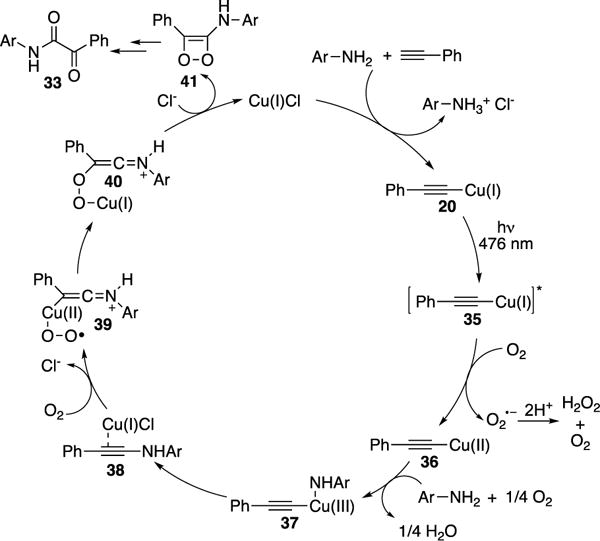
Mechanism of Hwang’s oxidative coupling of aryl amines and terminal alkynes.
The same group realized a sustainable and atom economical approach toward valuable 2,3-substituted indole derivatives 42 via a photoinduced Cu-catalyzed three-component coupling of terminal alkynes, aryl amines, and benzoquinone derivatives (Scheme 12).21 Though, similar strategies for formal C–H annulation of alkynes and aryl amines have been reported,22 most methods require either pre-functionalization of substrates or use of high temperature and stoichiometric oxidants. Hwang’s approach obviates these necessities with water being produced as the only by-product. The scope of the transformation was found to be quite general, as electron rich- (42b) and deficient phenyl acetylenes (42c), as well as alkyl alkynes (42d), reacted efficiently. Moreover, unsymmetrical benzoquinones worked well producing a single regioisomer of the C–H arylation adduct (42e). However, employment of secondary anilines led to a decrease of reaction efficiency (42f–g). Remarkably, halogen substituents were unaffected (42h), as no formation of potential competitive Sonogashira coupling byproducts were observed (vide supra).11
Scheme 12.
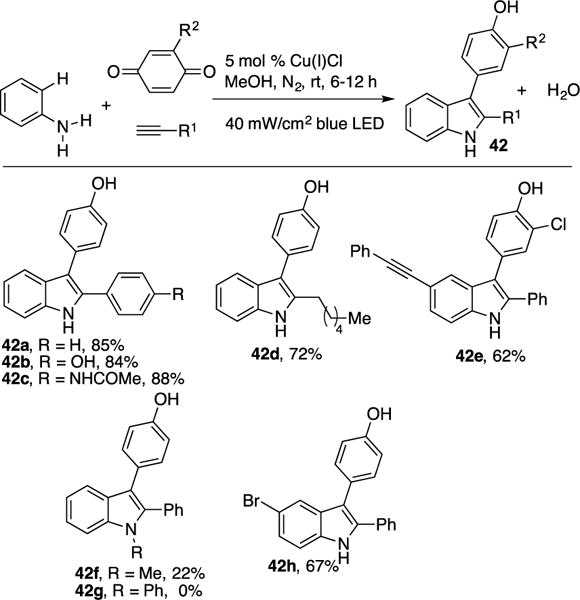
Hwang’s three-component coupling.
Control experiments demonstrated that light is required for this transformation. While irradiation with ambient light (white light/household bulb) worked, it was found that use of blue LEDs were more efficient. Evidence for radical intermediates were confirmed by the employment of radical scavengers, where the addition of TEMPO inhibited the reaction. Stoichiometric studies involving the reaction of copper acetylide complex 20 with aniline and benzoquinone generated product 42a in moderate yield (54%), thus indicating that complex 20 is indeed the active photocatalyst. The proposed mechanism by Hwang et al. (Scheme 13) occurs via excitation of complex 20 to yield triplet complex 35 (Scheme 11). Next, SET of complex 35 with benzoquinone produces Cu(II) complex and benzoquinone radical anion 43. The feasibility of this step was ascertained by matching the reduction potentials of the aforementioned species based on the cyclic voltammetry (CV) experiments. Recombination of 43 with the Cu(II) complex, followed by reductive elimination of the formed putative Cu(III) intermediate 44, produces 45. Subsequent intermolecular nucleophilic addition of the aryl amine results in vinyl copper complex 46, which then leads to intermediate 47 by extrusion of water. Finally, a Friedel-Crafts-type cyclization of the latter (47→48) followed by rearomatization produces the indole product 42a and closes the catalytic cycle.
Scheme 13.
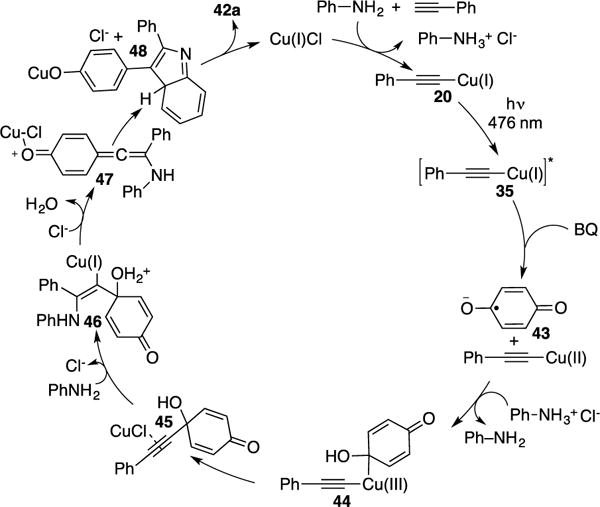
Catalytic-cycle for Hwang’s three-component coupling.
