

Abstract
Antigen-specific immunotherapy (ASIT) has been used to hyposensitize patients to allergens and offers an enticing approach for attenuating autoimmune diseases. Applying ASIT to mucosal surfaces such as the lungs may engage unique immune response pathways to improve efficacy. Pulmonary delivery of soluble antigen arrays (SAgAs) was explored in mice with experimental autoimmune encephalomyelitis (EAE), a multiple sclerosis model. SAgAs were designed to impede immune response to autoantigen epitopes and are composed of a hyaluronan backbone with peptides PLP139–151 and LABL, a disease-causing proteolipid peptide epitope and an ICAM-1 ligand, respectively. Pulmonary instillation of SAgAs decreased disease score, improved weight gain, and decreased incidence of disease in EAE mice compared to pulmonary delivery of HA polymer, LABL, or PLP. Interestingly, treating with PLP alone also showed some improvement. Splenocytes from SAgA-treated animals showed increased IFN-γ levels, and IL-6 and IL-17 were elevated in SAgA-treated animals compared to PLP treatments. IL-10, IL-2, and TNF-α levels showed no significant difference, yet trends across all cytokines suggested SAgAs induced a very different immune response compared to treatment with PLP alone. This work suggests that co-delivery of peptide components is essential when treating EAE via pulmonary instillation, and the immune response may have shifted towards immune tolerance. a
Keywords: experimental autoimmune encephalomyelitis, pulmonary instillation, pulmonary drug delivery, peptides, macromolecular drug delivery
1. Introduction
Inducing antigen-specific immune tolerance is an emerging avenue of treatment of autoimmune diseases1. The classical approach of antigen specific immunotherapy (ASIT) has been to desensitize the immune system through a series of subcutaneous injections. This approach has generally been applied to foreign antigens (e.g., hyposensitization in the form of “allergy shots”). The treatment regimen, however, requires injections over multiple years and disease improvement is often modest1–5. Similar strategies have been investigated for desensitizing patients to autoantigens including proinsulin peptide for type-1 diabetes6, spliceosomal peptide for Lupus7, and a pathogenic peptide for rheumatoid arthritis8. Alternative delivery approaches have appeared in an effort to improve the efficacy of ASIT. Recently, new products including Ragwitek® and Grastek® utilize sublingual administration to desensitize the immune system to foreign antigens through mucosal surfaces. Treating mucosal surfaces, such as nasal passages or lungs, may also improve performance or reduce the time required for ASIT, a concept that may also extrapolate to autoimmune diseases2,5,9. One particular autoimmune disease where this concept has been applied is multiple sclerosis.
Multiple sclerosis (MS) is an autoimmune disease in which immune cells attack and degrade the myelin sheath surrounding neurons, thus interrupting the transmission of electrical signals3,4,10,11. Many of the current therapies approved for MS focus on treating inflammation or neurological symptoms such as loss of balance, degrees of paralysis, and a loss of coordination. Recently, researchers have aimed to correct the underlying immune response using ASIT4,12,13. There are different forms of this disease: secondary progressive, primary progressive, progressive relapsing, and relapsing-remitting, the latter of which affects a majority of patients, approximately 80%4,10,11. If the autoimmunity is not corrected, patients can experience progressive cycles of remission and relapse, where periodic symptoms are followed by remission, but the patient does not fully recover to the previous baseline, as is experienced in relapsing-remitting MS. Experimental autoimmune encephalomyelitis (EAE), a murine model of relapsing and remitting MS, is induced by a single proteolipid protein epitope (PLP139–151) and serves as a useful model for studying ASIT approaches to autoimmune diseases3,4,14.
Recent research has suggested that the lungs may have a unique role in autoimmunity4,15. For example, the ‘Hub and Spoke’ hypothesis suggests that immune cells traffic through the lungs (‘hub’), acquiring information required to migrate to different parts of the body. Once these immune cells are imbued with this information (‘licensed’), they can pass to the particular ‘spoke’, such as the brain for MS, pancreas for type-1 diabetes, and the large intestine for irritable bowel disease16. In support of this, a recent publication showed that activated immune cells specific to CNS antigens did not elicit CNS symptoms until 5 days after injection when given intravenously. If these same cells were delivered directly into the CNS, no symptoms were observed. If administered directly into the lungs, however, these cells elicited CNS symptoms after approximately one day15,17.
The ‘Hub and Spoke’ hypothesis appears to be especially important for T cells, dictating migration to a particular location in the body by modifying transcription and translation of integrin molecules necessary for homing to target tissues. Inhibitors of tissue-specific integrins have proven to be successful as therapeutics (e.g., TYSABRI®). Thus, interfering with immune cells during licensing in the lungs of EAE mice may be a viable therapeutic approach. Researchers are currently elucidating the immune cell populations passing through the lungs and are tracking cells to determine how these cells affect disease. Modification of the immune response may hinge on the ability of cells to migrate through the lungs and to tissue compartments. Thus, the lungs provide an intriguing route of ASIT administration to influence immune responses throughout the body15–17.
Previous work has suggested that pulmonary delivery of soluble antigen arrays (SAgAs) may provide improved efficacy compared to other routes of administration4. In this paper, we explored ASIT using pulmonary delivery of multivalent SAgAs, which are composed of a hyaluronic acid (HA) polymer backbone with grafted myelin sheath epitope PLP139–151, and a grafted peptide inhibitor (LABL) of intracellular cell-adhesion molecule-1 (ICAM-1), the receptor for leukocyte integrins CD11a/CD183,18. In this paper, SAgAs, its components (HA, LABL, and PLP), and a bifunctional peptide inhibitor (BPI) containing LABL and PLP were compared when delivered to the lungs of mice. Clinical scores and cytokine responses were compared to elucidate mechanisms of inducing immune tolerance in mice with EAE.
