

Abstract
AIM
To investigate the role of suppressor of cytokine signaling 1 (SOCS1) in regulating MET-mediated invasive potential of hepatocellular carcinoma (HCC) cells.
METHODS
Stable derivatives of mouse (Hepa1-6) and human (hep3B, HepG2) HCC cell lines expressing SOCS1 or control vector were evaluated for their ability to migrate towards hepatocyte growth factor (HGF) in the transwell migration assay, invade extracellular matrix in response to HGF stimulation in a 3-D invasion assay by confocal microscopy, and to undergo anchorage-independent proliferation in semisolid agar. Following intravenous and intrasplenic inoculation into NOD.scid.gamma mice, the ability of Hepa cells to form othotopic tumors was evaluated. Following HGF stimulation of Hepa and Hep3B cells, expression of proteins implicated in epithelial-to-mesenchymal transition was evaluated by western blot and qRT-PCR.
RESULTS
SOCS1 expression in mouse and human HCC cells inhibited HGF-induced migration through matrigel. In the 3-D invasion assay, HGF stimulation induced invasion of HCC cells across type-I collagen matrix, and SOCS1 expression significantly reduced the depth of invasion. SOCS1 expression also reduced the number and size of colonies formed by anchorage-independent growth in semisolid agar. Following intravenous inoculation, control Hepa cell formed large tumor nodules that obliterated the liver whereas the SOCS1-expressing Hepa cells formed significantly smaller nodules. Tumors formed by SOCS1-expressing cells showed reduced phosphorylation of STAT3 and ERK that was accompanied by reduced levels of MET protein expression. HGF stimulated Hepa cells expressing SOCS1 showed increased expression of E-cadherin and decreased expression of EGR1, SNAI1 and ZEB1. Comparable results were obtained with Hep3B cells. SOCS1 expressing HCC cells also showed reduced levels of EGR1 and SNAI1 transcripts.
CONCLUSION
Our findings indicate that loss of SOCS1-dependent control over epithelial-to-mesenchymal transition may contribute to MET-mediated migration, invasion and metastatic growth of HCC.
Keywords: Migration, Invasion, MET, Hepatocellular carcinoma, Suppressor of cytokine signaling 1
Core tip: The suppressor of cytokine signaling 1 (SOCS1) gene is frequently repressed in primary hepatocellular carcinoma (HCC) specimens, and mice lacking SOCS1 are highly susceptible to experimental HCC. The tumor suppressor functions of SOCS1 in HCC are not yet fully understood. We have shown that SOCS1 regulates hepatocyte growth factor signaling via the MET receptor in HCC cells and inhibits their growth. In this study, we characterize SOCS1 as a regulator of MET-mediated migration and invasion of HCC cells. We propose that SOCS1 gene expression status could be exploited as a selection marker for precision therapies targeting MET in HCC.
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most prevalent cancers and a leading cause of worldwide cancer mortality[1]. HCC develops slowly over two to three decades and most cases present advanced disease at the time of diagnosis. Treatment options for HCC are constrained by the extent of disease, and are very limited and less effective in patients with advanced disease. Therefore, reducing HCC-associated mortality is critically dependent on the development of new treatment methods targeting molecular signaling pathways that promote HCC pathogenesis and diagnostic tools to facilitate targeted therapies[2-4].
Invasive intrahepatic dissemination is a key factor in malignant growth of HCC and its poor prognosis[5,6]. Up to 65% of HCC patients also present extrahepatic metastasis at autopsy[7-9]. HCC can also metastasize to stomach via direct invasion[10]. Detachment from the tumor mass and invasion of the extracellular matrix and the basement membrane are important steps in tumor cell invasion and metastasis. Cancer cells gain these abilities through epithelial-mesenchymal transition (EMT), a developmental genetic program that is crucial for embryogenesis and wound healing response[11,12]. A wide spectrum of paracrine (from the tumor stroma) and aurocrine cytokines and growth factors elicit and modulate the EMT program[12]. Even though TGFβ is the most important inducer of EMT, growth factor receptor tyrosine kinase (RTK) signaling induced by hepatocyte growth factor (HGF), epidermal growth factor and platelet-derived growth factor can also activate the EMT program in carcinomas[12,13].
The HGF receptor c-MET is overexpressed in many human cancers including HCC[14]. MET not only promotes neoplastic growth of HCC cells but also facilitates tumor metastasis by promoting EMT[14-16]. Recent studies have implicated the microRNAs miR-148a and miR-449a in regulating EMT in HCC cells by targeting the MET receptor[17,18]. Previously, we have shown that suppressor of cytokine signaling 1 (SOCS1) is an important regulator of HGF signaling hepatocytes. SOCS1 deficiency in primary mouse hepatocytes increases MET signaling and cell proliferation, whereas stable expression of SOCS1 in human HCC cells attenuates HGF-induced cell growth[19,20]. We have also shown that SOCS1 binds to the MET receptor and promotes its ubiquitination and proteasomal degradation[20]. The SOCS1 gene is frequently repressed in HCC, and Socs1-deficient mice show high susceptibility to experimental HCC with larger and more numerous tumor nodules[21-23], suggesting a role for SOCS1 in controlling MET-mediated tumor cell invasion.
In this study, we examined the role of SOCS1 in MET-mediated migration and invasion of HCC cells and their growth following delivery to the liver. Our findings show that SOCS1 inhibits the invasive potential of HCC cells and reduces orthotopic tumor growth that could result at least partly from modulating the expression of proteins implicated in EMT.
MATERIALS AND METHODS
Cell lines, antibodies and reagents
HCC cell lines of mouse (Hepa1-6; Hepa) and human origin (Hep3B, HepG2) were obtained from ATCC and grown in Dulbecco’s modified Eagles medium (DMEM) containing 10% fetal bovine serum (FBS, from Sigma). Cells stably expressing full-length HA-SOCS1 through lentiviral transduction have been previously described[20]. Recombinant mouse and human HGF were from Peprotech (Rocky Hill, NJ). Phospho-ERK (T202/Y204; #9106), phosphor-STAT3 (Y705; #9131), E-cadherin (24E10), ZO-1 (D7D12), N-cadherin (D4R1H), vimentin (D21H3), EGR1 (44D5), SNAI1 (C15D3) and ZEB1 (D80D3) antibodies were from Cell Signaling Technology (Beverly, MA). Antibodies (Ab) against total ERK (sc-93, sc-153), STAT3 (sc-483) and MET (sc-161) were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse mAb against β-actin (A4700) was from Sigma Aldrich (Oakville, ON, Canada). Secondary antibodies were from Jackson Immunoresearch Laboratories Inc. (Cedarlane, Burlington, ON, United States). Calcein was from Calbiochem (San Diego, CA, United States).