Classical approaches for the alkylation of amines with non-SN2 active alkyl halide substrates are notoriously difficult and often require extremely high reaction temperatures to promote the reaction.23 In 2014, the groups of Fu and Peters solved this problem, where alkylation of amines were efficiently achieved via Cu-catalysis under UV-light at ambient temperatures.3b–c In their recent follow-up work, the authors disclosed an elegant asymmetric variant, where efficient C–N coupling of tertiary alkyl chlorides and amines was achieved in high yields with high levels of enantioselectivity, Scheme 14.24 Notably, in contrast to their previous report, the use of benign visible light was sufficient to promote the reaction. Also, the coupling adducts were obtained with low catalyst loading and employed commercially available chiral phosphine ligand ((S)-L*)) at low reaction temperatures. The scope of the reaction was quite general; the reaction outcome was unaffected by use of different electronic substituted substrates (49a–e), the use of other N-nucleophiles (replacing carbazole with indole) (49d–e), and variation of the α-substituents of the alkyl halide (49f) and the amide (49g–h).
Scheme 14.
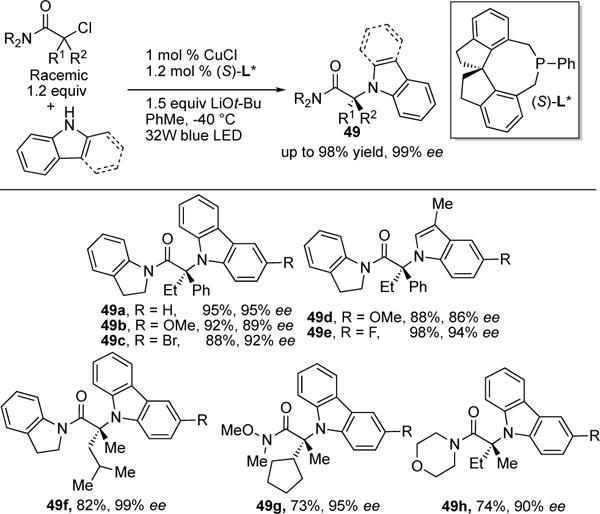
Scope of Fu and Peters’ asymmetric coupling.
Control studies revealed that both light and the Cu-catalyst were necessary for the reaction to proceed. The proposed mechanism involves a blue light irradiation of the copper complex 50 into excited complex 51, which undergoes SET with the alkyl chloride to form complex 52 (Scheme 15). The latter can either produce the coupling adduct directly (52→54) or can collapse into Cu(III) complex 53, a formal oxidative addition adduct of 51 and the alkyl halide. Subsequent reductive elimination of 53 occurs to generate the coupling adduct and Cu(I)X species 54. Ligand substitution of 54 with the amine nucleophile forms the active catalytic species. To confirm whether complex 50 is a viable intermediate, its enantiomeric complex (with ((R)-L*) ligand) was synthesized and characterized. Stoichiometric studies of complex 55 in the presence of alkyl halide 56 under visible light proceeded uneventfully, producing 57a with equivocal levels of enantioselectivity to that of the catalyzed version (49a). In contrast, no reaction occurred without photoirradiation (57b). Thus, these results indicate that complex 50 is indeed the active photocatalyst. Compared to the established asymmetric photocatalytic protocols which involve two cycles: 1) PC redox-cycle1 and 2) use of chiral co-catalyst for enantioinduction,1d Fu and Peters’ approach involves one cycle where the copper complex is responsible for both photo- and enantioinduction.
Scheme 15.
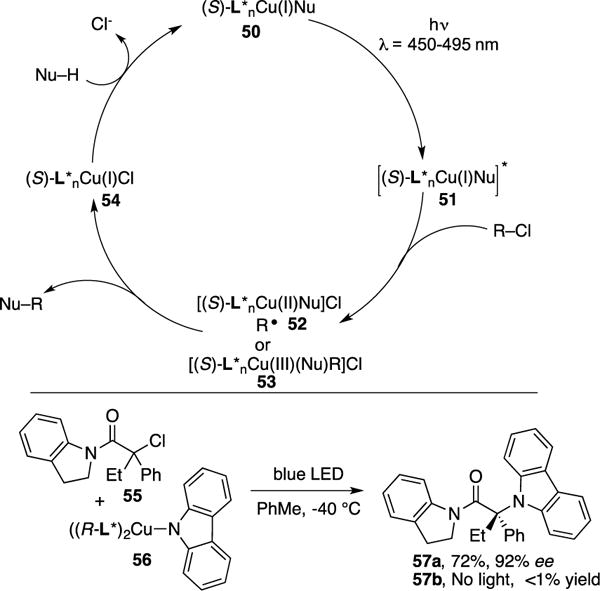
Mechanism of Fu and Peters’ photoinduced asymmetric coupling.
4.3. C≡C bond oxidation
Hwang and co-workers reported a mild visible light-induced Cu-catalyzed aerobic oxidation of ynamides and ynamines into synthetically valuable α-ketoimides (58) and α-ketoamides (59), respectively (Scheme 16).25 Although vast literature precedents exist for this exact synthetic approach, they typically employ: 1) expensive metal catalyst (Ru, Au); 2) stoichiometric oxidants; and are 3) conducted at high temperatures.26 Hwang’s method precludes the needs of the above requirements by utilizing cheap Cu-catalyst, abundant O2 as oxidant, and sustainable light sources. The scope of the reaction is quite general, as α-ketoimides and α-ketoamides possessing sensitive functionalities, such as halogens, cyano, ester groups, were well tolerated (58a–i, 59a–d). Even alkyl ynamides and ynamines produced their respective products in good yield (58h, 59d). As for the mechanism, UV-vis studies and ITC studies specified that the photoabsorbing species is the Cu(I)-corrdinated π-complex of ynamides/ynamines 60. Evidence of the oxygen source was obtained by 18O-labled studies, where the use of 18O2 produced a mixture of 18O- and 16O 58c with a ratio of 87.5%: 12.5%. Thus, confirming that source of oxygen is from the O2 employed. The mechanism of this transformation is analogous to that depicted in Scheme 11.
Scheme 16.
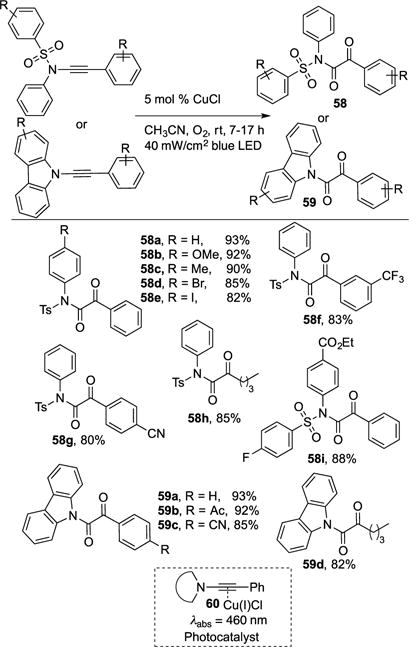
Hwang synthesis of α-ketoimides and α-ketoamides.