2. Materials and methods
2.1. Materials
Hyaluronic acid (HA) molecular weight 16 kDa was purchased from Lifecore Biomedical (Chaska, Minnesota). Aminooxy-LABL (AoLABL) and Aminooxy-PLP (AoPLP) peptides were obtained from PolyPeptide, Inc. (San Diego, CA). Incomplete Freund’s Adjuvant Oil and Mycobacterium tuberculosis were obtained from BD Difco Adjuvants (Franklin Lakes, New Jersey) and pertussis toxin was purchased from List Biological Laboratories, Inc. (Campbell, California). The mouse laryngoscope was purchased from Penn-Century (Wyndmoor, Pennsylvania). All water used was deionized (DI) water from a Labconco Pro PS system. All other chemicals and materials including 3500 Da molecular weight cutoff dialysis tubing, bent fine dissecting forceps, glacial acetic acid, sodium acetate, sodium phosphate monobasic monohydrate, sodium phosphate dibasic, and phosphate buffered saline were purchased from Fisher Scientific (Pittsburgh, Pennsylvania).
2.2. Soluble Antigen Array (SAgA) Synthesis
The purified AoPLP and AoLABL peptides were conjugated onto HA. In 20 mM acetate buffer pH 5.5, a concentration of 2 mg/mL of 16.9 kDa HA was made. AoLABL and AoPLP peptides were added in equal molar proportions, then were added to the 2 mg/mL HA solution using a ratio of 2 HA monomers to 1 Ao-peptide. The reaction was allowed to proceed for 24 hours at room temperature. After 24 hours, the reaction mixture was added to 3500 Da molecular weight cutoff dialysis tubing using DI water as the dialysate for 24 hours with the dialysate being changed every 6 hours. Samples were then frozen and lyophilized for 72 hours at a temperature of −72 °C at a vacuum of <300 millitorr (VirTis Feezemobile-12XL, The Virtis Company, NY).
SAgAs had their peptide loading analyzed using reverse phase high performance liquid chromatography (RP-HPLC) and size exclusion chromatography (SEC). SAgAs were dissolved in 0.1 M HCl pH 1.0 for 4 hours to hydrolyze peptides from the HA backbone. With the HPLC, a calibration curve of free peptide was employed to determine the amount of peptide that was conjugated to the HA for SAgAs, which was compared to the relative size ascertained from SEC. The HPLC method employed was performed using a Waters 2487 dual absorbance detector and a Waters 2796 bioseparation module. The HPLC method employed was a gradient system with mobile phase A composed of 94.9% DI water, 5% acetonitrile, and 0.1% trifluoroacetic acid and mobile phase B composed of 99.9% acetonitrile and 0.1% trifluoroacetic acid. The solvent system was 100% A from 0 to 15 minutes, 85% A/15% B at 15 minutes, 83% A/17% B at 20 minutes, 80% A/20% B at 33 minutes, 75% A/25% B at 42 minutes, 30% A/70% B at 43 minutes, and 100% A at 46 minutes until 60 minutes with a total flow rate of 1 mL/min. A C18 Higgins Analytical Proto200, 5 μm, 200 Å, 250 × 4.6 mm2 column was used with an injection volume of 30 μL using samples with a concentration of 1 mg/mL with wavelength detection at 220 nm.
SEC was employed to determine the increase in size of SAgA from HA due to the conjugation of peptides. The system used included a Waters e2695 separation module, Waters 2414 refractive index detector, and Waters 2489 UV/Vis detector with two columns in series: a PL Aquagel-OH 60 Analytical SEC (300 × 7.5 mm) then a PL Aquagel-OH 40 Analytical SEC (300 × 7.5 mm). The mobile phase employed for SEC was of 0.1 M ammonium acetate with 0.136M sodium chloride at pH 5 using an isocratic system. Samples injected were 80 μL at a concentration of 5 mg/mL with a total flow rate of 0.5 mL/min flow. Samples were dissolved in the mobile phase at a concentration of 5 mg/ml and chromatograms were analyzed using EMPower 3.
The hydrodynamic radius of the samples was determined using ZetaPALS (Brookhaven Instruments Corporation, Holtsville, NY). The samples were all at a concentration of 1 mg/mL and then dissolved in filtered PBS. Each of the samples was filtered once again immediately before the readings were taken. Three readings were taken per sample and then averaged for the values reported in the results (Table 1).
Table 1.
Characterization of treatments.
| Sample | Molar Ratio of LABL:PLP:HA | Approximate MW (kDa) | Dh in PBS (nm) | |
|---|---|---|---|---|
| Hyaluronic Acid (HA) |
|
0:0:1 | 16 | 2.4±1.0 |
| Proteolipid Protein (PLP139–151) |
|
0:10:0 | 1.5 | N/A |
| LABL |
|
10:0:0 | 1.0 | N/A |
| Soluble Antigen Array (SAgA) |
|
7.5:5.8:1 | 33 | 3.7±1.6 |
| Bifunctional Peptide Inhibitor (BPI) |
|
1:1:0 | 3.0 | N/A |
2.3. Animals
SJL/J mice (female, 4 weeks old) were supplied by Harlan Laboratories (Indianapolis, IN) for the study. Mice were approximately 13–16 g and were weighed throughout the course of the experiment. The mice were housed in a pathogen-free facility at the University of Kansas approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Mice were maintained on a 12 hour light/dark cycle and were fed standard rodent pellets with no dietary restrictions or withheld food with water ad libitum. All animal experiments were approved by the University of Kansas Institutional Animal Care and Use Committee (IACUC).
2.4. Induction of Experimental Autoimmune Encephalomyelitis (EAE) and Time Course of Study
Each SJL/J mouse had EAE induced on study day 0 with 4 injections of an emulsion of Incomplete Freund’s Adjuvant Oil, Mycobacterium tuberculosis, and 200 nmol of PLP. Each of the 4 injections was 50 μL given subcutaneously: 2 above the shoulder blades and 2 above the rear hip haunches. Each SJL/J mouse had 100 μL of 200 ng pertussis toxin injected intraperitoneally on day 0 as well as day 2. There were 6 mice in each of the groups split into 2 cages of 3 mice.
