

Abstract
Emerging evidence suggests that long-term exposure of insulin-secreting pancreatic β-cells to hyperglycemic [HG; glucotoxic] conditions promotes oxidative stress, which, in turn, leads to stress kinase activation, mitochondrial dysfunction, loss of nuclear structure and integrity and cell apoptosis. Original observations from our laboratory have proposed that Rac1 plays a key regulatory role in the generation of oxidative stress and downstream signaling events culminating in the onset of dysfunction of pancreatic β-cells under the duress of metabolic stress. However, precise molecular and cellular mechanisms underlying the metabolic roles of hyperactive Rac1 remain less understood. Using pharmacological and molecular biological approaches, we now report mistargetting of biologically-active Rac1 [GTP-bound conformation] to the nuclear compartment in clonal INS-1 cells, normal rat islets and human islets under HG conditions. Our findings also suggest that such a signaling step is independent of post-translational prenylation of Rac1. Evidence is also presented to highlight novel roles for sustained activation of Rac1 in HG-induced expression of Cluster of Differentiation 36 [CD36], a fatty acid transporter protein, which is implicated in cell apoptosis. Finally, our findings suggest that metformin, a biguanide anti-diabetic drug, at a clinically relevant concentration, prevents β-cell defects [Rac1 activation, nuclear association, CD36 expression, stress kinase and caspase-3 activation, and loss in metabolic viability] under the duress of glucotoxicity. Potential implications of these findings in the context of novel and direct regulation of islet β-cell function by metformin are discussed.
Keywords: Pancreatic β-cells, glucotoxicity, stress kinases, Rac1, CD36 and metformin
Introduction
Glucose-stimulated insulin secretion [GSIS] from the pancreatic β-cell involves a series of metabolic events including generation of soluble second messengers, such as adenine nucleotides [ATP], guanine nucleotides [GTP], cyclic nucleotides [cAMP, cGMP] and lipid hydrolytic products of phospholipases A2, C and D [diacyl glycerol, lysophospholipids, inositol triphosphates]. Furthermore, a precisely controlled increase in intracellular calcium concentrations via influx from the extracellular compartment as well as mobilization of intracellular pools from the endoplasmic reticulum [ER] have also been shown to be critical for insulin secretion to occur [1–4]. Together, these [metabolic, cationic and other] events facilitate the movement of insulin-laden secretory granules toward the membrane for fusion and exocytotic secretion of insulin. Several previous studies, including our own, have demonstrated that small G proteins [Arf6, Cdc42, Rac1 and Rab] play essential roles in GSIS including vesicular transport and cytoskeletal remodeling [5–10]. We have recently reported that alterations in the functional activation of these G proteins [Rac1] might represent a plausible mechanism underlying impaired insulin secretion, commonly associated with type 2 diabetes [11–16].
Considerable experimental evidence suggests that increases in intracellular generation of reactive oxygen species [ROS] triggers the metabolic dysfunction of the islet β-cell under the duress of glucotoxicity, lipotoxicity or exposure to proinflammatory cytokines or biologically-active sphingolipids, such as ceramide [11–19]. The phagocytic NADPH oxidase [Nox2] has been shown to be one of the contributors of ROS generation under these pathological conditions [20, 21]. Interestingly, Rac1 is an integral member of the Nox2 holoenzyme and, therefore, sustained activation of Rac1 under metabolic stress conditions is felt to be one of the signaling events necessary for the activation of Nox2. Using molecular biological [siRNA, antisense and dominant negative mutants] and pharmacological inhibitors of Rac1 [NSC23766, EHT1864, Ehop-016] and Nox2 [gp91-ds-tat peptide, VAS2870], recent studies have implicated requisite roles of accelerated Rac1-Nox2 module in islet β-cell dysfunction in in vitro and in vivo models of impaired GSIS and diabetes [8, 13, 14, 19–21].
As a logical extension to the above studies, we have recently reported that Rac1-Nox2 signaling axis promotes activation of specific stress kinases [p38MAPK, p53 and JNK1/2] in INS-1 832/13 cells, rodent islets and human islets under conditions of metabolic stress [13, 14, 16]. Small molecule inhibitors of Rac1 [NSC23766, EHT1864, Ehop-016] significantly attenuated stress kinase activation and associated metabolic dysfunction of the islet β-cell under these conditions, thus suggesting that Rac1-Nox2 module may be upstream to stress kinase activation. Based on the evidence on sustained activation of Rac1 under metabolic stress conditions, we recently proposed that potential defects in post-translational prenylation of Rac1 could lead to its activation in a constitutive manner, which, in turn, might lead to its localization in “inappropriate” cellular compartments [mislocalization], thus triggering pathways leading to cell dysfunction [22]. In addition, growing evidence suggests that metformin, which is commonly used in the clinic as an antidiabetic drug, exerts beneficial effects on islet β-cell against the noxious effects of glucolipotoxicity and ER stress [23–28]. Therefore, we sought to investigate potential cytoprotective effects of metformin against HG-induced metabolic dysfunction of insulin-secreting INS-1 832/13 cells. Specifically, we assessed the beneficial effects of metformin at a more clinically-relevant concentrations [15–30 μM] on HG-induced Rac1-stress kinase signaling pathway. Our results indicate significant protection by metformin of HG-induced metabolic dysfunction in pancreatic β-cells.
Materials and Methods
Materials
Rabbit polyclonal antibody for phospho-p38MAPK [Thr 180/Tyr 182] and total-p38MAPK and mouse monoclonal antibody for CD36 were from Santa Cruz Biotechnology [Santa Cruz, CA]. Phospho-p53, total-p53 and cleaved caspase-3 antibodies were from Cell Signaling [Danvers, MA]. IRDye® 800CW anti-rabbit and anti-mouse secondary antibodies were from LICOR [Lincoln, NE]. Metformin hydrochloride was purchased from Sigma-Aldrich. Rac1 antiserum was from BD Transduction Laboratories [San Jose, CA]. Antisera against cleaved caspase-3, lamin B, Phospho and total p53 and Myc tag were from Cell Signaling [Danvers, MA]. Mouse monoclonal β-actin antibody was obtained from Sigma-Aldrich [St. Louis, MO]. GGTI-2147 was from VDM biochemicals [Bedford Heights, OH]. EHT 1864 was from R&D Systems [Minneapolis, MN]. NE-PER® Nuclear and cytoplasmic extraction kit was from Thermo Scientific [Waltham, CA]. Lipofectamine 3000 was from Invitrogen [Carlsbad, CA]. All other reagents used in the studies were obtained from Sigma-Aldrich [St. Louis, MO].
Rac1 Constructs
Wild type GFP Rac-1 [GFP-Rac1] and non prenylatable and constitutively active Rac1 mutants [GFP-Rac1 SAAX] were kindly provided by Prof. Mark Philips [New York University School of Medicine, New York, NY]. Non prenylatable [GFP Rac1-C189A] Rac1 construct was prepared as described in [29]. Myc-Rac1 wild type and dominant negative N17 Myc-Rac1 constructs were from cDNA Resource Center [Bloomsberg, PA].