Very recently, Hwang reported a regioselective selective aerobic coupling of phenols and terminal alkynes under visible light in the presence of Cu-catalyst.27 The scope of the transformation is very broad. Terminal alkynes possessing aryl, vinyl-, and alkyl groups can be employed, resulting in ketone products 61a-e in good to excellent yields (Scheme 17). The novelty of the authors’ methodology was highlighted by the expeditious synthesis of drugs, pitofenone 62 (72%) and fenofibrate 63 (76%). Both were synthesized in two steps with much more superior overall yield over the state-of-the-art approaches (four steps, 37% for 62 and 71% for 63).28 Control studies revealed that replacing phenol with benzoquinone led to ketone 61a under the reaction conditions, thus implying that benzoquinone is a viable intermediate for the transformation. The radical nature of the transformation was ascertained by the addition of TEMPO to reaction conditions, which resulted in complete inhibition. Also, the quantum yield for the formation of 61a was found to be 0.75, which by the authors’ opinion rules out the possibility of a radical chain mechanism.29 The authors proposed the following mechanism (Scheme 18). Photoexcited Cu–acetylide complex 35 undergoes SET with molecular oxygen to yield superoxide radial anion and the oxidized Cu(II)-acetylide species 64. A Paterno-Buchi [2+2] cycloaddition of the latter with benzoquinone, formed via oxidation of phenol under the reaction conditions, generates oxetene adduct 65. Rapid ring opening of 65 produces acyl-Cu species 66 that undergoes rapid homolysis to form the active Cu(I) catalyst and acyl radical 67. The latter reacts with molecular oxygen to produce 68, followed by radical cleavage and aromatization (68→69), which extrudes CO2, to generate the ketone product 61.
Scheme 17.
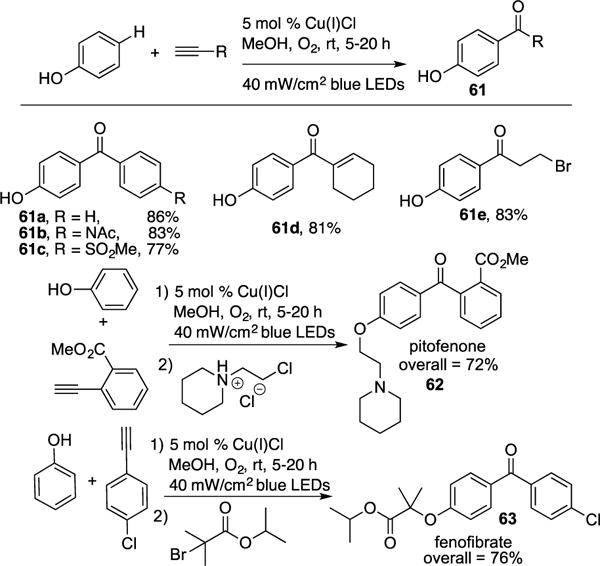
Hwang’s photocatalytic synthesis of aryl ketones.
Scheme 18.
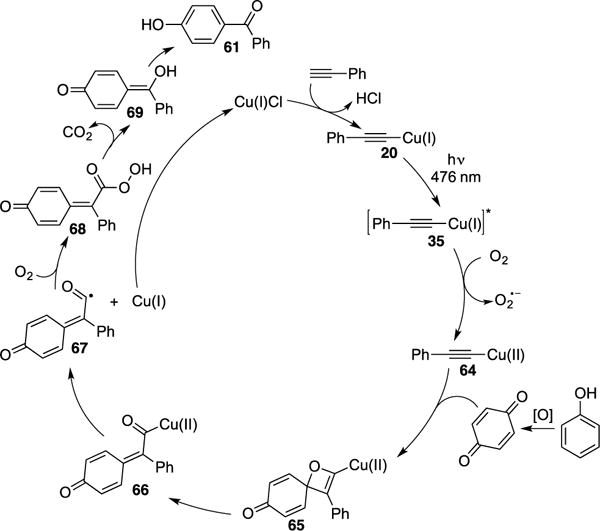
Mechanism of Hwang’s photocatalytic synthesis of aryl ketones.
5. Visible light-induced Pd-catalyzed reactions
5.1. Heck reaction
UV-induced Pd-catalyzed transformations involving alkyl halides have been established by Ryu and co-workers,3d however, reports on the use of visible light with Pd-catalysis are scarce. In 2009, Köhler and co-workers reported the first visible light-induced Heck reaction of bromobenzene with styrenes using simple Pd(OAc)2 catalyst.30 It was found that irradiation with visible light (150 W Xe (OF) lamp with a glass filter) dramatically increases the rate of the Heck reaction (Table 1, entries 1,2). Interestingly, the reaction proceeded without the use of photoactive ligands. The authors speculated that light can accelerate the reduction of Pd(OAc)2 to the active Pd(0) catalyst (Scheme 19, 70→71) via LMCT with the acetate ligands or other organics present in the reaction mixture (i.e. styrene). To probe this hypothesis, the authors added sodium formate, a well know reductant for Pd(II)→Pd(0), to the reaction mixture with and without the influence of light (Table 1, entries 3,4). No change in reaction rate was observed, which the authors interpreted that light irradiation accelerates the elementary reduction step. Further evidence for this was ascertained by 31P NMR studies with addition of PPh3 ligand (Table 1, entries 5,6). The authors reported that under visible light the precatalyst Pd(II)(OAc)2(PPh3)2 was consumed to the active Pd(0) species after 20 minutes, compared to 4.5 hours in the absence of light. Evidently, another reduction step of Pd(II)→Pd(0) occurs in the catalytic cycle of Heck reaction (Scheme 19, 72→71), which may also be accelerated by visible light. The authors tested this possibility by employing Pd(0) catalyst for which the initial reduction step is not applicable. It was found, however, that regardless of the effect of light, similar conversions and yields were obtained (Table 1, entries 7,8). Based on these results, the authors conclude that the sole role of light in this transformation is to accelerate the initial reduction step (Scheme 19, 70→71).