Over the course of the 25-day study, each mouse was weighed and given a clinical score from study day 7 to 25. The clinical scoring ranged from 0 to 5 with half integer values and a higher score correlated to an increase in disease progression. The advancement of the scoring was as follows: 0 corresponded to no symptoms of disease; 1 with a limp tail and waddling gait; 2 with partial leg paralysis and increased alteration of gait; 3 with paraplegia or complete hind leg paralysis; 4 with partial front leg paralysis; and 5 with complete front leg paralysis or moribund. Half integer values corresponded with disease progression between scoring levels3,4.
2.5. Treatment Schedule and Pulmonary Instillation of Compounds
Treatment was administered on study days 4, 7, and 10. Before treatment, each of the study compounds was dissolved in sterile phosphate buffered saline (PBS). The study groups were PBS, 16 kDa HA, LABL, PLP, SAgA, and bifunctional peptide inhibitor (BPI). Each animal had 50 μL of each compound corresponding to 200 nmol with the exception of BPI, which was 150 nmol administered via pulmonary instillation as described in details below3,4. The dose ranges of 16 kDa HA, PLP, LABL, SAgA, and BPI, respectively, were the following: 35.5 – 39.1 mg/kg, 16.6 – 20.3 mg/kg, 10.7 – 12.1 mg/kg, 65.1 – 74.5 mg/kg, and 24.7 – 29.4 mg/kg. All the statistical analyses were performed on Prism GraphPad 5 software, using ANOVA analysis. Tukey’s HSD post-hoc analysis was applied to the data.
For lung administration via pulmonary instillation, each animal was anesthetized with 2% isoflurane in an induction chamber for approximately 4 minutes. After the mouse was fully anesthetized, the mouse was positioned in dorsal recumbency using a dosing board at approximately 60° to a supine posit ion suspended by incisor teeth using a thin wire. A nose cone was used to maintain anesthesia. The mouth was opened and the tongue was gently pulled out and to the side of the mouth. A laryngoscope was then positioned to depress the tongue and visualize the vocal cords at the top of the trachea. A 50 μL solution of treatment was then pipetted at the top of the trachea. The tongue was withheld for at least 3 breaths, after which time the mouse was maintained under anesthesia for an additional 3 minutes on the dosing board. The mouse was then removed from the dosing board and allowed to recover from the anesthesia by being held vertically until movement was regained19.
2.6. Collection and Culturing of Splenocytes
The mice were euthanized via isoflurane overdose. Spleens were harvested under sterile conditions and were immediately transferred to a falcon tube with 5 mL sterile RPMI media with 1% Penicillin-Streptomycin (P/S). Within 30 minutes, the spleens were mashed under sterile conditions using a wire mesh in a petri dish and the rubber end of a 1-mL syringe. A 5-mL stripette was used to transfer the cell solution to a 15-mL falcon tube. The splenocytes were centrifuged at 3500 rpm for 4.5 minutes. The old media was decanted and the cells were resuspended in 1 mL of RPMI with 10% fetal bovine serum (FBS) and 1% P/S.
A fresh solution of Gey’s Lysis solution was made and 3.5 mL was added to the splenocytes. The cells were set on ice for 3.5 minutes to lyse the red blood cells. 10 mL RPMI with 10% FBS and 1% P/S was added to the cells. They were centrifuged once again at 3500 rpm for 4.5 minutes. The media was decanted and the cells were resuspended in 5 mL of fresh RPMI with 10% FBS and 1% P/S. The cells were counted using a hemocytometer and were plated at 5 × 106 cells per well at 500 μL per well in a 24-well plate. The splenocytes were incubated for 120 hours before the supernatant containing cytokines was collected and a resazurin assay was conducted.
2.7. Determination of Cytokine Profiles by ELISA
ELISAs from R&D Systems (Minneapolis, MN) were used to determine the following cytokine levels (IFN-γ, TNF-α, IL-2, IL-6, IL-10, and IL-17). A 100 μL aliquot of the collected supernatant was used for each of the ELISAs except for IFN-γ and IL-17, where the samples were diluted 2:5 using RPMI media with 10% FBS and 1% P/S. Dilution of IFN-γ and IL-17 samples was necessary to allow the samples to fall in the linear range of standards provided by the manufacturer. The ELISA was read using a SpectraMax plate reader (Molecular Devices, LLC, Sunnyvale, CA) at an absorbance of 450 nm. The protocol for each of the ELISAs was provided by R&D Systems.
2.8. Lung Histology
Mice were euthanized at the end of the study as described previously. After the spleen was excised from the mouse, the lungs were then removed. An incision was made in the ribcage to expose the trachea and lungs. A 10% formalin solution was injected into the trachea down into the lungs via a syringe until inflation was observed via visual inspection, which typically required 3 mL. The lungs were tied off using nylon sutures to keep the lungs inflated with 10% formalin and were suspended in 10% formalin in a 50 mL test tube. Enough formalin was used to cover the lungs and the test tube was inverted to maintain the lungs in formalin. After 24 hours, the remaining tissue was removed from the lungs, leaving just the lungs, which were then split into right and left and were placed in a histology cassette in 70% ethanol. After 72 hours, the 70% ethanol was replaced to remove excess red blood cells from solution.
For histology, the mice lungs were sectioned and submitted to the Pathology and Anatomic and Clinical pathology department at the Lawrence Memorial Hospital for formalin fixation. Briefly, the specimens were grossed and submitted in plastic cassettes. The cassettes then underwent additional processing within a Tissue-tek VIP Tissue Processor, at a temperature of 10° – 40° C. The tis sue went through further formalin fixation followed by 70% alcohol, 95% alcohol, 100% alcohol, and xylene. The tissue was then embedded in paraffin wax at 62° C.
The paraffin embedded tissue was then sectioned with a microtome at four microns per section. These slides containing both the paraffin and tissue were then placed into an oven to remove excess paraffin wax. After the slides and tissue were cooled, they were stained with hematoxylin and eosin. A cover slip was placed over the stained tissue samples allowing the tissue to be evaluated by Michael Thompson, M.D. of the Pathology and Anatomic and Clinical Pathology department at the Lawrence Memorial Hospital, who was blinded to the treatment groups.