Orthotopic tumor growth
Male NOD.scid.gamma (NSG) mice (8-12 wk old) purchased from the Jackson Labs (Bar Harbor, ME, United States) were used to evaluate tumor growth in vivo under protocols approved by the Université de Sherbrooke ethical committee on animal care and use. To evaluate the growth of hepatoma cells in the liver, cells were injected via intravenous or intrasplenic/portal route. For intravenous inoculation, 106 Hepa-vector or Hepa-SOCS1 cells in 100 μL volume were injected via the caudal vein. For intrasplenic inoculation, mice were anesthetized with ketamine (10 mg/kg) and the spleen was exposed through a small abdominal incision[24]. Tumor cells (106 cells in 100 μL) were injected into the spleen and mice were splenectomized 2 min later. Tumor nodules in the liver were examined 20 d later when the animals began to show distress. The images of hematoxylin and eosin (H and E) -stained sections of the liver were acquired using Nanozoomer Slide Scanner and analyzed by the Nanozoomer Digital Pathology (NDP) software (Hamamatsu Photonics, Japan).
Soft agar colony formation
After adding the bottom layer of 0.6% agar in 2 × DMEM containing 20% FBS to 100 mm Petri dishes, 5000 HCC cells suspended in 0.3% agar in 2 × DMEM containing 10% were added FBS as the top layer. Cells were fed every 3-5 d by overlying fresh 500 μL of DMEM containing 10% FBS. After 3 wk culture, colonies were stained with 1 mg/mL methylthiazol tetrazolium for 3 h, photographed and counted using the NIH ImageJ software.
Transwell migration assay
Migration of cells was assessed using transwells (Corning) with 8 μmol/L pore polycarbonated inserts, coated with growth factor-reduced matrigel (BD Pharmingen Biosciences, Palo Alto, CA, United States). The upper chamber contained 2 × 104 cells and the lower chamber contained 0.7 mL of complete medium with or without HGF. Migration through the membrane was determined after 24 h of incubation at 37 °C. Cells remaining on the top side of the transwell membrane were removed using a cotton swab, and cells migrated to bottom were stained with 0.5% crystal violet and examined by microscopy. Cells in five random fields of the lower side of the membrane were counted. Data represent means ± SEM of three independent experiments.
3-dimensional invasion assay
The wells of a 96-well microtiter plate was covered with 50 μL of 1% agarose containing 10% FBS as the bottom layer and overlaid with 50 μL of fibrillar type-I collagen (3 mg/mL) (R and D Systems, Minneapolis, MN, United States). Control and SOCS1-expressing HCC cells were serum starved overnight and layered on the top (20000 cells/well in 100 μL volume) in the presence or absence of HGF. After 48 h incubation, the cells were labeled with CellTraceTM Calcein Green, AM (Invitrogen, CA, United States) for 1 h, washed with PBS, fixed with 3% glutaraldehyde and confocal images were acquired using the FV1000 Olympus confocal microscope. The collagen matrix was scanned along the Z-axis at incremental 5 μm depths in order to reconstruct 3-D images. The images were analyzed to assess the depth of cell migration, which is expressed as ratio of the fluorescence intensity at each 5 μm layer over the fluorescence intensity of the non-invaded cells at the top 5 μm layer.
Western blot
Lysates of liver tissues harboring the tumor nodules were prepared in a buffer containing 150 mmol/L NaCl, 50 mmol/L Tris-HCl, 1 mmol/L EDTA, pH 8.0 and protease and phosphatase inhibitor cocktails (Roche, Indianapolis, IN) for 2 min using the bead mill MM 400 (Retsch, Hann, Germany). After adding equal volume of the same buffer containing 0.2% SDS, 1% Triton X-100 and 1% sodium deoxycholate, the lysates were kept on a shaker at 4 °C for 30 min. Following centrifugation at 15000 g for 20 min, the supernatant was collected and protein concentration determined using the RC-DC Protein Assay Kit (Bio-Rad, Mississauga, ON, United States). Cultured cells were lysed directly in 1 × SDS-PAGE sample buffer. Aliquots of 30-50 μg proteins were analyzed by western blot. Secondary antibodies and enhanced chemiluminescence reagents were from GE Healthcare Life Sciences (Pittsburg, PA, United States). Western blot images were captured by the VersaDOC 5000 imaging system (Bio-Rad). Densitometric quantification of the western blot bands was carried out using the NIH ImageJ 1.62 software.
Real-time RT-PCR
For RT-PCR analysis, total RNA was isolated from 1 × 106 cells, either unstimulated or stimulated with HGF for the indicated periods of time, using RiboZol™ (AMRESCO, Solon, OH, United States). After verifying the RNA quality by UV absorption, the first complementary strand was made from 1 μg total RNA using QuantiTect® reverse transcription kit (Qiagen). The primers for gene expression analysis are as follows: Mouse Egr1 (NM_007913): AGCGCCTTCAATCCTCAAG and TTTGGCTGGGATAACTCGTC; Snai1 (NM_011427): GTGAAGAGATACCAGTGCCAG and AAGATGCCAGCGAGGATG; Human EGR1 (NM_001964): TGTCACCAACTCCTTCAGC and TCCTGTCCTTTAAGTCTCTTGTG; SNAI1: TCTAGGCCCTGGCTGCTACAA and ACATCTGAGTGGGTCTGGAGGTG; housekeeping genes: mouse Gapdh (NM_008084.3): ATGACATCAAGAAGGTGGTGAA and GTCTTACTCCTTGGAGGCCATGT; Human GAPDH (NM_002046.5): GATGACATCAAGAAGGTGGTGAA and GTCTTACTCCTTGGAGGCCATGT. All primers showed more than 90% efficiency with a single melting curve in the MyQi5® cycler (Bio-Rad, Mississauga, ON, United States). Expression levels of the housekeeping gene were used to calculate fold induction of the specific genes modulated by the presence or absence of SOCS1.