Table 1.
A comparison study of the Heck reaction of bromobenzene with styrene with and without visible light irradiation.

| ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (0.1 %) | Irradiation | Additive (0.2 %) | T (° C) | Conversion (%) | Yield (%) |
| 1 | Pd(OAc)2 | none | none | 140 | 16 | 16 |
| 2 | Pd(OAc)2 | visible | none | 140 | 82 | 69 |
| 3 | Pd(OAc)2 | none | NaOOCH | 120 | 99 | 90 |
| 4 | Pd(OAc)2 | visible | NaOOCH | 120 | 99 | 91 |
| 5 | Pd(OAc)2 | none | PPh3 | 140 | 17 | 16 |
| 6 | Pd(OAc)2 | visible | PPh3 | 140 | 83 | 67 |
| 7 | Pd(PPh3)4 | none | none | 140 | 44 | 21 |
| 8 | Pd(PPh3)4 | visible | none | 140 | 41 | 21 |
Scheme 19.
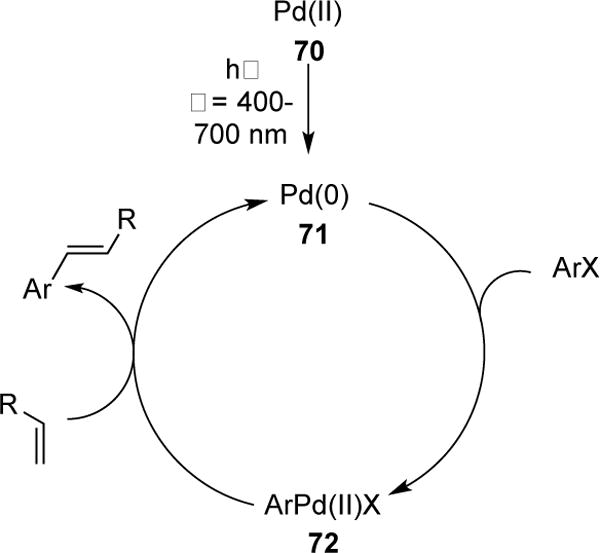
Catalytic Cycle of the photoinduced Heck reaction.
5.2. Remote desaturation reaction
Recently, Gevorgyan and co-workers disclosed a Pd-photocatalytic transformation involving the direct desaturation of silyl ethers 73 into silyl enol ethers 74, Scheme 20.31 Compared to Brookhart’s pioneering work on the direct desaturation of silyl ethers employing Rh/Ir-pincer catalyzed transfer hydrogenation,32 this approach is more general and occurs under milder conditions (room temperature). The authors suggested that the transformation operates via direct formation of a novel hybrid aryl Pd-radical species 75, exhibiting both radical and TM character, that enables a 1,5- hydrogen atom transfer (HAT) event (75→76), which is unprecedented for Pd–catalyzed transformations. Also, the formation and utilization of the hybrid aryl Pd-radical species 75 is noteworthy,33 as they are uncommon intermediates in the Pd(0)-catalyzed reactions with aryl iodides due to a facile outcompeting two electron oxidative addition step to form ArPd(II)I species. The scope of the transformation was found to be quite broad as both cyclic- (74a–e) and acylic silyl enols (74f–i), as well as complex silyl enol substrates (74j–k), reacted smoothly. An interesting retrosynthetic paradox was realized in desaturation of a system containing both protected and unprotected alcohol fragments, where oxidation of protected alcohol was obtained selectively (74l). Also, a double-fold desaturation was conceived; where 74m was obtained in high yield.
Scheme 20.
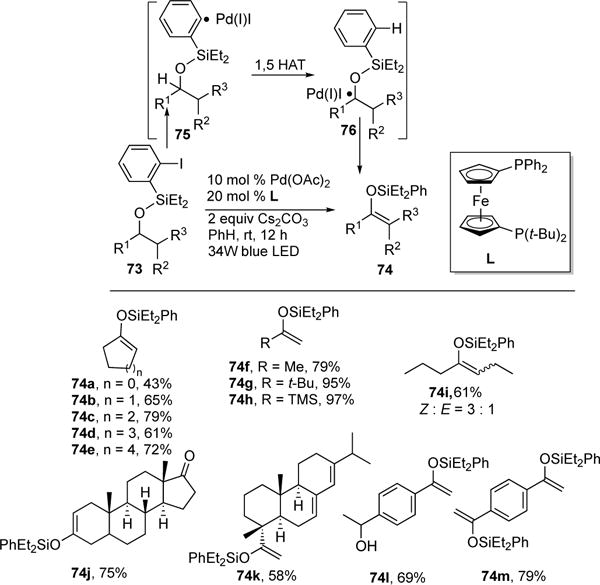
Scope of Gevorgyan’s photoinduced Pd-catalyzed desaturation reaction.
Control studies indicated that both the Pd-catalyst and blue light are essential for the reaction to proceed; under thermal conditions, only hydro-dehalogenation products were obtained. Evidence for a radical mechanism was obtained by radical quenching studies, where addition of TEMPO completely inhibited the reaction. In addition, a radical clock study was conducted, where the regioselectivity of the ring-opening product 77 was found to favor a radical pathway (Scheme 21a).34 The conventional concerted-metalation deprotonation (CMD) pathway35 was ruled out by the results of a deuterium-labeling experiment (78→79, Scheme 21a). The mechanism of this protocol (Scheme 21b) presumes formation of the photoinduced Pd(0), which undergoes SET with silyl ether 73c substrate to generate radical anion 80c and the Pd(I) species. Possible oxidative addition of Pd(0) with 73c into its respective Pd(II) intermediate was disproved through stoichiometric studies. Next, the homolysis of complex 80c furnishes hybrid intermediate 75c, which then generates complex 76c via a 1,5-HAT event. The latter subsequently produces silyl enol ether adduct 74c via recombination of the putative Pd(I)I species to form 81c followed by β–H elimination. Other endgame possibilities such as a direct H–atom abstraction by Pd(I)I, oxidation/elimination, or atom-transfer/HI-elimination have not been ruled out at this stage. It should be mentioned, however, that at this stage the nature of the proposed hybrid-Pd radical intermediates is still unclear.36 Overall, in this transformation, the authors showed the synergistic combination of two different reactivities; a typical for radical chemistry the HAT process, and the traditional for palladium, the β-hydride elimination event, which enabled remote C–H functionalization under benign visible light inducing conditions.