3. Results
3.1. Characterization of SAgAs
Reverse phase chromatography was utilized to determine the amount of peptide conjugated to HA. Based on the RP-HPLC results, there were approximately 5 to 6 PLP peptides and 7 to 8 LABL peptides conjugated per polymer chain. By determining the number of peptides conjugated, the approximate average molecular weight was calculated using both SEC and HPLC results (Table 1). The ZetaPALS readings for hydrodynamic radii are also reported in Table 1. The hydrodynamic radius of SAgA was larger than that of HA, which was most likely due to the addition of peptides to HA.
3.2. Efficacy of Co-Delivered Peptides
The co-delivery of peptides using SAgAs was necessary for ameliorating EAE symptoms. The mice were treated on study days 4, 7, and 10, as was established in earlier publications4,12,13,20–23. The PBS group had high clinical scores – in which high clinical scores depicted disease progression – that were similar to the group treated with LABL (Figure 1). Both groups had scores that increased as of day 10 with the PBS group having the highest scores between days 12–15 and peaking on 14, while LABL peaked slightly earlier at day 12. Scores for mice administered PBS or LABL remained high throughout the course of the study, ending with scores of approximately 1.0. The HA group had a slightly delayed onset of disease as disease was observed at day 11, peaking on day 14, and decreasing to near baseline values by day 18. The PLP group had similar scores to the BPI group with scores remaining around 0.5 after disease onset. The SAgA group had low scores throughout the length of the study with scores ranging between approximately 0 and 0.25, but were not statistically different from PLP and BPI. The statistical analysis of the clinical scores is reported in Supplemental Table 1.
Figure 1.

Clinical scores throughout the study from each of the 6 groups: PBS control, HA, LABL, PLP, SAgA, and BPI. Panel A depicts all 6 groups; Panel B shows PBS, HA, and SAgA (* p<0.05 for 16kD HA vs PBS Control; # p<0.05 for 16kD HA vs SAgA); Panel C represents PBS, PLP, and SAgA (* p<0.05 for PLP vs PBS Control); and Panel D depicts PBS, LABL, and SAgA (# p<0.05 for LABL vs SAgA). Each point is the average of 6 animals with the standard deviation within each group.
The area under the curve (AUC) from clinical scores in Figure 1 provides a holistic view of the disease state of EAE mice (Figure 2). The PBS group had the highest AUC with LABL being similar. HA showed a decreased AUC compared to LABL and PBS but was higher compared to both PLP and BPI treatments. BPI and PLP were quite similar, while SAgA had the lowest AUC value, although not statistically different from the BPI treatment.
Figure 2.
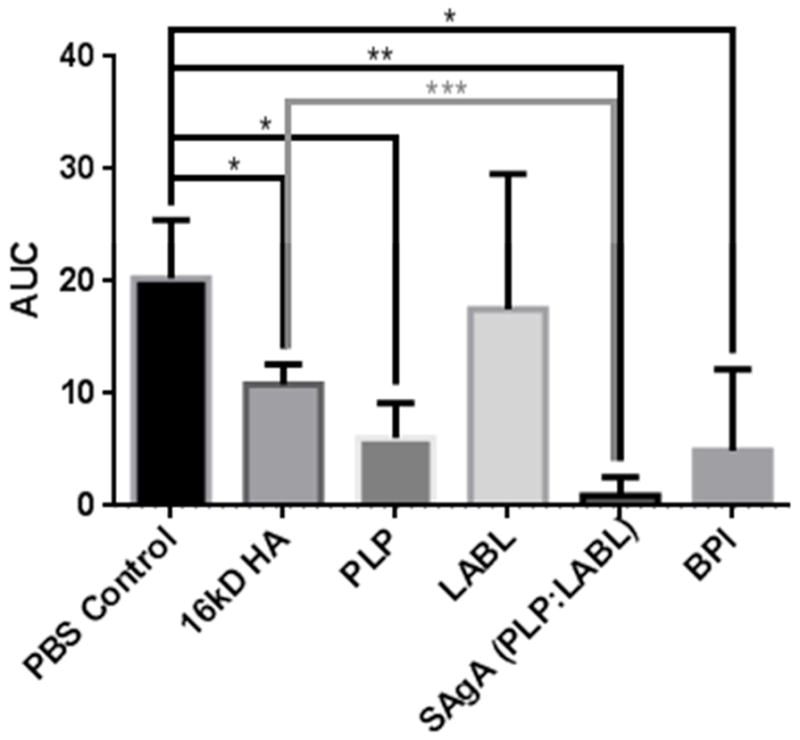
Area under the curve (AUC) of clinical scores throughout the study from each of the 6 groups. Each group is the aggregate total of the scores from each of the 6 animals. A one-way ANOVA test showed significances between the PBS control group and other groups and SAgA-treated group and other groups. * p<0.05, ** p<0.01, *** p<0.001
The incidence of disease was also determined (Figure 3). Mice are considered ‘diseased’ when progressing to a score of 1.0 or higher, corresponding to symptoms affecting the rear legs as well as the tail. The PBS, HA, and LABL groups had a rapid progression to disease. All HA and PBS-treated animals were sick by day 14 and all LABL-treated animals by day 17. The PLP group had only 1 animal remain disease free throughout the course of the study. The BPI group had 4 of 6 animals remain disease free, while the SAgA group had 5 of the 6 animals remain disease free.
Figure 3.

Incidence of disease, represented by a clinical score of 1.0, throughout the study from each of the 6 groups (PBS control, HA, LABL, PLP, SAgA, and BPI).
Changes in the weights of animals were also assessed throughout the study (Figure 4). The weight of the animal decreases around the onset of disease due to decreased mobility of the animal and general decline in health. Typically, animal weight begins to decrease around day 10 and as the disease resolves, will gradually increase. The PBS, LABL, and HA groups had similar decreases in weight starting on day 10 with the greatest decrease in weight occurring on days 14 and 15, corresponding to peak of disease. All three groups had similar weights by the end of the study, recovering to levels similar to the starting weights. The BPI and PLP groups were quite similar with decreases in weight from day 11 to 18 but with less weight loss compared to PBS, HA, and LABL groups. The SAgA group had almost continuous weight gain throughout the course of the study ending with ~10% weight gain on average. The statistical analysis of the animals’ change in weight is reported in Supplemental Table 1.