Insulin-secreting cells and treatment conditions
Pancreatic islets were isolated from 6 to 8 weeks old Sprague-Dawley male rats (ENVIGO, Indianapolis, IN) by the collagenase digestion method as we described previously [5, 18]. All protocols were approved by the Wayne State University’s Institutional Animal care and Use Committee. INS-1 832/13 cells were sub-cloned twice weekly following trypsinization and passages 53–61 were used for the study. INS-1 832/13 cells and rat islets were cultured in RPMI 1640 medium containing 10% heat–inactivated FBS supplemented with 100 IU penicillin and 100 IU/ml streptomycin, 1mM sodium pyruvate, 50 μM 2-mercaptoethanol [not included for culturing rat Islets] and 10mM HEPES [pH 7.4] at 37°C and 5% CO 2 in a humidified incubator. INS-1 832/13 cells were starved overnight in 2.5 mM glucose and 2.5% serum RPMI media, followed by glucose treatment [2.5 mM, low glucose; LG or 20 mM, high glucose; HG] in absence or presence of EHT 1864 [10 μM], GGTI-2147 [10 μM] or metformin [15 μM and 30 μM] for times indicated in the text. Rat islets were pre incubated with GGTI-2147 overnight along with LG prior to HG treatment.
Human islets from a normal donor [Caucasian male; 63 years; BMI= 21.8; HbA1c=5.8] were obtained from Prodo Laboratories, Inc. [Irvine, CA]. They were treated with LG (5.8mM) or HG [30mM] for 24 hours, and processed for studies to assess nuclear translocation of Rac1, which was accomplished by Western blotting [see below].
Isolation of nuclear fraction
Insulin-secreting cells were incubated with LG or HG with or without Rac1 inhibitors [EHT1864 and GGTI-2147] as described in the text. Nuclear fractions were isolated using NE-PER nuclear and cytoplasmic isolation kit as described earlier, and the purity of nuclear fractions was assessed using lamin B as a marker protein [30].
Cell transfections
INS-1 832/13 cells were transfected using Lipofectamine 3000 reagent with GFP-Rac1, Myc-Rac1 WT, N17 Myc-Rac1, GFP-S1987A, GFP-Rac1 SAAX mutants for 24 hrs and grown in low serum-LG containing media, followed by LG or HG treatment as indicated in the text.
Microscopy
INS-1 832/13 cells were grown on coverslips and transfected with constructs for 48 hours. Cells were washed with ice-cold PBS after transfection and fixation was done using 4% paraformaldehyde for 10 min at room temperature followed by washing with PBS. Subsequently, coverslips were mounted with DAKO anti-fade mounting medium with DAPI. Immunofluorescence was done using Olympus IX71 inverted fluorescent microscope.
Rac1 activation assay
Rac1 activation assay was performed using Rac1 pull-down activation assay kit [bead pull-down format; Cytoskeleton Inc.] as we described in [11, 13, 18]. Briefly, cells were grown to ~70% confluence in complete growth medium. Cells were then cultured in LG-low serum [2.5%] starvation media overnight followed by culture in LG and HG media in presence and absence of metformin [0–30 μM]. After 24 hours, growth media was aspirated and cells were washed with ice-cold PBS. After complete removal of PBS, ice-cold lysis buffer containing protease inhibitor cocktail were added to prevent protein degradation. Cell lysates were prepared and pull-down assay was performed the same day according to manufacturer’s instructions.
Immunoblotting
After 24-hour incubation with LG or HG in the absence and presence of metformin [15 or 30 μM), cells were lysed using RIPA buffer containing protease inhibitor cocktail, 1mM NaF, 1mM PMSF and 1mM Na3VO4. Cell lysates (~45 μg protein) were resolved by SDS-PAGE, and then transferred onto nitrocellulose membranes. Membranes were blocked in 5% non-fat dry milk in PBS-T buffer or 0.1% Casein in PBS-T and then incubated with appropriate primary antibody diluted with 5% non-fat dry milk in PBS-T buffer or 0.1% Casein in PBS-T, overnight at 4°C. The membranes were then washed [5 times for 5 minutes each] with PBS-T, and then probed with the appropriate secondary antibody IRDye ® 800CW anti-rabbit or anti-mouse. The immune complexes were then detected using Odyssey ® Imaging Systems. The band intensities were quantified using Carestream ® Molecular Imaging Software.
Statistical analysis of experimental data
Results are expressed as means with their standard errors as indicated. The statistical significance of differences between control and experimental groups was evaluated by ANOVA followed by SNK Post Hoc test where appropriate. P < 0.05 was considered to be statistically significant.
Results
Glucotoxicity model used in the current studies
We recently established a β-cell glucotoxicity [HG conditions] model in which chronic exposure of insulin-secreting INS-1 832/13 cells to HG [20 mM; 24 hrs.] resulted in a significant activation of caspase-3, nuclear lamin-B degradation and loss in GSIS [11, 13, 14, 16, 30]. We also reported accelerated β-cell apoptosis under those conditions as evidenced by increased Annexin V/propidium iodide staining. These observations were further confirmed in a cell death quantification assay [16]. Together, our observations indicated that long-term exposure of pancreatic β-cells to HG conditions leads to cell dysfunction [i.e., loss in their ability to secrete insulin in response to physiological glucose concentrations] and demise. In the context of the current investigation, we have recently reported that glucotoxic conditions promote sustained activation of Rac1 in pancreatic β-cells [11–15]. However, potential consequences of sustained activation of Rac1, including potential alterations in its subcellular distribution [mislocalization], have not been studied thus far. Therefore, we first examined nuclear association of Rac1 [a trigger for cell dysregulation]. Secondly, we examined protective effects of metformin against HG-induced apoptotic signaling pathways and dysregulation of INS-1 832/13 cells [i.e., our β-cell glucotoxicity model].
HG conditions promote nuclear accumulation of Rac-1 in INS-1 832/13 cells, normal rat islets and human islets
Previous observations from our laboratory have demonstrated sustained activation of Rac1 in INS-1 832/13 cells, normal rat islets and human islets exposed to HG conditions [11–13]. We provided evidence to indicate a significant reduction in geranylgeranyltransferase [GGTase] activity, which regulates post-translational prenylation of small G-proteins, including Rac1. As an index for decreased prenylation, we observed significant accumulation of unprenylated proteins in pancreatic β-cells exposed to HG conditions. Therein, we speculated that unprenylated, but paradoxically active G-proteins might translocate into “inappropriate” compartments [e.g., nucleus] to induce metabolic defects in the effete β-cell [22]. Therefore, we undertook the current investigation to determine potential targeting of Rac1 into the nuclear compartment in pancreatic β-cells exposed to HG conditions. Data depicted in Figure 1 [Panels A and B] demonstrate a marked increase in the nuclear localization of Rac1 in INS-1 832/13 cells exposed to glucotoxic conditions. This increase is not due to increase in the total Rac1 levels under HG conditions since we observed a corresponding decrease in the relative abundance of Rac1 in the non-nuclear fraction isolated from HG-treated cells [Figure 1; Panel A and B]. These findings were confirmed in normal rodent islets [Figure 1; Panels C and D] and human islets [Figure 1; Panel E]. Please note that the non-nuclear fraction is devoid of nuclear components as evidenced by lack of detectable lamin B, a nuclear marker, in these fractions isolated from INS-1 832/13 cells, rat islets and human islets [Panels A, C and E, respectively]. To further confirm our immunoblotting data on the nuclear accumulation of Rac1 under HG conditions, we transfected INS-1 832/13 cells with wild type GFP-Rac1 construct and treated the cells with HG for 24 hours post transfection. Data depicted in Figure 1 [Panel F] indicated even distribution of GFP-Rac1 in cells under basal conditions. However, nuclear association of Rac1 was observed in cells exposed to HG conditions further validating our Western blot data. Together, these observations validate our hypothesis that exposure of insulin-secreting cells to HG conditions leads to sustained activation of unprenylated Rac1 leading to its translocation to the nuclear compartment.