Scheme 21.
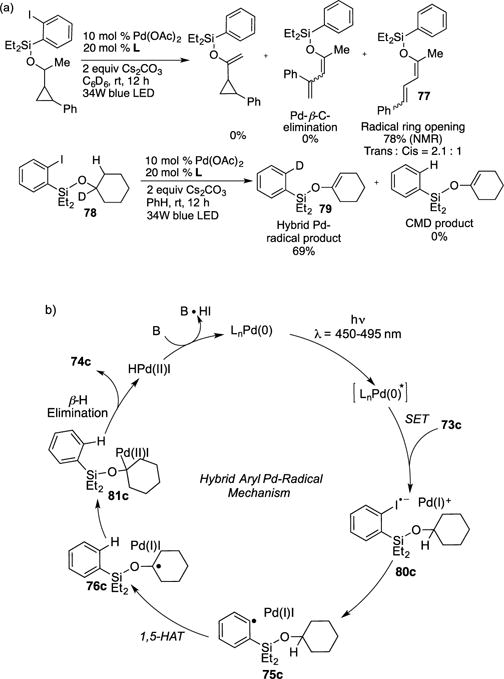
(a) Mechanistic studies for Gevorgyan’s photoinduced Pd-catalyzed desaturation reaction. (b) Proposed catalytic cycle.
6. Visible light-induced Pt-catalyzed reactions
6.1. Photocatalytic evolution of H2
In 2004, Tung and co-workers reported a visible light-induced Pt-catalyzed dehydrogenation of Hantzsch esters (Scheme 22).37 The Pt(II) terpyridyl complex 82 possesses a square-planar geometry, which contains a vacant coordination site for H-atoms to bind. Remarkably, employment of 0.1% of catalyst 82 was required for quantitative conversion of 83 to 84 with ca. 100% yield of H2 produced. For Hantzsch ester substrates, possessing a tertiary alkyl or benzyl group at the 4-position (85), H2 was not obtained under the reaction conditions; instead formation of dealkylation products 86 was observed. No reaction occurred when the 1-postion of the Hantzsch ester was substituted (86c). In addition, when D-1 labeled Hantzsch ester was submitted to the reaction conditions, the rate and quantum yield of the reaction decreased, indicating that abstraction of H-1 occurs first and is rate limiting. Control studies indicated that both light and the photocatalyst are required for this transformation. Analysis of the absorption spectrum of 82 and Hantzsch ester 83 revealed the lowest energy bands in region of 400–520 nm, indicative of MLCT of the Pt catalyst. Stern-Volmer studies supported that the photoexcited Pt-catalyst (82) is quenched by 83 and has a lifetime of 340 ns. Based on these studies, the following mechanism has been proposed (Scheme 23). The Pt catalyst 82 undergoes excitation to form triplet complex 87. The latter undergoes hydrogen atom abstraction of the Hantzsch ester 83 to produce Pt(III)-H and the nitrogen centered radical, complex 88. Then, the latter undergoes disproportionation to produce Pt-H2 complex 89 and pyridine 84. Facile elimination of H2 from complex 89 regenerates the cycle. The authors recently reported the photocatalytic evolution of H2 from 1,3,5-triaryl-2-pyrazolines, 3,4-diaryl-2,5-dihydropyrroles, and 3,4- Diaryl-2,5-dihydrothiophenes under the same reaction conditions.
Scheme 22.
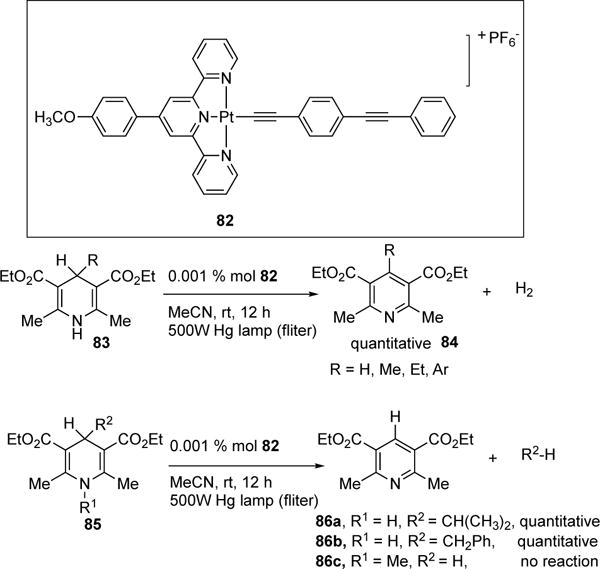
Tung’s Pt-catalyzed photochemical evolution of H2.
Scheme 23.
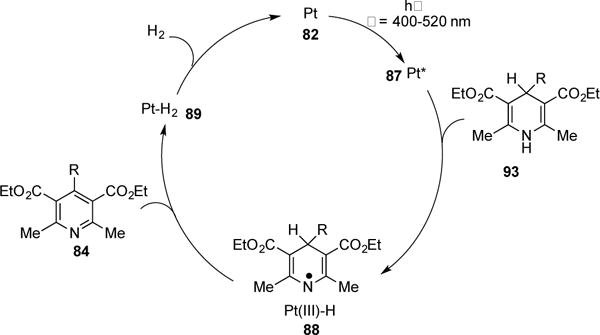
Catalytic cycle of the Pt-catalyzed photochemical evolution of H2.