Figure 4.

Change in weight based on normalization to the initial weight on Day 0 throughout the study with all 6 groups (PBS control, HA, LABL, PLP, SAgA, and BPI). Panel A depicts all 6 groups; Panel B shows PBS, HA, and SAgA (* p<0.05 for 16kD HA vs PBS Control; # p<0.05 for 16kD HA vs SAgA); Panel C represents PBS, PLP, and SAgA (* p<0.05 for PLP vs PBS Control; # p<0.05 for PLP vs SAgA); and Panel D depicts PBS, LABL, and SAgA (* p<0.05 for LABL vs SAgA). Each point is the average of 6 animals with the standard deviation within each group.
3.3. Cytokine Profiles from Splenocytes
Splenocytes were harvested from treated and control animals at the end of the study. Splenocytes were exposed to media with or without PLP and the cytokine response was determined. IFN-γ levels were significantly higher in splenocytes derived from animals treated with SAgAs compared to animals that were dosed with the individual components (Figure 5). IL-6 and IL-17 had significantly higher levels in splenocytes from animals treated with SAgAs compared to animals that only received PLP antigen. IL-2 had lower levels in animals treated with PLP, SAgA, and BPI compared to animals dosed with PBS, HA, or LABL. IL-10 and TNF-α levels in the SAgA and BPI groups trended slightly higher than other groups, but there was no statistical significance.
Figure 5.

Cytokine profiles with IFN-γ in Panel A, IL-10 in Panel B, IL-2 in Panel C, IL-6 in Panel D, IL-17 in Panel E, and TNF-α in Panel E. * p<0.05, ** p<0.01, *** p<0.001. Significance for BPI comparisons are not reported.
3.4. Lung Histology
Lung histology highlighted some intriguing trends, but it was difficult to draw firm conclusions based on the heterogeneity of the results within and between groups (Figures 6 and 7). Inflammation from the lungs was scored by differentiating into three distinct categories of mild, moderate, and severe inflammation, as is seen in Zhornitsky et al24. The treatment groups of PBS, LABL, and HA had a wide range of scoring with animals ranging from no inflammation (0) and mild focal inflammation (1) to severe diffuse inflammation with granulomas (6). Due to the wide range of scores, a quantitative analysis showed no statistical difference (Supplemental Table 2 and Figure 7). The treatment groups of SAgA and BPI had lower cumulative scores across the groups with maximum lung inflammation of 2 still qualifying as mild. The remaining group of PLP was more similar to the SAgA and BPI groups with a lower average and with most of the animals having 0 or 1.
Figure 6.

Representative histology of lungs from a mouse from each group. Each image was taken at 100X magnification and was performed after staining and fixing of tissue. Panel A is from the PBS group and shows severe inflammation; B is from the HA group and shows mild inflammation; C is from the LABL group and shows moderate inflammation; D is from the PLP group and shows moderate inflammation; E is from the SAgA group and shows no inflammation; F is from the BPI group and shows mild inflammation.
Figure 7.
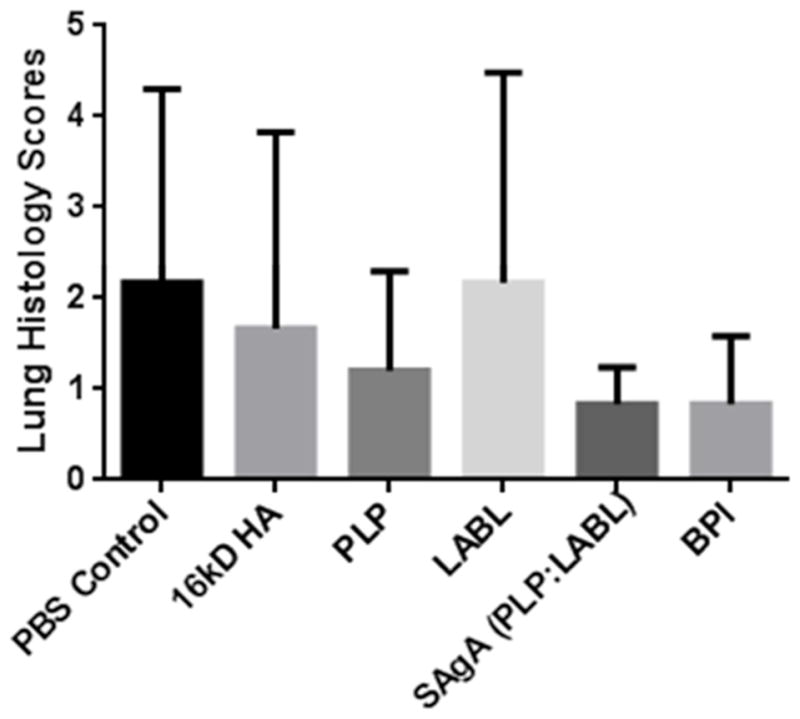
A summary of the scores given to tissues in Supplemental Table 2. These scores represent the level of inflammation of the tissues. After a one-way ANOVA statistical analysis, there was no significance seen between each of the groups.
4. Discussion
4.1. The Lungs and Immune Tolerance
Antigen-specific immunotherapies in the form of ‘allergy shots’ are traditionally administered via subcutaneous injections13,20,21. Injections of antigens can potentially transport by two mechanisms: via diffusion from the injection site to regional lymph nodes or into systemic circulation or via active cellular transport to lymph nodes1–5. Mucosal delivery, including buccal (e.g., Grastek®), nasal, and pulmonary delivery, offers a convenient route of administration and recent studies have suggested the potential for improved tolerization compared to subcutaneous injection2,5,9,25. Mucosal surfaces serve as the primary barrier to entry and are constantly inundated with foreign pathogens. These surfaces have their own mucosa-associated lymphoid tissue, which includes specialized epithelial cells that can take up antigens and direct the antigens to antigen presenting cells to elicit an immune response2,9. More recently, however, additional immunological mechanisms of the lungs have emerged.