Figure 1. HG conditions promote nuclear accumulation of Rac-1 in INS-1 832/13 cells, normal rat islets and human islets.

Panel A: Relative abundance of Rac1 in the nuclear and non-nuclear fractions isolated from INS-1 832/13 cells exposed to LG or HG conditions was determined by Western blotting.
Panel B: Pooled data from three independent experiments is shown. Accumulation of Rac1 was calculated as a ratio of Rac1 to lamin B in the nuclear fraction [loading control as well as marker] and represented as fold change over basal ** p<0.005 vs 2.5mM glucose.
Panel C: Relative abundance of Rac1 in the non-nuclear and nuclear fractions isolated from normal rat islets exposed to LG or HG conditions was determined by Western blotting.
Panel D: Pooled data from three independent experiments is represented herein. Accumulation of Rac1 was calculated as above. ** p<0.005 vs 2.5mM glucose alone,*p <0.05 vs 20 mM glucose alone.
Panel E: Human pancreatic islets were incubated with LG or HG for 24 hours and relative abundance of Rac1 in the non-nuclear and nuclear fractions was determined by Western blotting. A Western blot of one batch of human islet lysates is provided here.
Panel F: WT GFP Rac1 was transfected 24 hours treated with LG and HG for 24 hours. DAPI was used to counter stain the nucleus. Nuclear association of Rac1 is indicated by arrows.
Active [GTP-bound] conformation of Rac1 is required for its nuclear translocation
To further affirm our hypothesis, we next asked if biologically-active conformation of Rac1 [Rac1-GTP] is essential for its translocation to the nuclear fraction. We addressed this question via molecular biological and pharmacological approaches. In the first approach, we utilized a dominant negative mutant [myc-tagged] Rac1 or WT-Rac1 to determine if HG conditions would promote nuclear association of Rac1 in cells transfected with the inactive mutant. Data in Figure 2 [Panels A and B] demonstrate significant translocation of WT-Rac1 into the nuclear compartment in INS-1 832/13 cells exposed to HG-conditions. However, there was a 50% reduction in nuclear association of N17 mutant in cells under the same conditions [Figure 2; Panels A and B]. In the second approach, we utilized EHT1864, a known inhibitor of Rac1 [31, 32] to functionally inactivate endogenous Rac1 and then assessed nuclear association of Rac1 under LG and HG conditions. Data in Figure 2 [Panels C and D] demonstrate decreased localization of Rac1 in INS-1 832/13 cells exposed to EHT1864. It is noteworthy that, we consistently noticed an increase in Rac1 accumulation in cells treated with EHT inhibitor under LG conditions even though HG-induced Rac1 translocation was more potently inhibited by EHT-1864. Together, data in Figure 2 [Panels A–D] suggest that GTP-bound configuration [active form] is preferentially translocated to the nuclear fraction following exposure to glucotoxic conditions.
Figure 2. Active configuration of Rac-1 [GTP-bound form] is necessary for nuclear accumulation under HG exposure conditions.

Panel A: INS-1 832/13 cells were transfected with either myc-tagged wild type [WT] or dominant negative Rac1 [N17] prior to exposure to HG conditions. Accumulation of Rac1 was assessed by Western blotting.
Panel B: Pooled data from three independent experiments are shown in the graph ** p<0.005 vs WT Rac1.
Panel C: INS-1 832/13 cells were treated with LG or HG in presence or absence of EHT1864 (10 μM), a specific small molecule inhibitor of Rac1, for 24 hrs. Cells were then pelleted and subjected to nuclear fractionation using NE-PER kit according to manufacturer’s instructions.
Panel D: Inhibition of HG-induced nuclear translocation by EHT 1864, was quantified by densitometric analysis. Results were expressed as mean fold change from three independent experiments *p <0.05 vs 20 mM glucose.
Post-translational geranylgeranylation is not essential for nuclear translocation of Rac1
We next asked if post-translational geranylgeranylation is required for the GTP-bound form of Rac1 to associate with the nuclear fraction. To address this, we first determined nuclear association of Rac1 in INS-1 832/13 cells exposed to either HG conditions or to GGTI-2147, a known inhibitor of geranylgeranylation of Rac1 [11, 12, 18]. Data in Figure 3 [Panels A and B] demonstrate significantly higher nuclear localization of Rac1 in INS-1 832/13 cells exposed to HG and GGTI-2147, thus suggesting that post-translational geranylgeranylation is not requisite for nuclear association of Rac1. In a second approach, we transfected INS-1 832/13 cells with two unprenylatable, but biologically-active, mutants of Rac1 [C189A and SAAX] to further determine their association with nuclear fraction. Data in Figure 3 [Panel C] demonstrate a significantly high abundance of these Rac1 mutants in the nuclear fraction compared to the WT-Rac1. These data further confirm our findings in Figure 3 [Panel A]. In the second approach, we overexpressed non-prenylatable and constitutively active Rac1 mutants [GFP-C189A and GFP-Rac1 SAAX] in INS-1 832/13 cells and analyzed the localization of these mutants by immunofluorescence. Compatible with data from studies described in Panels A and B, our microscopic evidence [Figure 3; Panel D] indicates that both non prenylatable and constitutively active mutants mostly localize to nuclear compartment, thus suggesting that defects in prenylation lead to increased nuclear association of Rac1 with the nuclear fraction. Based on the data accrued thus far, we conclude that HG conditions promote nuclear association of unprenylated, but active Rac1 in a variety of insulin-secreting cells. These findings further support our recently proposed model that metabolic stress promotes defective prenylation and aberrant activation of Rac1 to initiate downstream signaling events leading to the metabolic dysfunction and demise of the β-cell.
Figure 3. Prenylation is not requisite for nuclear localization of Rac 1.

Panel A: Rat pancreatic islets were isolated and pre-incubated with GGTI-2147 for overnight prior to treatment with LG or HG for 24 hours. Nuclear accumulation of Rac1 was determined by Western blotting.
Panel B: Pooled data from three independent experiments are shown in the graph. ** p<0.005 vs WT Rac1.
Panel C: INS-1 832/13 cells were transfected with GFP wild type [WT] or unprenylatable and constitutively active GFP Rac-1 [Rac1 SAAX], unprenylatable Rac1 [Rac1 C189A] and relative abundance of Rac1 in the non-nuclear and nuclear fractions was assessed by Western blotting followed by densitometry. Lamin B was used as marker for the nuclear fraction.
Panel D: INS-1 832/13 cells were transfected with GFP wild type [WT] or unprenylatable and constitutively active GFP Rac-1 [Rac1 SAAX], unprenylatable Rac1 [Rac1 C189A] and localization of Rac1 was analyzed using fluorescent microscope. DAPI was used to counterstain the nucleus.
In the following set of studies, we investigated protective effects of metformin, a biguanide derivative, against HG-induced metabolic defects in pancreatic β-cells including sustained activation of Rac1 and its downstream signaling pathways, such as stress kinase activation, mitochondrial dysregulation and loss in cell viability under HG conditions.