7. Visible light-induced Au-catalyzed reactions
7.1. Difunctionalization of alkynes
Recently, Hashmi and co-workers reported the first photosensitizer-free visible light-induced Au-catalyzed reaction involving 1,2-difunctionalization of alkynes (Scheme 24a).38 Blue light irradiation of aryl diazonium salts and internal alkynes in the presence of Au(I) catalyst and methanol generated the α-arylated ketone products in good yields. Using other light sources, such as CFLs or UVA lamps, led to lower efficiency. Interestingly, addition of external photosensitizers2 to the reaction mixtures and use of UV-induced dimeric gold species3e resulted in diminished yields. Investigation of the scope of this transformation indicated that substrates bearing both electron releasing- (90b) and withdrawing substituents (90c), as well as sensitive functionalities (90d-e and 90g), were well tolerated. Remarkably, alkyl-substituted alkynes reacted well (90f and 90h). Also, both electron rich (90i) and deficient substituted aryl diazonium salts (90j–k) worked efficiently, however substrate possessing a trifluoromethyl substituent (90l) failed to react due to a rapid decomposition. The authors extended their photoinduced concept toward synthesis of α-aryl-α-fluoro ketones (91) and to other transformations, such as a Meyer-Schuster arylation (93), and Sonogashira cascade arylation (92, Scheme 24b). All of which have been previously reported under dual gold/photoredox catalysis.2 Interestingly, oxyarylation of alkene 94 was performed under photoirradiation with Au-catalyst and resulted in moderate yield of 95 (Scheme 24c). In sharp contrast, Glorius previously reported that under visible light-induced Au-catalysis, oxyarylation of 94 required a PC in order to promote the reaction (96a), without which only 4% of the oxyarylation product (96b) was obtained.39 This discrepancy underscores the importance of the photoinduced reaction components, including ligands, nature of the TM-catalyst, and light sources, which can significantly affect the reaction outcome.
Scheme 24.

(a) Hashmi’s visible light-induced Au-catalyzed 1,2-difunctionalization of alkynes. (b) Further applications of Hashmi’s method. (c) Oxyarylation of alkenes under photoinduced Au-catalysis with (right) and without (left) exogenous photosensitizers.
The performed mechanistic studies indicated that addition of 10 equiv of H2 18O to reaction conditions generates the 18O-incorporated ketone product 97, thus implying a hydrolysis step (Scheme 25a). It was speculated that the transformation may proceed via a ketone intermediate (hydrolysis of the alkyne), however, this possibility was disproven, since no reaction occurred using ketone 98 under the reaction conditions. The authors proposed the following mechanism shown in Scheme 25b. SET from Au(I) complex to the aryl diazonium salt produces Au(II) intermediate 99 and aryldiazonium radical. Recombination of the aforementioned species forms Au(III) intermediate 100, previously reported by Chan and Shi.40 With the influence of light N2 is released, affording the formal oxidative addition adduct 101. The authors mentioned that the efforts to isolate 101 were unsuccessful. Fortunately in their follow-up work,41 Hashmi reported a general access to similar Au(III) complexes via a visible light-induced oxidative addition of aryl diazonium salt with P,N-bidentate ligands and other monodentate ligands, which indirectly confirms 101 as a valid intermediate. Coordination of alkyne substrate with 101 results in formation of complex 102, which upon intermolecular attack by methanol affords vinyl Au complex 103. Reductive elimination of the latter yields alkyl ether 104 and the active catalyst. Subsequent hydrolysis of 104 generates the arylated ketone substrate 90. Notably, Hashmi’s proposed mechanism is distinct from the other photoinduced transformations (vide supra) since it does not involve visible light excitation of the TM-complex. An alternative route toward 101 can be envisioned, where the SET of a photoinduced Au(I) complex 105 with the aryl diazonium salt generates a hybrid Au-radical complex 106. A subsequent recombination would result in intermediate 101.
Scheme 25.

a) Mechanistic studies and b) Proposed mechanism of Hashmi’s visible light-induced Au-catalyzed 1,2-difunctionalization of alkynes.
7.2. C–C cross-coupling reaction
Very recently, the same group reported a visible light-induced Au-catalyzed Suzuki-type cross-coupling of boronic acids and aryl diazonium salts (Scheme 26).42 The scope of the transformation with respect to the boronic acids and aryl diazonium salts substrates was broad, however, substrates possessing an electron-releasing group did not work well (107e–f,l). Notably, both boronic acid-based and aryl diazonium salt-based heterocycles can be utilized, resulting in acceptable yields of their respective cross-coupling adducts (107m–n). Employment of aryl boronic pinacol esters also worked well (107o). Control studies indicated that BF4− counter-ion from the aryl diazonium salts was critical for the success of this reaction. The authors hypothesize that the counter-ion serves as a fluoride source that in situ activates the aryl boronic acid toward a transmetalation event. Also, a quantum yield of 0.302 was obtained, which disfavors the possibility of a radical chain pathway.29 The authors proposed the following mechanism (Scheme 27). The Au(I) catalyst undergoes a SET process with the aryl diazonium salt to form the electrophilic Au(II) complex 99 and the aryl diazo radical. The latter recombines with 99 to generate Au(III) complex 100. A subsequent photoinduced extrusion of N2 from 100 yields Au(III) intermediate 101. Next, transmetalation of the in situ activated boronic acid complex with 101 produces diarylated Au complex 108. Finally, reductive elimination of the latter delivers the desired cross-coupling products (107) and closes the catalytic cycle.
Scheme 26.
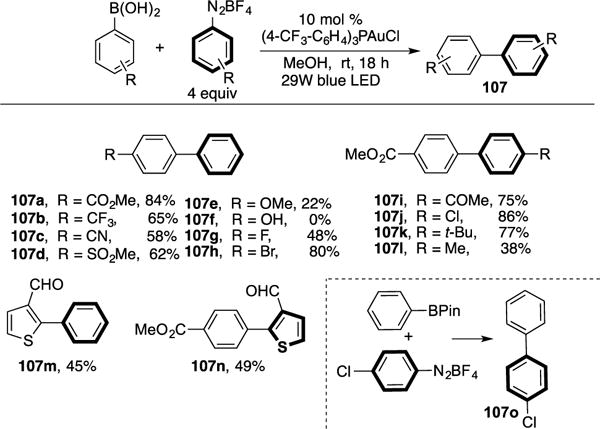
Hashmi’s visible light-induced Au-catalyzed cross-coupling reaction.
Scheme 27.