4.3. Potential Immune Modulation Mechanism for SAgAs
Pulmonary delivery of SAgA components indicated the need for co-delivery of the PLP antigen epitope with LABL peptide to ameliorate EAE in mice. HA has some inherent immune activity as it can bind to CD44 on immune cells yet no therapeutic efficacy was observed with the delivery of HA in control studies. The delivery of LABL also did not change the disease state suggesting blocking ICAM-1, a known mediator of immune cell adhesion and co-stimulation, was not therapeutic. It is possible that LABL was quickly eliminated from the lung tissue via clearance or absorption or that LABL was digested quickly by peptidases in the lungs. Treating with PLP alone had increased efficacy based on the AUC of the clinical scores, but most EAE mice treated with PLP became sick. Animals treated with PLP had a delayed onset of disease and exhibited decreased scores compared to HA and LABL hinting that PLP may function as conventional ASIT. If this is the case, repeated, escalating doses may hyposensitize EAE mice to PLP. Ultimately, the co-delivery of the peptides practically eliminated disease as seen with both BPI and SAgA suggesting that efficacy depends on coincident delivery of both PLP and LABL.
4.4. Cellular and Cytokine Response in EAE with SAgA Treatment
Cytokine profiles provide some insight regarding possible mechanisms of action for SAgA therapy. Some cytokines have been classically considered to be pro-inflammatory or anti-inflammatory, but with further study the picture becomes more complex. For example, IFN-γ has traditionally been identified as a deleterious inflammatory response when elevated in autoimmune diseases26,27. New studies, however, have shown that local, elevated levels of IFN-γ may not always correspond to inflammatory responses22.
Cytokine profiles differ kinetically throughout the course of the disease and spatially depending on the tissue compartment. Circulating cytokines will differ from cytokines gathered from splenocytes, which are also different from cytokines expressed in affected tissues (e.g., MS brain lesions). We observed a pronounced IFN-γ response in splenocytes from mice that received SAgA treatment, which is similar to the response previously seen in treatment with BPI28. Splenocytes were collected at day 25, during remission of EAE, thus, PLP-specific T cells may have returned to the spleen, which caused the spike in IFN-γ levels. In related studies, Kobayashi et al. reported the levels of IFN-γ seem to be significantly higher in the group of mice treated with PLP-BPI in comparison to the PBS group20,22. The levels of IFN-γ were significantly higher in mice treated with SAgAs than that of mice treated with PBS, 16kD HA, PLP, or LABL. Following the trends seen with Kobayashi et al., it is possible that the increased levels of IFN-γ from splenocytes corresponded to improved treatment efficacy.
IL-6 responses from splenocytes were also high in mice treated with SAgA. IL-6 may cause EAE to progress, suppressing Fox3P+ Treg cells, causing a pro-inflammatory response29. Similar to the rationale for IFN-γ, IL-6-producing cells may have returned to the spleen during remission, instead of being present in the CNS. A similar rationale may apply to the observed increase in IL-17. The increased levels of IFN-γ and IL-6 may also suggest antigen-specific B cells recognizing PLP were present in the spleen. Their activity as antigen presenting cells (APCs) may have been inhibited by SAgAs in vivo, but these cells responded to PLP when isolated and rechallenged with PLP ex vivo30.
The IL-2, IL-10, and TNF-α results in Figure 5 were not statistically different. IL-2 is known for differentiation of T cells, and is typically pro-inflammatory, as shown by Saskida, et al., suggesting this cytokine should increase as did IFN-γ31. But, no change was seen in these cytokines, as we have shown in literature3,28. All three cytokine levels, however, seem to be similar to previous studies21. One major reason for the variation in cytokine levels reported in literature is because of comparisons made between differing time points of the study. Cytokines in the spleen will differ at the point before disease symptoms were prominent, at the peak of disease, and at the end of the study, when symptoms decrease. Assays of cytokines secreted from splenocytes also use time points from 0 hours to 120 hours after starting the primary splenocyte culture. Future studies should compare cytokine levels in different tissue samples over the course of the study, as well as compare cytokines collected at varying time points after the primary splenocyte culture is established.
Some of the results may be explained by looking at potential immune interactions of SAgAs compared to earlier studies of BPI treatment. Like SAgA, BPI is also composed of the PLP peptide and the LABL peptide, but with a short linker connecting the two peptides. When antigen presenting cells such as dendritic cells interact with T cells, they form an immunological synapse where the cell surface receptors coalesce into a distinct pattern32,33. It was previously shown that a BPI molecule called GAD-BPI with glutamic acid decarboxylate (GAD) peptide antigen could effectively colocalize both MHC-II and ICAM-1 on the surface of APCs from NOD mice compared to a mixture of GAD peptide and LABL peptide34. The results suggested that GAD-BPI molecule could bind simultaneously to MHC-II and ICAM-1 on the surface of APCs. As a result, the BPI molecule prevented clustering of TCR/MHC-II-Ag and LFA-1/ICAM-1 inhibited formation of the immunological synapse at the interface between T cells and APCs35. Blocking the formation of the immunological synapse can alter the differentiation and clonal expansion of T cells.
Besides possible cytokine and cellular mechanisms, transport of SAgA molecules may be substantially different than BPI and control peptides. Previous work on SAgAs also reported a range of physical sizes as determined by the HA polymer size and the amount of peptide conjugated. The size of SAgA constructs has typically ranged from 3–10 nm4. This size range may facilitate access to different physiological compartments as the largest of these, 10 nm, can potentially be excluded or slowed from absorption into systemic circulation and passively drain to the lymphatics36–38. Active binding to immune cells and transport to secondary lymphoid organs may be a likely mechanism of SAgA efficacy, which could favor smaller, more mobile SAgAs or BPI. Future studies identifying the local and systemic biodistribution of SAgAs would help define potential mechanisms when comparing SAgAs to conventional ASIT.