Statistical analysis
Statistical significance of the differences in cell proliferation, tumor growth and protein expression was evaluated by student’s t-test.
RESULTS
Previously we have shown that SOCS1 inhibited HGF-induced MET signaling, cell proliferation and migration in mouse (Hepa) and human (Hep3B, HepG2, SNU-423) HCC cell lines[19,20]. Here, we investigated whether SOCS1 also inhibits HGF-induced invasion of the extracellular matrix in some of these cell lines. At the concentration previously found to be effective in inducing cell proliferation and migration[19], HGF induced cell scattering in murine Hepa cells and human Hep3B cells, which was attenuated by SOCS1 (Figure 1A). In a trans-well migration assay, Hepa cells expressing the control vector showed marked ability to invade the matrigel in response to HGF, whereas the SOCS1-expressing cells showed significantly reduced ability (Figure 1B). Similar reduction of HGF-induced matrix invasion by SOCS1 was also observed with the human HCC cell line Hep3B (Figure 1C).
Figure 1.
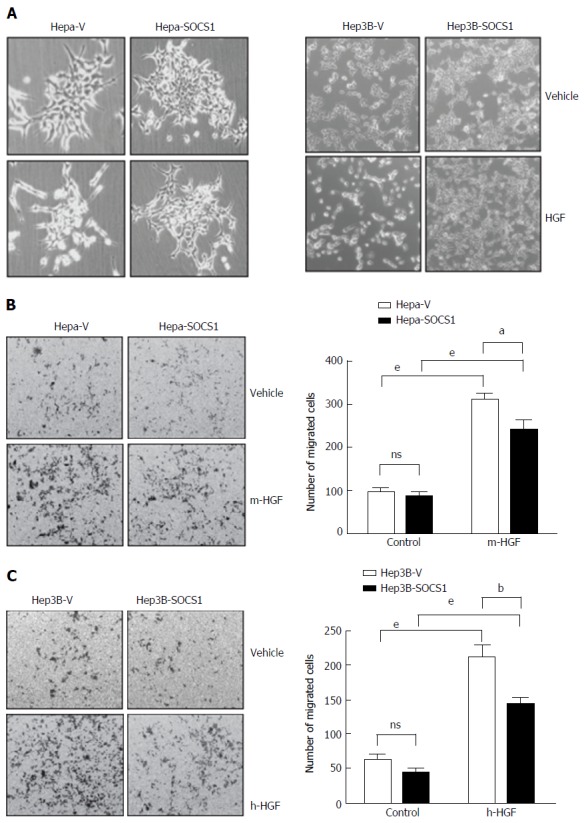
Suppressor of cytokine signaling 1 attenuates hepatocyte growth factor-induced scattering and migration of hepatocellular carcinoma cells. A: Hepa and Hep3B cells expressing SOCS1 (Hepa-SOCS1, Hep3B-SOCS1) or control vector (Hepa-V, Hep3B-V) were grown to less than 50% confluent state, starved in serum-free medium for 16 h and exposed to murine or human HGF (HGF, 25 ng/mL), respectively. After 48 h, the culture plates were examined by phase contrast microscopy to visualize HGF-induced cell scattering; B: Hepa-SOCS1 and Hepa-V cells were seeded onto Matrigel-coated trans-well migration chambers, and migration towards m-HGF was evaluated after 48 h. Representative images of cells that had migrated across the membrane and the number of migrated cells in five random fields from three separate experiments (mean ± SE) are shown; C: Migration of Hep3B cells expressing SOCS1 (Hep3B-SOCS1) or control vector (Hepa-V) towards human HGF (h-HGF, 10 ng/mL). aP < 0.05, bP < 0.01,eP < 0.001. SOCS1: Suppressor of cytokine signaling 1.
Next, we used a 3-D matrix invasion assay using collagen as matrix, where the depth of invasion was evaluated by confocal microscopy and quantified (Figure 2A and B). HGF stimulation increased the ability of Hepa, Hep3B and HepG2 cells to invade the collagen matrix, and this invasive potential was markedly pronounced in Hep3B and HepG2 cells, as indicated by the maximum depth of invasion (Figure 2A and C). In all the three cell lines, SOCS1 reduced the HGF-induced invasive potential (Figure 2A and C). Even though the SOCS1-mediated inhibition of HGF-induced invasion was only marginal in Hepa cells, Hep3B-SOCS1 and HepG2-SOCS1 cells showed significant reduction in HGF-induced invasive potential (Figure 2D). These results indicated that SOCS1 is an important regulator of MET signaling that promotes cell migration and invasion.
Figure 2.