Metformin prevents sustained activation of Rac1 and its nuclear localization under the duress of glucotoxicity
At the outset, we asked if HG-induced sustained activation of Rac1 is prevented by clinically-relevant concentrations of metformin. To address this, activation of Rac1 was quantified by a pull-down assay in INS-1 832/13 cells exposed to HG in the absence or presence of metformin [30 μM]. Data depicted in Figure 4 demonstrate significant [~3.5 fold] activation of Rac1 in INS-1 832/13 cells exposed to glucotoxic conditions. Co-provision of metformin markedly attenuated HG-induced activation of Rac1 under these conditions. It is noteworthy, however, that metformin modestly, but significantly, increased Rac1 activation in cells exposed to basal glucose [normal] conditions [Figure 4; Panel A]. Pooled data from multiple experiments are provided in Figure 4 [Panel B]. Furthermore, as shown in Figure 4 [Panels C and D], metformin treatment prevented nuclear association of Rac1 under HG conditions. Together, these findings suggest that metformin significantly inhibits HG-induced Rac1 activation and nuclear association in pancreatic β-cells.
Figure 4. Metformin suppresses HG-induced Rac1 activation and nuclear association in INS-1 832/13 cells.

Panel A: INS-1 832/13 cells were cultured in LG [2.5mM] or HG [20mM] for 24 hours in the absence and presence of Metformin [30 μM]. Rac1 activation assay was performed using pull-down activation assay biochem kit [see Methods for additional details]. Cell lysates were separated by SDS-PAGE followed by Western blotting.
Panel B: Band intensities of Rac1 were quantified by densitometric analysis. Abundance of active Rac1 in pull-down samples was normalized by total Rac1. Pooled data from three experiments was represented in this panel. *p < 0.05 vs. 2.5mM glucose alone, **p < 0.05 vs. 20mM glucose alone.
Panels C and D: INS-1 832/13 cells were incubated with LG [2.5mM] and HG [20mM] in the absence and presence of metformin for 24 hrs. Cell lysates were analyzed for Rac1 by Western blotting. Purity of the nuclear fractions was verified by probing with Lamin B. Band intensities for Rac1 were measured using densitometry and the ratios were calculated over lamin B in the presence of metformin [n=3]. *p < 0.05 vs. 2.5 mM glucose alone, **p < 0.05 vs 20mM glucose alone.
Sustained activation of Rac1 under glucotoxic conditions leads to increased expression of the fatty acid transporter CD36: Metformin inhibits HG-induced CD36 expression
Several recent studies have established novel roles for CD36, a membrane-associated fatty acid transporter protein, in the onset of β-cell dysfunction and demise under chronic hyperglycemic conditions [19, 28]. Therefore, we asked if HG conditions increase expression of CD36 in INS-1 832/13 cells, and if so, whether Rac1 activation represents an upstream signaling mechanism for HG-induced expression of CD36. To address this question, we quantified HG-induced expression of CD36 in INS-1 832/13 cells exposed to EHT1864. Data in Figure 5 [Panel A] indicate a significant increase in the expression of CD36 in INS-1 832/13 cells exposed to glucotoxic conditions. Moreover, HG-induced expression of CD36 was reduced following inhibition of Rac1, thus suggesting that Rac1 activation may be upstream to CD36 expression in β-cells exposed to HG conditions [Figure 5; Panels A and B]. We next questioned whether metformin exerts any protective effects on HG-induced CD36 expression. Data in Figure 5 [Panels C and D] demonstrate a significant increase in the expression of CD36 in INS-1 832/13 cells following exposure to HG conditions [as in Panel A]. In addition, co-provision of metformin markedly suppressed HG-induced expression of CD36. Note that in a manner akin to Rac1 activation [Figure 4], metformin treatment slightly increased the expression of CD36 under basal glucose conditions [Figure 5; Panel C; lanes 1 vs.2]. Pooled data from multiple experiments are included in Figure 5 [Panel D]. Compatible with data described above [Figure 2], findings from this experiment suggest that HG-induced activation and nuclear association of Rac1 and downstream CD36 expression are sensitive to metformin.
Figure 5. Rac1 mediates HG-induced expression of CD36 in INS-1 832/13 cells: Protection by metformin.

Panel A: Quantification of the CD36 and actin bands was done by densitometric analysis and ratios were calculated over actins in the presence and absence of EHT 1864. *p < 0.05 vs 2.5mM glucose alone [n=3].
Panel B: INS-1 832/13 cells were treated with LG [2.5mM] and HG [20mM] with or without Metformin [30μM] for 24 hrs. Relative abundance of CD36 was determined by Western blotting. Actin was used as a loading control. Data are representative of three experiments.
Panel C: Quantification of the CD36 and actin bands was done by densitometry and ratios were calculated over actins in the presence and absence of metformin. *p < 0.05 vs 2.5mM glucose alone [n=3].
HG-induced stress kinase [p38MAPK and p53] activation is prevented by metformin
In the next series of studies, we assessed the protective effects of metformin against HG-induced stress kinase [p38MAPK and p53] in INS-1 832/13 cells. As we reported recently [14], data in Figure 6 [Panels A and B] demonstrate a significant stimulation on p38MAPK activity in these cells following exposure to HG conditions. Metformin treatment significantly alleviated such effects. Interestingly, however, as in the case of Rac1 activation [Figure 4] and CD36 expression, [Figure 5] metformin-treatment [30 μM] consistently increased p38MAPK activation under basal glucose conditions despite its protective effects against high glucose-induced Rac1 activation, CD36 expression and p38MAPK activation.
Figure 6. Metformin inhibits HG- induced p38MAPK and p53 activation in INS-1 832/13 cells.

Panel A: INS-1 832/13 cells were incubated with LG [2.5mM] and HG [20mM] in the presence and absence of metformin [0–30 μM] for 24 h. Relative abundance of phopho-p38MAPK and total-p38MAPK was determined by Western blotting.
Panel B: Quantification of the phopho-p38 bands was done by densitometry and the ratios were calculated over total-p38 in the presence of metformin [n=5]. *p < 0.05 vs 2.5mM glucose alone, **p < 0.05 vs 20mM glucose alone; NS: not significant.
Panel C: INS-1 832/13 cells were treated with LG [2.5mM] and HG [20mM] in the absence or presence of metformin [0–30 μM] for 24 h. Cell lysates were separated and analyzed for phosphorylated and total p53 by Western blotting.
Panel D: Band intensities for phospho-p53 were measured using densitometry and the ratios were calculated over total-p53 in the presence of metformin. *p < 0.05 vs 2.5mM glucose alone, **p < 0.05 vs 20mM glucose alone [n=3].
In the next series of studies we examined protective effects, if any, on HG-induced activation of p53. Data in Figure 6 [Panels C and D] confirm our recently published observations of activation of p53 by glucotoxic conditions [16]. More importantly, we observed marked protection by metformin of INS-1 832/13 cells against HG-induced p53 activation [Figure 6; Panels C and D]. It is noteworthy that, unlike in the case of Rac1 activation, CD36 expression and p38MAPK activation, metformin did not exert any effects of p53 activation under basal glucose conditions.
Metformin treatment attenuates HG-induced caspase-3 activation in INS-1 832/13 cells
In the last series of experiments, we determined the degree of caspase-3 activation in INS-1 832/13 cells exposed to HG conditions in the absence or presence of metformin. Our findings demonstrated a high degree of caspase-3 activation in cells exposed to HG conditions. This is evidenced by emergence of the cleaved [biologically- active] caspase-3 band under these conditions [Figure 7; Panel A]. We also observed a significant reduction in high glucose-induced caspase-3 activation in cells exposed to metformin. A modest increase in caspase-3 activation was also seen in cells under normal culture [basal] conditions. Pooled data from multiple experiments are provided in Figure 7 [Panel B].