5. Conclusion
Restoring immune tolerance in autoimmune diseases may one day reverse the root cause of disease. ASIT has been used for decades to desensitize patients to allergens, but similar approaches applied to autoimmune diseases have yet to achieve robust clinical success. Traditional allergy desensitization strategies have employed injections, while new approaches have achieved immune tolerance by applying ASIT to mucosal membranes (e.g., Grastek® and Ragwitek®), suggesting the lungs may also be a viable target tissue. SAgA molecules exhibiting grafted antigen and a grafted inhibitor of immune cell adhesion may amplify the effect of traditional ASIT. SAgAs with grafted PLP and LABL delivered into the lungs of EAE mice showed increased efficacy based on clinical scoring, weight changes, and incidence of disease compared to the individual components. The cytokine response of splenocytes indicated a shift towards increased levels of IFN-γ when treated with SAgA compared to the individual components and elevated IL-6 and IL-17 with SAgA compared to PLP. Cytokines, such as IL-10, IL-2, and TNF-α showed no significant difference and yet co-delivery of PLP and LABL using SAgAs or BPI provided a potent ASIT when delivered to the lungs. The distinct mechanisms of these therapeutics merits further exploration.
Supplementary Material
Acknowledgments
This work was supported by the NIH (R56 AI091996), PhRMA Foundation Pre-Doctoral Fellowship, and University of Kansas Department of Pharmaceutical Chemistry. Christopher Kuehl acknowledges receiving financial support provided by Dynamic Aspects of Chemical Biology Training Grant (T32 GM08545).
Footnotes
Antigen-specific immunotherapy (ASIT), soluble antigen arrays (SAgAs), experimental autoimmune encephalomyelitis (EAE), multiple sclerosis (MS), proteolipid protein (PLP), hyaluronic acid (HA), intracellular cell-adhesion molecule-1 (ICAM-1), bifunctional peptide inhibitor (BPI), reverse phase high performance liquid chromatography (RP HPLC), size exclusion chromatography (SEC), phosphate buffered saline (PBS), penicillin-streptomycin (P/S), fetal bovine serum (FBS), area under the curve (AUC), antigen presenting cells (APCs), glutamic acid decarboxalate (GAD)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Current opinion in immunology. 2010;22(5):609–615. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nature medicine. 2005;11(4 Suppl):S45–53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 3.Sestak JO, Sullivan BP, Thati S, Northrup L, Hartwell B, Antunez L, Forrest ML, Vines CM, Siahaan TJ, Berkland C. Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Molecular Therapy - Methods & Clinical Development. 2014 doi: 10.1038/mtm.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thati S, Kuehl C, Hartwell B, Sestak J, Siahaan T, Forrest ML, Berkland C. Routes of administration and dose optimization of soluble antigen arrays in mice with experimental autoimmune encephalomyelitis. Journal of pharmaceutical sciences. 2015;104(2):714–721. doi: 10.1002/jps.24272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aasbjerg K, Backer V, Lund G, Holm J, Nielsen NC, Holse M, Wagtmann VR, Wurtzen PA. Immunological comparison of allergen immunotherapy tablet treatment and subcutaneous immunotherapy against grass allergy. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2014;44(3):417–428. doi: 10.1111/cea.12241. [DOI] [PubMed] [Google Scholar]
- 6.Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, Van-Krinks C, Lozanoska-Ochser B, Marquesini L, Brown S, Wong FS, Dayan CM, Peakman M. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clinical and experimental immunology. 2009;155(2):156–165. doi: 10.1111/j.1365-2249.2008.03814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller S, Monneaux F, Schall N, Rashkov RK, Oparanov BA, Wiesel P, Geiger JM, Zimmer R. Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: results of an early phase II clinical trial. Arthritis and rheumatism. 2008;58(12):3873–3883. doi: 10.1002/art.24027. [DOI] [PubMed] [Google Scholar]
- 8.Koffeman EC, Genovese M, Amox D, Keogh E, Santana E, Matteson EL, Kavanaugh A, Molitor JA, Schiff MH, Posever JO, Bathon JM, Kivitz AJ, Samodal R, Belardi F, Dennehey C, van den Broek T, van Wijk F, Zhang X, Zieseniss P, Le T, Prakken BA, Cutter GC, Albani S. Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis and rheumatism. 2009;60(11):3207–3216. doi: 10.1002/art.24916. [DOI] [PubMed] [Google Scholar]
- 9.Ye YL, Chuang YH, Chiang BL. Strategies of mucosal immunotherapy for allergic diseases. Cellular & molecular immunology. 2011;8(6):453–461. doi: 10.1038/cmi.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nature reviews Immunology. 2009;9(6):440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- 11.Steinman L. Immunology of relapse and remission in multiple sclerosis. Annual review of immunology. 2014;32:257–281. doi: 10.1146/annurev-immunol-032713-120227. [DOI] [PubMed] [Google Scholar]
- 12.Buyuktimkin B, Manikwar P, Kiptoo PK, Badawi AH, Stewart JM, Jr, Siahaan TJ. Vaccinelike and prophylactic treatments of EAE with novel I-domain antigen conjugates (IDAC): targeting multiple antigenic peptides to APC. Molecular pharmaceutics. 2013;10(1):297–306. doi: 10.1021/mp300440x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sestak J, Mullins M, Northrup L, Thati S, Forrest ML, Siahaan TJ, Berkland C. Single-step grafting of aminooxy-peptides to hyaluronan: a simple approach to multifunctional therapeutics for experimental autoimmune encephalomyelitis. Journal of controlled release: official journal of the Controlled Release Society. 2013;168(3):334–340. doi: 10.1016/j.jconrel.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annual review of immunology. 2002;20:101–123. doi: 10.1146/annurev.immunol.20.081701.141316. [DOI] [PubMed] [Google Scholar]
- 15.Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schlager C, Lodygin D, Heckelsmiller K, Nietfeld W, Ellwart J, Klinkert WE, Lottaz C, Nosov M, Brinkmann V, Spang R, Lehrach H, Vingron M, Wekerle H, Flugel-Koch C, Flugel A. T cells become licensed in the lung to enter the central nervous system. Nature. 2012;488(7413):675–679. doi: 10.1038/nature11337. [DOI] [PubMed] [Google Scholar]