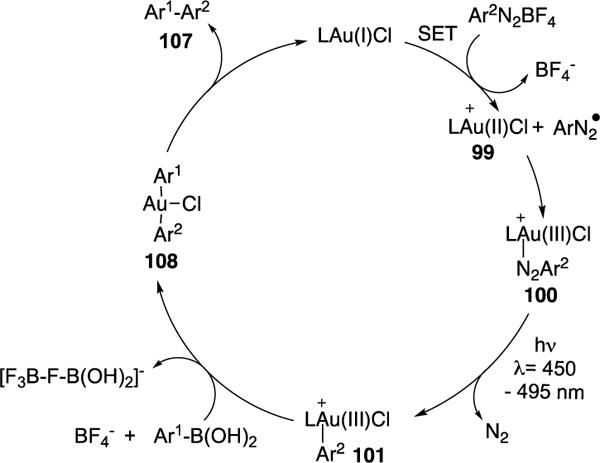
Proposed mechanism of Hashmi’s visible light-induced Au-catalyzed cross-coupling reaction.
8. Conclusions
Compared to the established areas of conventional photocatalysis (Figure 1A) and cooperative/dual photocatalysis (Figure 1B), this emerging area employing unconventional transition metal photocatalysts (Figure 1C) has particular inherent advantages. For instance, this approach allows for the use of a single-TM-catalyst that serves as photo-absorbing species and enables catalysis, thus, making this a cost-effective alternative. Moreover, these transition metal photocatalysts are involved in the bond-forming event via substrate TM-interaction, a rarely seen aspect in prior art. Furthermore, the employment of new unconventional transition metal photocatalysts already has led to the discovery of new reactivity and development of exciting methodology that are, in most cases, unachievable by conventional transition metal catalysis or by photocatalytic means. Seemingly, the fine-tuning of the single TM-photocatalytic systems (Figure 1C) is more difficult compared to that of the cooperative/dual systems (Figure 1B). On the other hand, the former approach does not suffer from a potential compatibility issue of merging two catalytic cycles in one transformation. Future directions of this field may rely on the development and fine-tuning of photoactive ligands, and the utilization of cheap and abundant first row metals, such as Fe, Mn, and Ni, for TM-photocatalysis. Also, further development of enantioselective TM-photocatalytic methods taking advantage of the versatility and abundance of Pd- and Cu-compatible with P- and N-based chiral ligands can greatly enhance this emerging area of photocatalysis. Apparently, for each case, in depth mechanistic studies are required to elucidate the operative mode of activation (radical-chain, charge transfer, electron transfer, etc.), which in turn, could help designing new chemical transformations.
Key Learning Points.
The readers of this review will learn about:
Employment of new photocatalyst for unprecedented synthetic transformations.
Mechanistic details involving unconventional TM (Co, Fe, Cu, Pd, Pt, Au)-based PCs.
Photophysical properties of some of the catalyst employed.
Importance of the photoactive ligands employed for photoinduced synthetic transformations.
Unexplored photoinduced novel asymmetric and C–H functionalization reactions.
Acknowledgments
Financial support from the National Science Foundation (CHE-1663779) and the National Institutes of Health (GM120281) are gratefully acknowledged.
Biographies

Marvin Parasram was born and raised in the Bronx, New York. He received his BS degree in Chemistry from Stony Brook University in 2010. Later that year, he joined Prof. Gevorgyan’s group at the University of Illinois at Chicago as a Ph.D. student. During his Ph.D. studies, he was involved in the development of novel Pd-catalyzed synthetic methodologies.

Vladimir Gevorgyan received his Ph.D. from the Latvian Institute of Organic Synthesis in 1984. After postdoctoral research (1992–1994, JSPS- and Ciba-Geigy International Fellowships) at Tohoku University, Japan, and a visiting professorship (1995) at CNR, Italy, he joined the faculty at Tohoku University. In 1999, Prof. Gevorgyan moved to UIC as an Associate Professor. Since 2012, he was promoted to Distinguished Professor and has won numerous awards such as Honorary Professor of St. Petersburg State University (2012), UIC University Scholar (2012), and Foreign Member of Latvian Academy of Sciences (2016). His group is interested in the development of TM-catalyzed synthetic methodology for C–H functionalization reactions.
Footnotes
Footnotes relating to the title and/or authors should appear here.
Notes and references
- 1.For reviews on reactions involving organometallic photocatalyst, see:; (a) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]; (b) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schultz DM, Yoon TP. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review on asymmetric photoredox catalysis, see:; (d) Meggers E. Chem Commun. 2015;51:3290. doi: 10.1039/c4cc09268f. [DOI] [PubMed] [Google Scholar]; For a review on reactions involving organic photocatalyst, see:; (e) Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 2.For reviews on reactions involving dual/cooperative photocatalysis, see:; (a) Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA. Acc Chem Res. 2016;49:1429. doi: 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skubi KL, Blum TR, Yoon TP. Chem Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lang X, Zhao J, Chen X. Chem Soc Rev. 2016;45:3026. doi: 10.1039/c5cs00659g. [DOI] [PubMed] [Google Scholar]