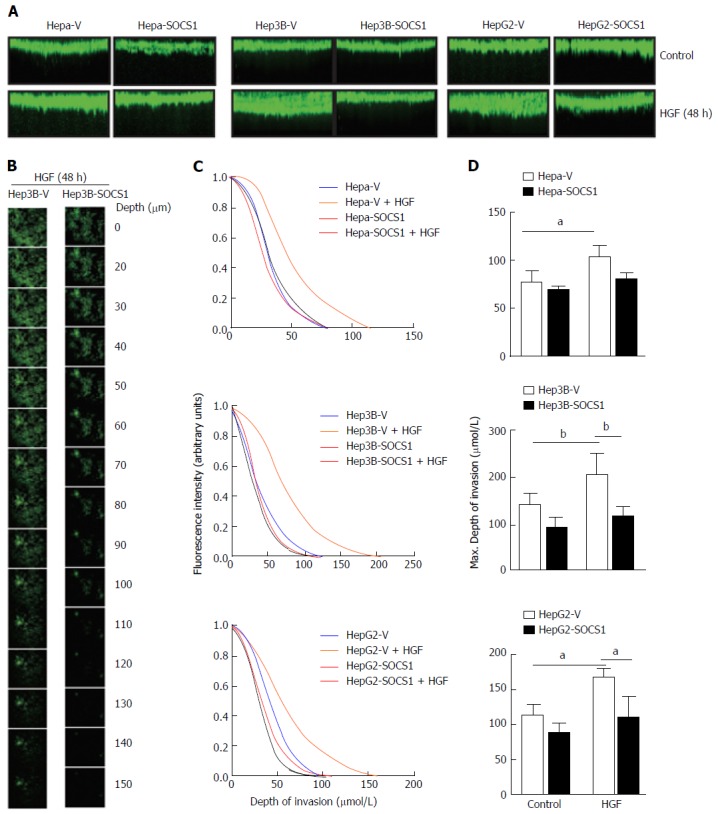
Inhibition of hepatocyte growth factor-induced invasion of collagen matrix by suppressor of cytokine signaling 1 in hepatocellular carcinoma cells. Hepa-V and Hepa-SOCS1, Hep3B-V and Hep3B-SOCS1, and HepG2-V and HepG2-SOCS1 cell lines were evaluated for their ability to invade collagen matrix in a 3-D invasion assay in the presence or absence of mouse HGF (25 ng/mL for Hepa cells) or human HGF (30 ng/mL for Hep3B, HepG2 cells). After 48 h, cells were fluorescently labeled with calcein Green, fixed and examined by confocal microscopy. A: The reconstructed cross-sectional images of cell migration, representative of two independent experiments, are shown; B: Representative images of Hep3B-V and Hep3B-SOCS1 cells migrated to different planes of the collagen-agarose matrix along the z-axis; C: Efficiency of migration (ratio of the fluorescence intensity at each 5 μm layer over the fluorescence intensity of the non-invaded cells at the top 5 μm); D: Maximum depth of invasion by control and SOCS1-expressing cells in the absence or presence of HGF. aP < 0.05, bP < 0.01. SOCS1: Suppressor of cytokine signaling 1.
Tumor dissemination requires not only the ability to invade but also the ability to survive and undergo anchorage-independent proliferation. Therefore, we compared the ability of control and SOCS1-expressing HCC cells to form colonies in semi-solid agar. As shown in Figure 3A, Hepa-SOCS1 cells form fewer and smaller colonies than Hepa-V cells. Similar reduction of colony forming ability was also observed in Hep3B-SOCS1 and HepG2-SOCS1 cells compared to controls (Figure 3B).
Figure 3.
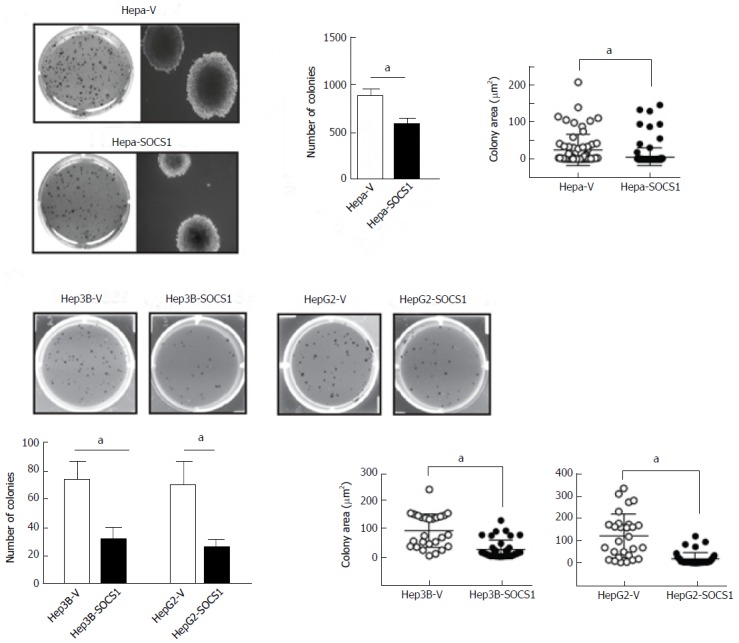
Reduced anchorage-independent proliferation of hepatocellular carcinoma cells expressing suppressor of cytokine signaling 1. SOCS1-expressing and control mouse Hepa (A) and human Hep3B and HepG2 (B) HCC cell lines were evaluated for their ability to form colonies in soft agar. After 3 wk culture, the colonies were photographed, and the number and size of colonies (area; mean ± SE) were evaluated from triplicates of two separate experiments. aP < 0.05. SOCS1: Suppressor of cytokine signaling 1.
To evaluate whether SOCS1 attenuates invasive tumor growth within the liver, we delivered control and SOCS1-expressing HCC cells via parenteral inoculation to the liver. For this purpose, we used murine Hepa1-6 cells, which can utilize endogenous HGF and other mouse growth factors better than human hepatoma cells[25]. Following intravenous injection, the control cells formed numerous orthotopic tumor nodules that obliterated the liver, whereas Hepa-SOCS1 cells induced fewer nodules (Figure 4A). The lungs of these mice did not show macroscopically visible tumor, suggesting that Hepa cells preferentially colonized the liver. We also delivered the HCC cells to the liver via intrasplenic route through the portal circulation. In the latter setting, Hepa-vector cells obliterated the liver with numerous macroscopically visible nodules, whereas Hepa-SOCS1 cells formed markedly smaller nodules (Figure 4B and C). Orthotopic tumors formed by Hepa-SOCS1 showed markedly diminished phosphorylation of STAT3 and ERK (Figure 4D), presumably through SOCS1-mediated inhibition of HGF and other cytokine and growth factor signaling[19,26,27]. Moreover, the SOCS1 expressing tumors showed significantly reduced levels of MET expression (Figure 4E), in agreement with our previous report that SOCS1 targets the activated MET receptor to the proteasomal degradation machinery[20]. These results indicated that SOCS1 inhibits the invasive growth potential of HCC cells and that at least part of this invasive growth could result from attenuation of HGF-induced MET signaling.
Figure 4.