- 16.Steinman L. ‘Hub-and-spoke’ T cell traffic in autoimmunity. Nat Med. 2013;19(2):139–141. doi: 10.1038/nm.3088. [DOI] [PubMed] [Google Scholar]
- 17.Ransohoff RM. Immunology: Licensed in the lungs. Nature. 2012;488(7413):595–596. doi: 10.1038/488595a. [DOI] [PubMed] [Google Scholar]
- 18.Cheng CY, Lee YC. Anti-Inflammatory Effects of Traditional Chinese Medicines against Ischemic Injury in In Vivo Models of Cerebral Ischemia. Evid Based Complement Alternat Med. 2016;2016:16. doi: 10.1155/2016/5739434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rayamajhi M, Redente EF, Condon TV, Gonzalez-Juarrero M, Riches DW, Lenz LL. Non-surgical intratracheal instillation of mice with analysis of lungs and lung draining lymph nodes by flow cytometry. Journal of visualized experiments: JoVE. 2011;(51) doi: 10.3791/2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ridwan R, Kiptoo P, Kobayashi N, Weir S, Hughes M, Williams T, Soegianto R, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor: structure optimization and pharmacokinetics. The Journal of pharmacology and experimental therapeutics. 2010;332(3):1136–1145. doi: 10.1124/jpet.109.161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Northrup L, Sestak JO, Sullivan BP, Thati S, Hartwell BL, Siahaan TJ, Vines CM, Berkland C. Co-delivery of autoantigen and b7 pathway modulators suppresses experimental autoimmune encephalomyelitis. The AAPS journal. 2014;16(6):1204–1213. doi: 10.1208/s12248-014-9671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi N, Kiptoo P, Kobayashi H, Ridwan R, Brocke S, Siahaan TJ. Prophylactic and therapeutic suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. Clinical immunology. 2008;129(1):69–79. doi: 10.1016/j.clim.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badawi AH, Kiptoo P, Siahaan TJ. Immune Tolerance Induction against Experimental Autoimmune Encephalomyelitis (EAE) Using A New PLP-B7AP Conjugate that Simultaneously Targets B7/CD28 Costimulatory Signal and TCR/MHC-II Signal. Journal of multiple sclerosis. 2015;2(1) doi: 10.4172/2376-0389.1000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhornitsky S, Johnson TA, Metz LM, Weiss S, Yong VW. Prolactin in combination with interferon-beta reduces disease severity in an animal model of multiple sclerosis. Journal of neuroinflammation. 2015;12:55. doi: 10.1186/s12974-015-0278-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ZH, Cao XH, Du XG, Feng HB, Di W, He S, Zeng XY. Mucosal and systemic immunity in mice after intranasal immunization with recombinant Lactococcus lactis expressing ORF6 of PRRSV. Cellular immunology. 2014;287(2):69–73. doi: 10.1016/j.cellimm.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Cieslak M, Wojtczak A, Cieslak M. Role of pro-inflammatory cytokines of pancreatic islets and prospects of elaboration of new methods for the diabetes treatment. Acta biochimica Polonica. 2015;62(1):15–21. doi: 10.18388/abp.2014_853. [DOI] [PubMed] [Google Scholar]
- 27.Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M. The immunoproteasome: a novel drug target for autoimmune diseases. Clinical and experimental rheumatology. 2015;33(4 Suppl 92):S74–79. [PubMed] [Google Scholar]
- 28.Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. The Journal of pharmacology and experimental therapeutics. 2007;322(2):879–886. doi: 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
- 29.Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine & growth factor reviews. 2011;22(2):83–89. doi: 10.1016/j.cytogfr.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Naradikian MS, Hao Y, Cancro MP. Age-Associated B Cells: key mediators of both protective and autoreactive humoral responses. Immunol Rev. 2016;269:118–129. doi: 10.1111/imr.12380. [DOI] [PubMed] [Google Scholar]
- 31.Saksida T, Miljkovic D, Timotijevic G, Stojanovic I, Mijatovic S, Fagone P, Mangano K, Mammana S, Farina C, Ascione E, Maiello V, Nicoletti F, Stosic-Grujicic S. Apotransferrin inhibits interleukin-2 expression and protects mice from experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2013;262(1–2):72–78. doi: 10.1016/j.jneuroim.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 32.Friedl P, den Boer AT, Gunzer M. Tuning immune responses: diversity and adaptation of the immunological synapse. Nature reviews Immunology. 2005;5(7):532–545. doi: 10.1038/nri1647. [DOI] [PubMed] [Google Scholar]
- 33.Merwe Avd. Formation and function of the immunological synapse. Current opinion in immunology. 2002;14(3):293–298. doi: 10.1016/s0952-7915(02)00350-3. [DOI] [PubMed] [Google Scholar]
- 34.Murray JS, Oney S, Page JE, Kratochvil-Stava A, Hu Y, Makagiansar IT, Brown JC, Kobayashi N, Siahaan TJ. Suppression of type 1 diabetes in NOD mice by bifunctional peptide inhibitor: modulation of the immunological synapse formation. Chemical biology & drug design. 2007;70(3):227–236. doi: 10.1111/j.1747-0285.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 35.Manikwar P, Kiptoo P, Badawi AH, Buyuktimkin B, Siahaan TJ. Antigen-specific blocking of CD4-specific immunological synapse formation using BPI and current therapies for autoimmune diseases. Medicinal Research Reviews. 2011;32(4):727–764. doi: 10.1002/med.20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao DA, Forrest ML, Alani AW, Kwon GS, Robinson JR. Biodegradable PLGA based nanoparticles for sustained regional lymphatic drug delivery. Journal of pharmaceutical sciences. 2010;99(4):2018–2031. doi: 10.1002/jps.21970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagby TR, Cai S, Duan S, Thati S, Aires DJ, Forrest L. Impact of molecular weight on lymphatic drainage of a biopolymer-based imaging agent. Pharmaceutics. 2012;4(2):276–295. doi: 10.3390/pharmaceutics4020276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan A, Mudassir J, Mohtar N, Darwis Y. Advanced drug dlivery to the lymphatic sustem: lipid-based nanoformulations. International Journal of Nanomedicine. 2013;8(1):2733–2744. doi: 10.2147/IJN.S41521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.