- 3.For a review on photoinduced (mainly UV) homogenous catalyst featuring Co, Fe, Cr, Cu, Pd, Re, Ru, Ir metals, see:; (a) Hoffmann N. ChemSusChem. 2012;5:352. doi: 10.1002/cssc.201100286. [DOI] [PubMed] [Google Scholar]; For selected examples of UV-induced Cu-catalyzed reactions, see:; (b) Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647. doi: 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]; (c) Ratani TS, Bachman S, Fu GC, Peters JC. J Am Chem Soc. 2015;137:13902. doi: 10.1021/jacs.5b08452. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review on UV-induced Pd-catalyzed reactions, see:; (d) Sumino S, Fusano A, Fukuyama T, Ryu I. Acc Chem Res. 2014;47:1563. doi: 10.1021/ar500035q. [DOI] [PubMed] [Google Scholar]; For a selected example of UV-induced Au-catalyzed reactions, see:; (e) Xie J, Shi S, Zhang T, Mehrkens N, Rudolph M, Hashmi ASK. Angew Chem, Int Ed. 2015;54:6046. doi: 10.1002/anie.201412399. [DOI] [PubMed] [Google Scholar]
- 4.Kisch H. Semiconductor Photocatalysis, Wiley-VCH Verlag GmbH & Co KGaA. 2014:85–210. [Google Scholar]
- 5.Busato S, Tinembart O, Zhang Z-D, Scheffold R. Tetrahedron. 1990;46:3155. [Google Scholar]
- 6.Gupta BD, Roy S. Inorg Chim Acta. 1988;146:209. [Google Scholar]
- 7.Weiss ME, Carreira EM. Angew Chem, Int Ed. 2011;50:11501. doi: 10.1002/anie.201104681. [DOI] [PubMed] [Google Scholar]
- 8.Weiss ME, Kreis LM, Lauber A, Carreira EM. Angew Chem, Int Ed. 2011;50:11125. doi: 10.1002/anie.201105235. [DOI] [PubMed] [Google Scholar]
- 9.Kreis LM, Krautwald S, Pfeiffer N, Martin RE, Carreira EM. Org Lett. 2013;15:1634. doi: 10.1021/ol400410m. [DOI] [PubMed] [Google Scholar]
- 10.Castro LCM, Bézier D, Sortais JB, Darcel C. Adv Synth Catal. 2011;353:1279. [Google Scholar]
- 11.Sagadevan A, Hwang KC. Adv Synth Catal. 2012;354:3421. [Google Scholar]
- 12.Majek M, Jacobi von Wangelin A. Angew Chem, Int Ed. 2013;52:5919. doi: 10.1002/anie.201301843. [DOI] [PubMed] [Google Scholar]
- 13.Sagadevan A, Lyu PC, Hwang KC. Green Chem. 2016;18:4526. [Google Scholar]
- 14.Sheldon RA. Chem Commun. 2008:3352. doi: 10.1039/b803584a. [DOI] [PubMed] [Google Scholar]
- 15.Yin W, He C, Chen M, Zhang H, Lei A. Org Lett. 2009;11:709. doi: 10.1021/ol8027863. [DOI] [PubMed] [Google Scholar]
- 16.Sagadevan A, Charpe VP, Hwang KC. Catal Sci Technol. 2016;6:7688. [Google Scholar]
- 17.Sagadevan, Ragupathi A, Lin C-C, Hwu JR, Hwang KC. Green Chem. 2015;17:1113. [Google Scholar]
- 18.Zhang C, Jiao N. J Am Chem Soc. 2010;132:28. doi: 10.1021/ja908911n. [DOI] [PubMed] [Google Scholar]
- 19.Patel RDJGDV, Webb H, Heather K, Anandan SK, Aavula BR. WO 2008073623. PCT Int Appl. 2008
- 20.Sagadevan A, Ragupathi A, Hwang KC. Photochem Photobiol Sci. 2013;12:2110. doi: 10.1039/c3pp50186h. [DOI] [PubMed] [Google Scholar]
- 21.Sagadevan A, Ragupathi A, Hwang KC. Angew Chem, Int Ed. 2015;54:13896. doi: 10.1002/anie.201506579. [DOI] [PubMed] [Google Scholar]
- 22.Song G, Wang F, Li X. Chem Soc Rev. 2012;41:3651. doi: 10.1039/c2cs15281a. [DOI] [PubMed] [Google Scholar]
- 23.Zhou G, et al. WO CN102241645. PCT Int Appl. 2013
- 24.Kainz QM, Matier CD, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681. doi: 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ragupathi A, Charpe VP, Sagadevan A, Hwang KC. Adv Synth Catal. 2017;359:1138. [Google Scholar]
- 26.Al-Rashid ZF, Johnson WL, Hsung RP, Wei Y, Yao PY, Liu R, Zhao K. J Org Chem. 2008;73:8780. doi: 10.1021/jo8015067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sagadevan A, Charpe VP, Ragupathi A, Hwang KC. J Am Chem Soc. 2017;139:2896. doi: 10.1021/jacs.6b13113. [DOI] [PubMed] [Google Scholar]
- 28.(a) Bal-Tembe S, Blumbach J, Dohadwana A, Punekar NS, Rajgopalan R, Rupp RH, Bickel M. 5821252. M US pat. 1998; (b) Guazzi G. US2004073058(A1) US pat. 2004
- 29.Cismesia MA, Yoon TP. Chem Sci. 2015;6:5426. doi: 10.1039/c5sc02185e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fredricks MA, Drees M, Köhler K. ChemCatChem. 2010;2:1467. [Google Scholar]
- 31.Parasram M, Chuentragool P, Sarkar D, Gevorgyan V. J Am Chem Soc. 2016;138:6340. doi: 10.1021/jacs.6b01628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenges CP, White PS, Brookhart M. J Am Chem Soc. 1999;121:4385. [Google Scholar]
- 33.For an example of formation of aryl radical species with Pd, see:; Manolikakes G, Knochel P. Angew Chem, Int Ed. 2009;48:205. doi: 10.1002/anie.200803730. [DOI] [PubMed] [Google Scholar]
- 34.Satoh T, Miura M. Top Organomet Chem. 2005;14:1. [Google Scholar]
- 35.Kefalidis CE, Davi M, Holstein PM, Clot E, Baudoin O. J Org Chem. 2014;79:11903. doi: 10.1021/jo501610x. and references therein. [DOI] [PubMed] [Google Scholar]
- 36.(a) Liu Q, Dong X, Li J, Xiao J, Dong Y, Liu H. ACS Catal. 2015;5:6111. [Google Scholar]; (b) Parasram M, Iaroshenko VO, Gevorgyan V. J Am Chem Soc. 2014;136:17926. doi: 10.1021/ja5104525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, Wu LZ, Zhou L, Han X, Yang QZ, Zhang LP, Tung CH. J Am Chem Soc. 2004;126:3440. doi: 10.1021/ja037631o. [DOI] [PubMed] [Google Scholar]
- 38.Huang L, Rudolph M, Rominger F, Hashmi ASK. Angew Chem, Int Ed. 2016;55:4808. doi: 10.1002/anie.201511487. [DOI] [PubMed] [Google Scholar]
- 39.Sahoo B, Hopkinson MN, Glorius F. J Am Chem Soc. 2013;135:5505. doi: 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]
- 40.Cai R, Lu M, Aguilera EY, Xi Y, Akhmedov NG, Petersen JL, Chen H, Shi X. Angew Chem, Int Ed. 2015;54:8772. doi: 10.1002/anie.201503546. [DOI] [PubMed] [Google Scholar]
- 41.Huang L, Rominger F, Rudolph M, Hashmi ASK. Chem Commun. 2016;52:6435. doi: 10.1039/c6cc02199a. [DOI] [PubMed] [Google Scholar]
- 42.Witzel S, Xie J, Rudolph M, Hashmi ASK. Adv Synth Catal. 2017;359:1522. [Google Scholar]