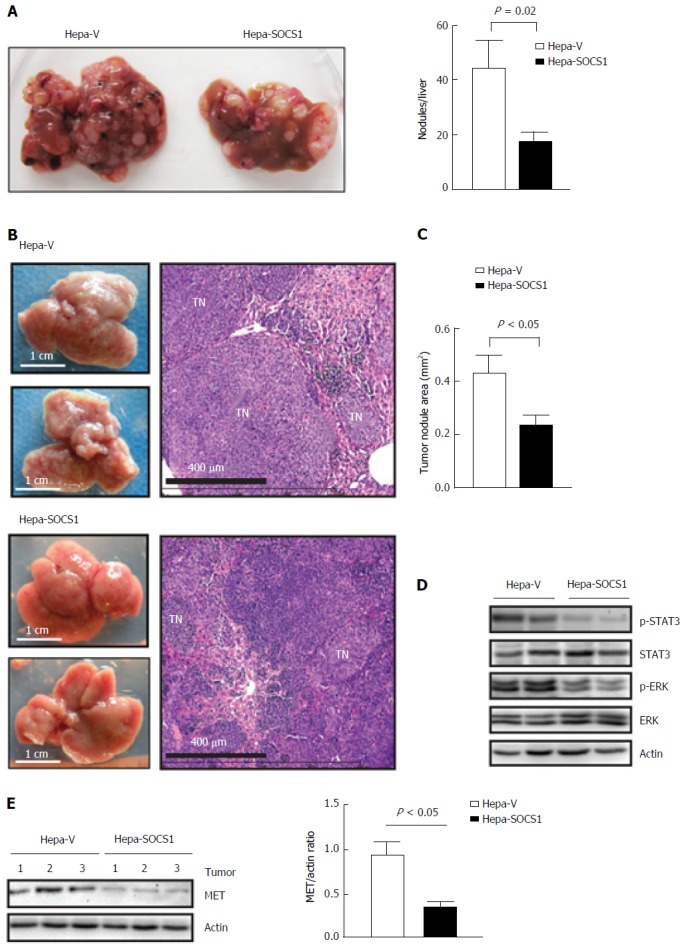
Suppressor of cytokine signaling 1 attenuates invasive growth of orthotopic Hepa cell tumors. A: Hepa-V and Hepa-SOCS1 cells were injected via intravenous route into NOD.scid.gamma (NSG) mice and the livers were examined macroscopically after 20 d. Representative images (left) and quantification of the liver tumor nodules (right) from four mice per group are shown; B-D: Hepa-vector and Hepa-SOCS1 were injected via intrasplenic route into NSG mice (1 × 106 cells per mouse; n = 6 per group from two separate experiments) and the mice were splenectomized. After 20 d, the liver tissues were examined macroscopically for tumor nodules. The dorsal and ventral views of representative livers (B, left) and H and E-stained sections of a representative liver for each group (B, right) are shown; C: For each group, the areas of thirty random tumor nodules from three different mice were measured digitally using the NDP software and compared by Student’s paired t test; D: Western blot evaluation of STAT3 and ERK phosphorylation in the tumors from 2 mice per group; E: Western blot evaluation of MET expression and its quantification in the tumors from 3 mice per group. SOCS1: Suppressor of cytokine signaling 1.
To understand the mechanisms underlying SOCS1-mediated attenuation of the invasive growth of HCC cells, we evaluated the expression of proteins implicated in EMT that promote tumor cell spreading following HGF stimulation[28-30]. Even though HGF stimulation did not appreciably affect the expression of E-Cadherin in control or SOCS1-expressing Hepa and Hep3B cells, the SOCS1-expressing cells showed increased basal expression of E-Cadherin (Figure 5A). SOCS1-expresing Hepa cells but not Hep3B cells showed discernible increase in ZO-1 expression, whereas the expression of N-Cadherin was not affected by SOCS1 in both cell lines. Analysis of gene expression showed marked reduction in the induction Egr1 and Snai1 genes in Hepa-SOCS1 cells, and EGR1 and SNAI2 genes in Hep3B-SOCS1 cells (Figure 5B). Western blot analysis showed reduced expression of EGR1, SNAI1 and ZEB1 in Hepa-SOCS1 cells compared to Hepa-V cells, whereas only SNAI1 showed appreciable reduction in Hep3B cells (Figure 5A). These findings suggest that SOCS1 may modulate diverse components of the EMT signaling machinery during MET-mediated invasion of HCC cells.
Figure 5.
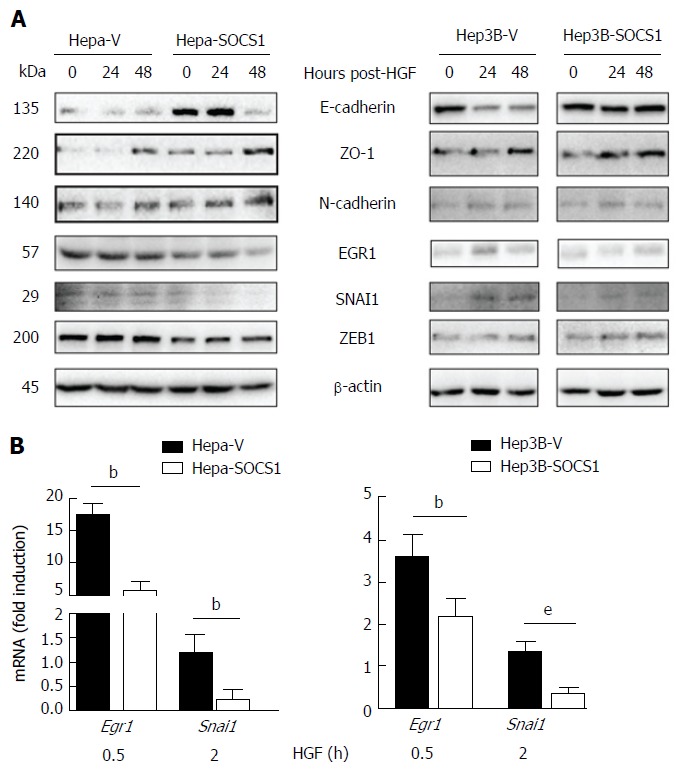
Effect of suppressor of cytokine signaling 1 on the expression of proteins modulated during epithelial-mesenchymal transition in hepatocyte growth factor-stimulated hepatocellular carcinoma cells. A: Hepa-V and Hepa-SOCS1, and Hep3B-V and Hep3B-SOCS1 were cultured in low-serum medium (0.25%) overnight and stimulated or not with m-HGF or h-HGF for 24 or 48 h. Unstimulated and HGF-stimulated cells were lysed in SDS sample buffer and analyzed by western blot for the indicated proteins. Representative data from two experiments are shown; B: Cells were stimulated with HGF for 30 min or 2 h and the expression of Egr1 and Snai1 genes was evaluated by qRT-PCR. Fold-induction relative to the housekeeping gene Gapdh was evaluated from triplicates of two separate experiments (mean ± SE). bP < 0.01,eP < 0.001. SOCS1: Suppressor of cytokine signaling 1.