Figure 7. HG- mediated caspase-3 activation is reduced by metformin.
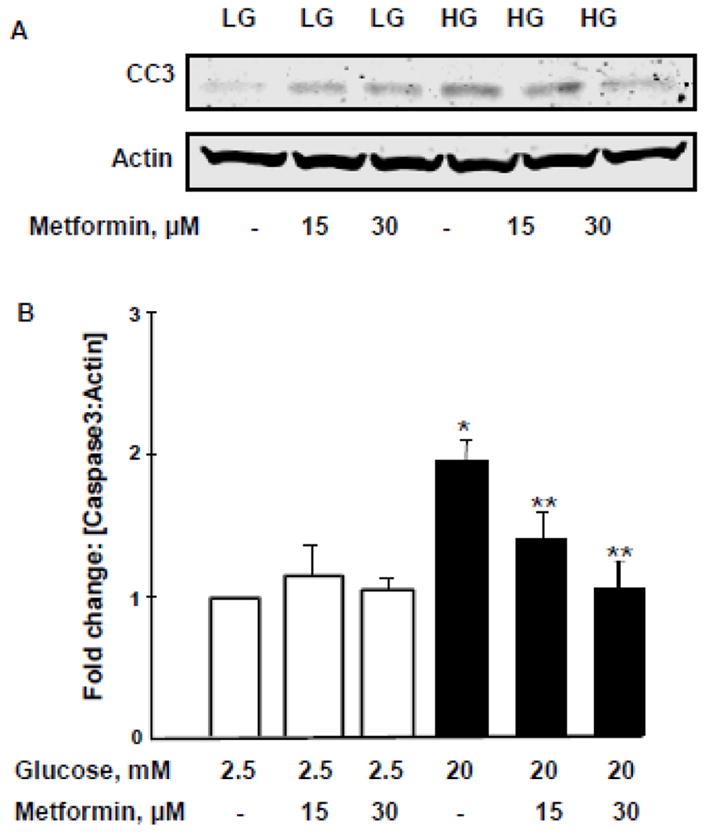
Panel A: INS-1 832/13 cells were incubated with LG [2.5mM] and HG [20mM] in the absence and presence of metformin [0–30 μM] for 24 h. Cell lysates were analyzed for caspase-3 [active fragment] by Western blotting.
Panel B: Densitometry was used to quantify the bands and the ratios were calculated over actin in the presence of metformin. *p < 0.05 vs 2.5mM glucose alone, **p < 0.05 vs 20mM glucose alone [n=3].
Metformin treatment relieves HG-induced loss in metabolic cell viability in INS-1 832/13 cells
Compatible with above findings [Figure 7], we noticed significant protection by metformin of HG-induced loss in metabolic cell viability in these cells [Figure 8]. Our findings demonstrated a significant reduction in metabolic cell viability in INS-1 832/13 cells incubated under HG conditions. Co-provision of metformin [30 μM] significantly protected these cells from metabolic alterations. Compatible with data described in the above sections, metformin treatment alone decreased cell viability under basal glucose concentrations. These data clearly imply dual regulatory roles of metformin [see Discussion].
Figure 8. Metformin protects HG-induced loss in cell viability in INS-1 832/13 cells.
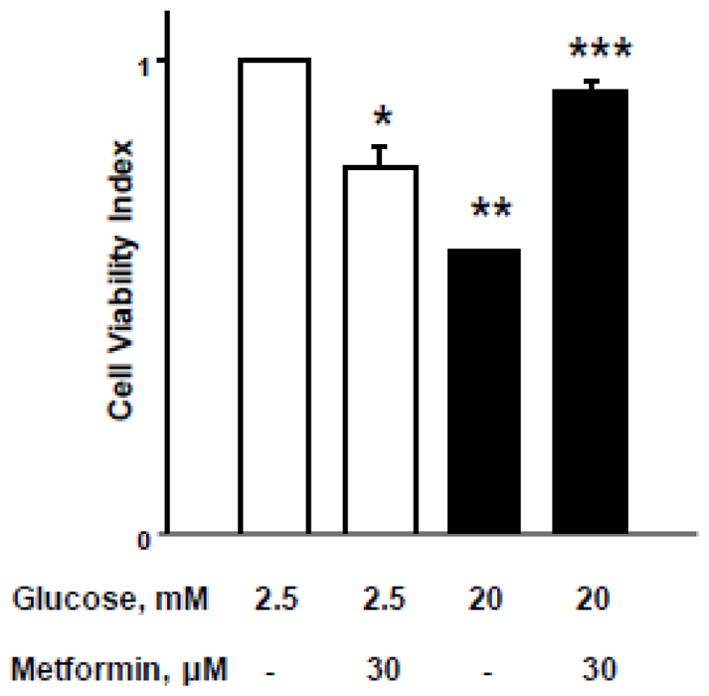
INS-1 832/13 cells were incubated with low [2.5 mM] or high [20mM] glucose for 24 hrs. in the absence or presence of metformin [30 μM]. After 24 hrs. of glucose treatment, the cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide [MTT] reagent for 4 hrs. and absorbance was measured at 540 nm. Data are represented as mean ± SEM from 8–10 determinations in each condition. *p=0.033, ** p=0.0001; and *** p=0.78 [not significant] vs. basal conditions.
Based on the findings described in these studies, we conclude that HG conditions promote sustained activation and nuclear association [mislocalization] of Rac1 and metabolic dysfunction [CD36 expression, stress kinase activation, caspase-3 activation and loss in metabolic cell viability] in pancreatic islet β-cells. We also provide evidence in support of significant protection of these metabolic defects by metformin. Together, these data provide evidence for novel targets for metformin, specifically at the level of pancreatic β-cell [see below].
Discussion
It is well established that chronic exposure of pancreatic β-cells to HG conditions results in significant metabolic dysfunction, including loss in cell proliferation and GSIS leading to apoptotic demise of the β-cell [17]. More recent findings from our laboratory have demonstrated novel regulatory roles for Rac1, a small G-protein, in the induction of islet β-cell dysfunction under the duress of glucotoxicity [8, 12–14]. Specifically, we demonstrated that, under glucotoxic conditions, sustained activation of Rac1 results in accelerated Nox2 signaling leading to increased oxidative stress [ROS production], stress kinase [p38MAPK and p53], caspase 3 activation and nuclear lamin degradation culminating in cell death [8, 12–14, 30]. During these investigations, we also identified two guanine nucleotide exchange factors [Tiam1 and Vav2] that mediate activation of Rac1 in eliciting damaging effects on β-cells [15]. As a logical extension to these studies, we undertook the current studies to examine potential alterations, if any, in the subcellular distribution [mislocalization] of Rac1 in pancreatic β-cells exposed to glucotoxic conditions. Furthermore, we assessed the efficacy of metformin, an antidiabetic drug, against HG-mediated effects on β-cell function. Salient findings from these studies are: [i] exposure of INS-1 832/13 cells, normal rodent islets and human islets to HG-conditions result in nuclear association of Rac1; and [ii] biologically-active [GTP-bound] conformation of Rac1, but not its post-translational prenylation, is required for its association with the nuclear fraction. We also demonstrated that clinically-relevant concentrations of metformin prevent HG-induced; [i] Rac1 activation and nuclear translocation; [ii] CD36 expression; [iii] stress kinase and caspase-3 activation; and [iv] loss in cell viability. Implications of these findings in the context of regulatory roles of constitutively-active Rac1 in the pathology of islet dysfunction, and its prevention by metformin are discussed below.