DISCUSSION
The SOCS1 gene is frequently repressed in HCC by promoter CpG methylation[21,31,32]. Recent meta-analyses of published works on DNA methylation status of several HCC-associated genes have shown that SOCS1 gene promoter hypermethylation is an important risk factor and predictive biomarker for HCC[33,34]. Consistent with the role of SOCS1 as a putative tumor suppressor in HCC, others and we have shown that loss of Socs1 gene expression in mice increases susceptibility to HCC induction[22,23]. The loss of SOCS1 expression might contribute to HCC progression via multiple mechanisms. We have shown that SOCS1 is an important regulator of oncogenic MET RTK signaling and suppresses the oncogenic potential of P21CIP in hepatocytes[19,20,23]. These reports suggested that the loss of SOCS1 expression in HCC by epigenetic mechanisms could promote cancer cell survival, migration and proliferation. The findings of the present study indicate that the loss of SOCS1-dependent regulation of HGF/MET signaling may also contribute to invasive growth of HCC cells through, at least partly, via promoting EMT.
Aberrant MET signaling that facilitate tumor progression and metastasis occurs in many cancers mainly via receptor overexpression, gene amplification or through the loss of MET-targeting microRNAs, although activating mutations and increased autocrine HGF stimulation can also amplify MET signaling[14]. While gain-of-function mutations and gene amplification of MET are rare in HCC, MET overexpression has been frequently reported[14]. The elevated MET expression would not only lead to ligand-independent activation but also confer increased responsiveness to des-gamma-carboxy prothrombin, which is highly expressed in HCC[35]. The molecular basis of MET overexpression in HCC has not been elucidated yet[14]. The high frequency of SOCS1 promoter methylation in HCC[33,34] and the ability of SOCS1 to promote MET degradation by proteasomes[20] suggest that MET overexpression in HCC could result at least partly from the loss of SOCS1 mediated control of MET expression.
The MET signaling pathway is an important mediator of EMT in HCC progression via intra- and extra- hepatic metastases[36,37]. EMT is mediated via induction of transcription factors (SNAI1, SNAI2, ZEB1, ZEB2, TWIST1), which repress genes coding for proteins that maintain epithelial cell integrity[12,13]. During EMT, progressive loss of epithelial markers (E-cadherin, occludin, ZO-1) and acquisition of mesenchymal markers (N-cadherin, vimentin) cause tumor cells to disrupt intercellular junctions, become motile, remodel cell-matrix interactions and become invasive. However, these changes may not be absolute and intermediate phenotypes may represent partial EMT[37]. Reduced expression of E-cadherin occurs in 50%-60% of primary HCC that correlates with increased SNAI1 expression and poor prognosis[38]. Similar changes were reported in human and murine HCC cell lines with high invasive potential[36,38]. Indeed, elevated MET expression has been associated with mesenchymal phenotype characterized by reduced expression of E-Cadherin and increased expression of the CDH1 gene repressor ZEB2 in human HCC cell lines MHCC97-L and MHCC97-H that display increased metastatic potential[16]. Similar findings have been reported in murine HCC cell lines[15]. A murine HCC line-derived circulating tumor cells isolated following intrahepatic implantation displayed elevated MET expression with reduced levels of E-Cadherin, implicating the EMT process in HCC dissemination[39]. Our findings that SOCS1 expression reduces HGF-induced invasive growth in HCC cells that is accompanied by increased expression of E-cadherin and discernibly reduced expression of EGR1, SNAI1 and ZEB1, as well as the markedly diminished orthotopic growth of SOCS1-expressing HCC cells associated with reduced MET expression support the idea that SOCS1 is an important regulator of MET-mediated cancer progression via EMT. Thus, our findings add SOCS1 to the growing list of endogenous regulators of MET-mediated EMT, namely miR-148a, miR449a, SENP1[17,30,40].
Molecular mechanisms underlying MET-mediated EMT are not yet fully elucidated. In HepG2 human HCC cells, HGF-induced cell scattering requires ERK activation, leading to induction of EGR1 (Early growth response factor), a transcriptional activator of SNAI1, which represses CDH1 encoding E-cadherin[28]. The HGF-induced morphological and molecular changes of EMT in HCC cells could be reversed by blocking ERK activation using sorafenib that targets RTKs, or by U0126 that inhibits the ERK upstream kinase MEK1[29]. Given that the MET signaling pathways are implicated in multiple aspects of cancer progression, attenuating MET signaling will also disrupt MET-mediated EMT[37]. Several therapeutics targeting the MET signaling pathway including multikinase inhibitors, selective MET inhibitors, antibodies targeting HGF or MET, agents that neutralize HGF and molecules that disrupt MET signaling[14]. Given the possible emergence of drug resistance to agents that target signaling pathways, and that MET can be activated by alternate ligands such as des-gamma-carboxy prothrombin and heparin[35,41], alternate strategies are needed to effectively target MET-mediated HCC invasion and metastasis. In this context, anti-MET Ab such as DN-30 that promotes MET degradation[42] and strategies to disrupt MET receptor expression[14] could represent promising approaches. However, this would require identification of HCC patients who would benefit from aggressive MET-targeting therapy[14]. Although MET overexpression could be a direct marker, clinical grade MET Ab are still under development[43]. Given that SOCS1 is an important regulator of MET protein expression, and that loss of SOCS1 expression predominantly occur by promoter methylation in HCC, assessing SOCS1 methylation status could be a useful approach to identify HCC patients who would benefit from MET-targeting therapies.
In conclusion, our findings show that SOCS1 attenuates migration and invasion properties of HCC cells at least partly via modulation of MET-mediated EMT, and controls invasive tumor growth. Based on these findings, we propose that the SOCS1 gene methylation and expression in primary HCC may serve as useful markers to identify patients for targeted MET therapy.
COMMENTS
Background
Invasive intrahepatic dissemination is a key factor in malignant growth of hepatocellular carcinoma (HCC) and its poor prognosis. HCC patients also present extrahepatic metastasis that may occur via direct invasion. Cancer cells gain invasive potential through epithelial-mesenchymal transition (EMT) induced by cytokines and growth factors that stimulate receptor tyrosine kinases (RTK). An important RTK implicated in EMT is the HGF receptor MET. Suppressor of cytokine signaling 1 (SOCS1) is an important regulator of MET signaling in hepatocytes.
Research frontiers
Emerging data implicate EMT in cancer growth and metastasis in HCC and many other cancers. Among the strategies to target EMT, the MET signaling pathway holds promise. The present study investigates the role of SOCS1 in regulating MET signaling involved in the invasive tumor growth of HCC.
Innovations and breakthroughs
This study shows that SOCS1 inhibits the invasive growth of HCC cells in vitro and in vivo, and adds SOCS1 to the growing list of endogenous regulators of MET-mediated EMT.
Applications
SOCS1 expression in HCC biopsies could be a useful biomarker of invasive and metastatic potential to identify patients suitable for treatment with MET-targeting therapies. Development of therapeutics to restore SOCS1 expression in cancer cells could be another approach to attenuate the invasive potential and metastatic growth of HCC.
Terminology
SOCS1 is a negative feedback regulator of cytokine and growth factor signaling. SOCS1 inhibits HGF-induced MET signaling pathways that promote invasive growth, which can be assessed in vitro using a 3-dimensional invasion assay and in vivo by orthotopic tumor growth.
Peer-review
In this study, the authors investigated the role of SOCS1 in the invasion ability of HCC. Extending the findings of their previous work, the authors show that SOCS1 inhibits the invasive growth of HCC cells by targeting MET signaling.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Canada
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional animal care and use committee statement: Tumor growth studies in mice were carried out under protocols approved by the Université de Sherbrooke ethics committee in accordance with Canadian Council on Animal Care guidelines (Protocol number 226-13B).
Conflict-of-interest statement: The authors do not have any conflicts of interest to disclose.
Peer-review started: March 2, 2017
First decision: April 21, 2017
Article in press: July 4, 2017
P- Reviewer: Huang C S- Editor: Gong ZM L- Editor: A E- Editor: Li D
Contributor Information
Yirui Gui, Department of Pediatrics, Immunology Division, Faculty of Medicine and Health Sciences, University of Sherbrooke, Sherbrooke, Québec J1H 5N4, Canada.
Md Gulam Musawwir Khan, Department of Pediatrics, Immunology Division, Faculty of Medicine and Health Sciences, University of Sherbrooke, Sherbrooke, Québec J1H 5N4, Canada.
Diwakar Bobbala, Department of Pediatrics, Immunology Division, Faculty of Medicine and Health Sciences, University of Sherbrooke, Sherbrooke, Québec J1H 5N4, Canada.
Claire Dubois, Department of Pediatrics, Immunology Division, Faculty of Medicine and Health Sciences, University of Sherbrooke, Sherbrooke, Québec J1H 5N4, Canada.
Sheela Ramanathan, Department of Pediatrics, Immunology Division, Faculty of Medicine and Health Sciences, University of Sherbrooke, Sherbrooke, Québec J1H 5N4, Canada.
Caroline Saucier, Department of Anatomy and Cell Biology, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Québec J1H 5N4, Canada.
Subburaj Ilangumaran, Department of Pediatrics, Immunology Division, Faculty of Medicine and Health Sciences, University of Sherbrooke, Sherbrooke, Québec J1H 5N4, Canada. subburaj.ilangumaran@Usherbrooke.ca.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–1763. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 3.Lachenmayer A, Alsinet C, Chang CY, Llovet JM. Molecular approaches to treatment of hepatocellular carcinoma. Dig Liver Dis. 2010;42 Suppl 3:S264–S272. doi: 10.1016/S1590-8658(10)60515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12:408–424. doi: 10.1038/nrclinonc.2015.103. [DOI] [PubMed] [Google Scholar]
- 5.Tang ZY. Hepatocellular carcinoma--cause, treatment and metastasis. World J Gastroenterol. 2001;7:445–454. doi: 10.3748/wjg.v7.i4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu L, Lau SH, Tzang CH, Wen JM, Wang W, Xie D, Huang M, Wang Y, Wu MC, Huang JF, et al. Association of Vimentin overexpression and hepatocellular carcinoma metastasis. Oncogene. 2004;23:298–302. doi: 10.1038/sj.onc.1206483. [DOI] [PubMed] [Google Scholar]
- 7.Nakashima T, Okuda K, Kojiro M, Jimi A, Yamaguchi R, Sakamoto K, Ikari T. Pathology of hepatocellular carcinoma in Japan. 232 Consecutive cases autopsied in ten years. Cancer. 1983;51:863–877. doi: 10.1002/1097-0142(19830301)51:5<863::aid-cncr2820510520>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 8.Kummar S, Shafi NQ. Metastatic hepatocellular carcinoma. Clin Oncol (R Coll Radiol) 2003;15:288–294. doi: 10.1016/s0936-6555(03)00067-0. [DOI] [PubMed] [Google Scholar]