Recent studies from our laboratory have reported sustained activation of Rac1 in clonal INS-1 832/13 β-cells, normal rodent islets, and human islets under the duress of metabolic stress, including glucotoxicity, lipotoxicity, exposure to proinflammatory cytokines, and biologically-active sphingolipids, such as ceramide [8, 11–16, 18]. These observations were also confirmed in islets derived from type 2 DM animal models and human donors with T2DM. Furthermore, pharmacological inhibition [NSC23766] of Tiam1, a GEF for Rac1, attenuated Rac1 activation in all the above experimental conditions, thus suggesting that Tiam1 represents one of the GEFs that mediate hyper-activation of Rac1. More importantly, inhibition of Tiam1-Rac1 signaling axis also prevented HG-induced, Nox2 activation and downstream stress kinase activation and mitochondrial dysfunction in pancreatic β-cells exposed to HG conditions [15]. Together, these findings implicate Rac1 as a key mediator of islet β-cell dysfunction in metabolic stress and diabetes.
What then are the underlying mechanisms for Rac1 activation in metabolic stress? We recently proposed, and provided experimental evidence to suggest that non-canonical activation of Rac1 under these conditions might, in part, be due to defects in requisite prenylation. In this context, we reported a significant reduction in protein prenyltransferase [farnesyltransferase and geranylgeranyltransferases] activities in INS-1 832/13 cells exposed to glucotoxic, lipotoxic and ER stress conditions. Consequentially, a significant increase in the relative abundance of unprenylated proteins was seen under these conditions [22]. As noted above, sustained activation was demonstrable in islet β-cells even at the height of defective prenylation, thus validating our original hypothesis that metabolic stress induces defective prenylation and sustained activation of G-proteins. We also proposed, but not provided evidence for, that inhibition of prenylation leads to mistargetting of G-proteins resulting in pathology. Indeed, data from our current investigations support such a formulation since we reported prenylation-independent nuclear translocation of active Rac1 under glucotoxic conditions. Evidence is also emerging in other cell models to suggest that defects in protein prenylation lead to aberrant activation and mislocalization of unprenylated proteins. For example, Khan et al. have recently reported hyper-activation of macrophages and associated induction of erosive arthritis in mice lacking GGTase-1, thus raising potential for regulatory roles of geranylgeranylation in inflammatory cell signaling [33]. It is noteworthy that GGTase deficiency in these cells is manifested by significantly elevated levels of active [GTP-bound] Rac1, Cdc42 and RhoA, thus suggesting an alternate prenylation-independent mechanism for the activation of these unprenylated G proteins under pathological conditions. Along these lines, Dunford et al have demonstrated constitutive activation of Rac, Cdc42 and Rho G-proteins in J774 macrophages following inhibition of prenylation by nitrogen-containing bisphosphonates [34]. Taken together, these findings demonstrate inhibition of prenylation results in sustained activation and translocation of G-proteins to inappropriate cellular compartments, thus raising an important question if such a signaling step leads to cellular pathology including islet dysfunction, especially under HG conditions [35].
Several recent studies have investigated beneficial effects of metformin against islet β-cell function. It is noteworthy that these in vitro investigations utilized a wide range of metformin concentrations [10 μM-1 mM]. For example, Simon-Szabo and associates [36] have reported significant attenuation of palmitate-induced [lipoapoptosis] ER stress [elF2α phosphorylation and CHOP expression] and stress kinase [JNK1/2] activation by metformin [10–100 μM] in rat insulinoma cells. Using murine islets and human islets Lundquist et al [37] have demonstrated a marked reduction by metformin [20 μM] in nitric oxide synthase-derived nitric oxide, insulin secretory dysfunction and loss in cell viability under conditions of long-term exposure to glibenclamide and HG. Using rodent islets, Hashemitabar and associates have demonstrated beneficial effects of metformin [15 μM] on insulin gene expression, insulin secretion and islet cell viability [38]. Natalichhio and coworkers have shown significant restoration of GLP-1 receptor impairment by metformin [0.5–1.0 mM] in murine islets following exposure to palmitate [39]. Together, the above studies provide supporting evidence for beneficial/protective effects of metformin against gluco-, or lipotoxicity and ER stress. Our current findings demonstrate marked protection of INS-1 832/13 cells, by metformin, against HG-induced metabolic defects at concentrations as low as 30 μM.
A growing body of evidence implicates CD36, a fatty acid transport protein, in cell apoptosis under glucolipotoxic conditions [19, 28]. Data from our current studies have provided evidence to suggest that Rac1 activation is upstream to CD36 expression since EHT1864, a known inhibitor of Rac1 [31, 32], attenuated HG-induced CD36 expression in INS-1 832/13 cells. Our findings are also compatible with recent observations of Elumalai and associates demonstrating regulatory roles for Rac1-Nox2 signaling axis promotes CD36 expression in INS-1 cells under the duress of glucotoxic conditions [19]. Using specific inhibitors of Tiam1-Rac1 [NSC23766] and Nox2 [VAS2870] these researchers were able to identify Tiam1-Rac1-Nox2 signaling steps as upstream modulators of CD36 expression under HG glucose exposure conditions. It should be noted that the findings of Elumalai et al [19] further validate our original proposal that Tiam1-Rac1-Nox2 signaling pathway contributes to islet β-cell dysfunction under metabolic stress conditions [8, 12, 15, 21]. Data from our current investigations involving a structurally distinct inhibitor of Rac1 [EHT1864] further support this working model. Our current observations also demonstrated a significant reduction in HG-induced CD36 expression by metformin at 30 μM concentration. Further, inhibition of Rac1-CD36 pathway appears to regulate the downstream stress kinase [p38MAPK and p53] activation and mitochondrial dysregulation [caspase-3 activation] in INS-1 832/13 cells. In further support of our findings are the recent observations of Moon and associates demonstrating significant protective effects of metformin [0.5 mM] against oxidative- and endoplasmic reticulum stress-induced CD36 expression in clonal β-cells and rodent islets [28].
It is noteworthy that metformin appears to exert dual regulatory roles in pancreatic β-cells. For example, in the current studies, we consistently noted that under basal glucose conditions, metformin increased Rac1-CD36-Stress kinase activation to a modest, but significant degree while affording protection against HG-induced effects on these signaling steps. Along these lines, using insulin-secreting MIN6 cells, Jiang and associates have provided evidence to suggest dual regulatory roles for metformin in pancreatic β-cell function. First, under normal growth conditions metformin significantly suppressed MIN6 cell proliferation and triggered apoptosis via a mechanisms involving AMPK-activation and autophagy-related signaling steps [40]. Interestingly, however, metformin significantly protected MIN6 cells against palmitate-induced mitochondrial dysfunction [caspase activation] and cell death. While these data appear to support our findings of significant protective effects of metformin on HG-induced effects in INS-1 832/13 cells, it should be noted that studies of Jiang and associates [40] used relatively high concentrations of metformin [2 mM] compared to much less concentration of metformin we used in our current studies [15–30 μM].
Conclusions and working model
Based on available evidence, we propose [Figure 9] that HG conditions promote functional inactivation of protein prenyltransferases leading to defective prenylation, and paradoxical activation of G-proteins such as Rac1. Hyper-activation of Rac1 may, in part, be due to weak association of Rac1 with its regulatory guanine nucleotide dissociation inhibitor [GDI] and/or increased activity of Tiam1 [a known GEF for Rac1]. Unprenylated, and yet functionally active Rac1 is targeted to nuclear fraction [mislocalization; ref. 35] leading to accelerated apoptotic signaling [p53 activation] and the onset of cell dysfunction [16]. It is also proposed that hyperactive Rac1 might regulate other apoptotic function including CD36 expression, other stress kinase [p38MAPK and JNK1/2] activation to initiate signaling events leading to caspase-3 activation and nuclear lamin-B degradation terminating in loss in GSIS, inhibition of proliferation and cellular apoptosis [11, 13, 14, 30]. We also propose that metformin affords protection against above mentioned glucotoxic effects [Figure 9 indicated by red arrows] at clinically relevant concentrations [15–30 μM]. Future studies will determine potential targets for metformin, specifically regulatory factors for Rac1 activation including GEFs, GTPase-activating proteins and the Rho GDP dissociation inhibitor in the islet β-cell, the interplay of which is expected to retain Rac1 in its active, GTP-bound conformation to promote downstream signaling events that could contribute to metabolic dysregulation and onset of type 2DM [8, 41].
Figure 9. A proposed model for metabolic stress induced dysfunction of pancreatic islet β-cells: Reversal by metformin.

Based on the data accrued thus far, we propose that exposure of pancreatic islet β-cells to metabolic stress conditions [e.g., glucotoxicity results in functional inactivation of prenyltransferases, and consequently, a significant accumulation of unprenylated Rac1 occurs. Our findings suggest a paradoxical stimulation of G protein activity even at the height of inhibition of requisite prenylation. We presented data herein to suggest a significant association of unprenylated, but active [GTP-bound] Rac1 with the nuclear fraction in a variety of insulin-secreting cells under glucotoxic conditions. This, in turn, promotes phosphorylation of pro-apoptotic proteins/factor, including p53 [15]. Along these lines, we also demonstrated that constitutively activated Rac1 promotes activation of Nox2 resulting in the generation of oxidative stress and activation of downstream signaling kinases including p38MAPK. These events singly, or in combination, promote caspase activation, nuclear lamin degradation leading to cell dysfunction. Note that specific signaling events/steps that are regulated by metformin [current studies] are indicated by red arrows.
Highlights.
Chronic exposure of pancreatic β-cells to glucotoxic conditions induces sustained activation and nuclear translocation of Rac1 in clonal β-cells, normal rodent islets and human islets.
Post-translational prenylation is not essential for nuclear association of active Rac1.
Glucotoxic conditions induce Rac1-mediated expression of CD36, a fatty acid transporter, which is involved in cell apoptosis.
Metformin, a biguanide anti-diabetic compound, prevents high glucose-induced Rac1 activation, nuclear translocation, CD36 expression, stress kinase [p38MAPK and p53] activation, caspase-3 activation, and loss in metabolic cell viability.
Acknowledgments
Funding
This research is supported [to AK] by a MERIT Review Award from the US Department of Veterans Affairs [BX002801] and the National Institutes of Health [DK-74921 and EY-022230]. AK also received a Senior Research Career Scientist Award [13S-RCS-006] from the Department of Veterans Affairs.
This work is conducted for the partial fulfillment of an MS degree in Pharmaceutical Sciences [to SB] from Wayne State University. The authors thank Drs. Jairus Reddy, Anil Poudel, Naveen Mekala, Vino Cheriyan and Ms. Gurbani Bedi for technical assistance in certain aspects of these studies.
Abbreviations used
- AMPK
AMP-activated protein kinase
- Arf6
ADP-ribosylation factor 6
- CC3
Cleaved caspase-3
- Cdc42
Cell division control protein 42
- CD36
Cluster of Differentiation36
- ER stress
Endoplasmic reticulum stress
- GEFs
Guanine nucleotide exchange factors
- GGTase
Geranylgeranyl transferase
- GGTI-2147
Geranylgeranyl transferase inhibitor
- GSIS
Glucose-stimulated insulin secretion
- HG
High glucose
- LG
Low glucose
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Nox2
NADPH oxidase 2
- p38MAPK
p38 mitogen activated protein kinase
- Rac1
Ras-related C3 botulinum toxin substrate 1
- ROS
Reactive oxygen species
- siRNA
Small (or short) interfering RNA
- T2DM
Type 2 diabetes mellitus
- and Tiam1
T-lymphoma invasion and metastasis-inducing protein 1
Footnotes
Conflict of interests
The authors declare no conflict of interest
Authors contributions
SB and AC conducted studies and analyzed the experimental data. DLH developed specific experimental tools, reviewed and edited the manuscript. AK planned and supervised studies, and wrote and edited the manuscript.
References
- 1.Berggren PO, Leibiger IB. Novel aspects on signal-transduction in the pancreatic beta-cell. Nutr Metab Cardiovasc Dis. 2006;16(1):S7–10. doi: 10.1016/j.numecd.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia. 2010;53(6):1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komatsu M, Takei M, Ishii H, Sato Y. Glucose-stimulated insulin secretion: A newer perspective. J Diabetes Investig. 2013;4(6):511–516. doi: 10.1111/jdi.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18(2):162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Kowluru A, Li G, Rabaglia ME, Segu VB, Hofmann F, Aktories K, Metz SA. Evidence for differential roles of the Rho subfamily of GTP-binding proteins in glucose- and calcium-induced insulin secretion from pancreatic beta cell. Biochem Pharmacol. 1997;54(10):1097–1108. doi: 10.1016/s0006-2952(97)00314-6. [DOI] [PubMed] [Google Scholar]
- 6.Aizawa T, Komatsu M. Rab27a: a new face in beta cell metabolism-secretion coupling. J Clin Invest. 2005;115(2):227–230. doi: 10.1172/JCI24269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122(Pt 7):893–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowluru A. Small G proteins in islet beta-cell function. Endocr Rev. 2010;31(1):52–78. doi: 10.1210/er.2009-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayaram B, Syed I, Kyathanahalli CN, Rhodes CJ, Kowluru A. Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 beta-cells and rat islets. Biochem Pharmacol. 2011;81(8):1016–1027. doi: 10.1016/j.bcp.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arora DK, Syed I, Machhadieh B, McKenna CE, Kowluru A. Rab-geranylgeranyl transferase regulates glucose-stimulated insulin secretion from pancreatic beta cells. Islets. 2012;4(5):354–358. doi: 10.4161/isl.22538. doi:0.4161/isl.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Syed I, Jayaram B, Subasinghe W, Kowluru A. Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol. 2010;80(6):874–883. doi: 10.1016/j.bcp.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol. 2011;81(8):965–975. doi: 10.1016/j.bcp.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, Kowluru A. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes. 2011;60(11):2843–2852. doi: 10.2337/db11-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sidarala V, Veluthakal R, Syeda K, Vlaar C, Newsholme P, Kowluru A. Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic beta-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1) Biochem Pharmacol. 2015;95(4):301–310. doi: 10.1016/j.bcp.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowluru A. Tiam1/Vav2-Rac1 axis: A tug-of-war between islet function and dysfunction. Biochem Pharmacol. 2017;132:9–17. doi: 10.1016/j.bcp.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sidarala V, Kowluru A. Exposure to chronic hyperglycemic conditions results in Ras-related C3 botulinum toxin substrate 1 (Rac1)-mediated activation of p53 and ATM kinase in pancreatic beta-cells. Apoptosis. 2017;22(5):597–607. doi: 10.1007/s10495-017-1354-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl 1):S119–124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 18.Subasinghe W, Syed I, Kowluru A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic beta-cells: evidence for regulation by Rac1. Am J Physiol Regul Integr Comp Physiol. 2011;300(1):R12–20. doi: 10.1152/ajpregu.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elumalai S, Karunakaran U, Lee IK, Moon JS, Won KC. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017;11:126–134. doi: 10.1016/j.redox.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newsholme P, Morgan D, Rebelato E, Oliveira-Emilio HC, Procopio J, Curi R, Carpinelli A. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia. 2009;52(12):2489–2498. doi: 10.1007/s00125-009-1536-z. [DOI] [PubMed] [Google Scholar]
- 21.Kowluru A, Kowluru RA. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem Pharmacol. 2014;88(3):275–283. doi: 10.1016/j.bcp.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veluthakal R, Arora DK, Goalstone ML, Kowluru RA, Kowluru A. Metabolic Stress Induces Caspase-3 Mediated Degradation and Inactivation of Farnesyl and Geranylgeranyl Transferase Activities in Pancreatic beta-Cells. Cell Physiol Biochem. 2016;39(6):2110–2120. doi: 10.1159/000447907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sreenan S, Sturis J, Pugh W, Burant CF, Polonsky KS. Prevention of hyperglycemia in the Zucker diabetic fatty rat by treatment with metformin or troglitazone. Am J Physiol. 1996;271(4 Pt 1):E742–747. doi: 10.1152/ajpendo.1996.271.4.E742. [DOI] [PubMed] [Google Scholar]
- 24.Lupi R, Del Guerra S, Tellini C, Giannarelli R, Coppelli A, Lorenzetti M, Carmellini M, Mosca F, Navalesi R, Marchetti P. The biguanide compound metformin prevents desensitization of human pancreatic islets induced by high glucose. Eur J Pharmacol. 1999;364(2–3):205–209. doi: 10.1016/s0014-2999(98)00807-3. [DOI] [PubMed] [Google Scholar]
- 25.Patane G, Piro S, Rabuazzo AM, Anello M, Vigneri R, Purrello F. Metformin restores insulin secretion altered by chronic exposure to free fatty acids or high glucose: a direct metformin effect on pancreatic beta-cells. Diabetes. 2000;49(5):735–740. doi: 10.2337/diabetes.49.5.735. [DOI] [PubMed] [Google Scholar]
- 26.Lupi R, Del Guerra S, Fierabracci V, Marselli L, Novelli M, Patane G, Boggi U, Mosca F, Piro S, Del Prato S, Marchetti P. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes. 2002;51(Suppl 1):S134–137. doi: 10.2337/diabetes.51.2007.s134. [DOI] [PubMed] [Google Scholar]
- 27.Yang X, Xu Z, Zhang C, Cai Z, Zhang J. Metformin, beyond an insulin sensitizer, targeting heart and pancreatic beta cells. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbadis.2016.09.019. pii: S0925–4439(16)30243–30250. [DOI] [PubMed] [Google Scholar]
- 28.Moon JS, Karunakaran U, Elumalai S, Lee IK, Lee HW, Kim YW, Won KC. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J Diabetes Complications. 2017;31(1):21–30. doi: 10.1016/j.jdiacomp.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Reddy JM, Samuel FG, McConnell JA, Reddy CP, Beck BW, Hynds DL. Non-prenylatable, cytosolic Rac1 alters neurite outgrowth while retaining the ability to be activated. Cell Signal. 2015;27(3):630–637. doi: 10.1016/j.cellsig.2014.11.033. [DOI] [PubMed] [Google Scholar]
- 30.Syeda K, Mohammed AM, Arora DK, Kowluru A. Glucotoxic conditions induce endoplasmic reticulum stress to cause caspase 3 mediated lamin B degradation in pancreatic beta-cells: protection by nifedipine. Biochem Pharmacol. 2013;86(9):1338–1346. doi: 10.1016/j.bcp.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahman A, Davis B, Lovdahl C, Hanumaiah VT, Feil R, Brakebusch C, Arner A. The small GTPase Rac1 is required for smooth muscle contraction. J Physiol. 2014;592(5):915–926. doi: 10.1113/jphysiol.2013.262998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sidarala V, Veluthakal R, Syeda K, Kowluru A. EHT 1864, a small molecule inhibitor of Ras-related C3 botulinum toxin substrate 1 (Rac1), attenuates glucose-stimulated insulin secretion in pancreatic beta-cells. Cell Signal. 2015;27(6):1159–1167. doi: 10.1016/j.cellsig.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan OM, Ibrahim MX, Jonsson IM, Karlsson C, Liu M, Sjogren AK, Olofsson FJ, Brisslert M, Andersson S, Ohlsson C. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J Clin Invest. 2011;121(2):628–639. doi: 10.1172/JCI43758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunford JE, Rogers MJ, Ebetino FH, Phipps RJ, Coxon FP. Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. J Bone MIner Res. 2006;21(5):684–694. doi: 10.1359/jbmr.060118. [DOI] [PubMed] [Google Scholar]
- 35.Kowluru A. Inappropriate movement of Rac1 contributes to glucotoxicity of the islet β-cell. Cell Cycle. 2017 doi: 10.1080/15384101.2017.1345229. (in press). Doi org/10.1080/15384101. [DOI] [PMC free article] [PubMed]
- 36.Simon-Szabo L, Kokas M, Mandl J, Keri G, Csala M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PloS one. 2014;9(6):e97868. doi: 10.1371/journal.pone.0097868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundquist I, Mohammed Al-Amily I, Meidute Abaraviciene S, Salehi A. Metformin Ameliorates Dysfunctional Traits of Glibenclamide- and Glucose-Induced Insulin Secretion by Suppression of Imposed Overactivity of the Islet Nitric Oxide Synthase-NO System. PloS one. 2016;11(11):e0165668. doi: 10.1371/journal.pone.0165668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashemitabar M, Bahramzadeh S, Saremy S, Nejaddehbashi F. Glucose plus metformin compared with glucose alone on beta-cell function in mouse pancreatic islets. Biomed Rep. 2015;3(5):721–725. doi: 10.3892/br.2015.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Natalicchio A, Biondi G, Marrano N, Labarbuta R, Tortosa F, Spagnuolo R, D’Oria R, Carchia E, Leonardini A, Cignarelli A. Long-Term Exposure of Pancreatic beta-Cells to Palmitate Results in SREBP-1C-Dependent Decreases in GLP-1 Receptor Signaling via CREB and AKT and Insulin Secretory Response. 2016;157(6):2243–2258. doi: 10.1210/en.2015-2003. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X, Zhou Z, Zhang J. Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK-dependent autophagy. Int J Biol Sci. 2014;10(3):268–277. doi: 10.7150/ijbs.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kowluru A, Kowluru RA. Protein prenylation in islet beta-cell function in health and diabetes: Putting the pieces of the puzzle together. Biochem Pharmacol. 2015;98(3):363–370. doi: 10.1016/j.bcp.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]