- 9.Kim HS, Shin JW, Kim GY, Kim YM, Cha HJ, Jeong YK, Jeong ID, Bang SJ, Kim DH, Park NH. Metastasis of hepatocellular carcinoma to the small bowel manifested by intussusception. World J Gastroenterol. 2006;12:1969–1971. doi: 10.3748/wjg.v12.i12.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korkolis DP, Aggeli C, Plataniotis GD, Gontikakis E, Zerbinis H, Papantoniou N, Xinopoulos D, Apostolikas N, Vassilopoulos PP. Successful en bloc resection of primary hepatocellular carcinoma directly invading the stomach and pancreas. World J Gastroenterol. 2009;15:1134–1137. doi: 10.3748/wjg.15.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 12.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol. 2014;60:442–452. doi: 10.1016/j.jhep.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Ogunwobi OO, Liu C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin Exp Metastasis. 2011;28:721–731. doi: 10.1007/s10585-011-9404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology. 2011;54:879–889. doi: 10.1002/hep.24450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM. MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling. Oncogene. 2014;33:4069–4076. doi: 10.1038/onc.2013.369. [DOI] [PubMed] [Google Scholar]
- 18.Chen SP, Liu BX, Xu J, Pei XF, Liao YJ, Yuan F, Zheng F. MiR-449a suppresses the epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma by multiple targets. BMC Cancer. 2015;15:706. doi: 10.1186/s12885-015-1738-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gui Y, Yeganeh M, Ramanathan S, Leblanc C, Pomerleau V, Ferbeyre G, Saucier C, Ilangumaran S. SOCS1 controls liver regeneration by regulating HGF signaling in hepatocytes. J Hepatol. 2011;55:1300–1308. doi: 10.1016/j.jhep.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 20.Gui Y, Yeganeh M, Donates YC, Tobelaim WS, Chababi W, Mayhue M, Yoshimura A, Ramanathan S, Saucier C, Ilangumaran S. Regulation of MET receptor tyrosine kinase signaling by suppressor of cytokine signaling 1 in hepatocellular carcinoma. Oncogene. 2015;34:5718–5728. doi: 10.1038/onc.2015.20. [DOI] [PubMed] [Google Scholar]
- 21.Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, Harris CC, Herman JG. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. 2001;28:29–35. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida T, Ogata H, Kamio M, Joo A, Shiraishi H, Tokunaga Y, Sata M, Nagai H, Yoshimura A. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J Exp Med. 2004;199:1701–1707. doi: 10.1084/jem.20031675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeganeh M, Gui Y, Kandhi R, Bobbala D, Tobelaim WS, Saucier C, Yoshimura A, Ferbeyre G, Ramanathan S, Ilangumaran S. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21(CIP1/WAF1) in the liver. Oncogene. 2016;35:4200–4211. doi: 10.1038/onc.2015.485. [DOI] [PubMed] [Google Scholar]
- 24.Bernier J, Chababi W, Pomerleau V, Saucier C. Oncogenic engagement of the Met receptor is sufficient to evoke angiogenic, tumorigenic, and metastatic activities in rat intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2010;299:G677–G686. doi: 10.1152/ajpgi.00315.2009. [DOI] [PubMed] [Google Scholar]
- 25.Francone TD, Landmann RG, Chen CT, Sun MY, Kuntz EJ, Zeng Z, Dematteo RP, Paty PB, Weiser MR. Novel xenograft model expressing human hepatocyte growth factor shows ligand-dependent growth of c-Met-expressing tumors. Mol Cancer Ther. 2007;6:1460–1466. doi: 10.1158/1535-7163.MCT-06-0466. [DOI] [PubMed] [Google Scholar]
- 26.Kazi JU, Kabir NN, Flores-Morales A, Rönnstrand L. SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell Mol Life Sci. 2014;71:3297–3310. doi: 10.1007/s00018-014-1619-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol. 2013;2:1–29. [PMC free article] [PubMed] [Google Scholar]
- 28.Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006;25:3534–3545. doi: 10.1038/sj.emboj.7601213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagai T, Arao T, Furuta K, Sakai K, Kudo K, Kaneda H, Tamura D, Aomatsu K, Kimura H, Fujita Y, et al. Sorafenib inhibits the hepatocyte growth factor-mediated epithelial mesenchymal transition in hepatocellular carcinoma. Mol Cancer Ther. 2011;10:169–177. doi: 10.1158/1535-7163.MCT-10-0544. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Sun H, Shi X, Wang H, Cui C, Xiao F, Wu C, Guo X, Wang L. SENP1 regulates hepatocyte growth factor-induced migration and epithelial-mesenchymal transition of hepatocellular carcinoma. Tumour Biol. 2016;37:7741–7748. doi: 10.1007/s13277-015-4406-y. [DOI] [PubMed] [Google Scholar]
- 31.Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163:1101–1107. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nomoto S, Kinoshita T, Kato K, Otani S, Kasuya H, Takeda S, Kanazumi N, Sugimoto H, Nakao A. Hypermethylation of multiple genes as clonal markers in multicentric hepatocellular carcinoma. Br J Cancer. 2007;97:1260–1265. doi: 10.1038/sj.bjc.6604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Li J, Huang T, Duan S, Dai D, Jiang D, Sui X, Li D, Chen Y, Ding F, et al. Meta-analysis of DNA methylation biomarkers in hepatocellular carcinoma. Oncotarget. 2016;7:81255–81267. doi: 10.18632/oncotarget.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu M, Cui LH, Li CC, Zhang L. Association of APC, GSTP1 and SOCS1 promoter methylation with the risk of hepatocellular carcinoma: a meta-analysis. Eur J Cancer Prev. 2015;24:470–483. doi: 10.1097/CEJ.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki M, Shiraha H, Fujikawa T, Takaoka N, Ueda N, Nakanishi Y, Koike K, Takaki A, Shiratori Y. Des-gamma-carboxy prothrombin is a potential autologous growth factor for hepatocellular carcinoma. J Biol Chem. 2005;280:6409–6415. doi: 10.1074/jbc.M406714200. [DOI] [PubMed] [Google Scholar]
- 36.Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC, Stiles B, Rountree CB. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology. 2010;52:945–953. doi: 10.1002/hep.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65:798–808. doi: 10.1016/j.jhep.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY, Peng WL, Wu JC. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology. 2009;50:1464–1474. doi: 10.1002/hep.23221. [DOI] [PubMed] [Google Scholar]
- 39.Ogunwobi OO, Puszyk W, Dong HJ, Liu C. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS One. 2013;8:e63765. doi: 10.1371/journal.pone.0063765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozen E, Gozukizil A, Erdal E, Uren A, Bottaro DP, Atabey N. Heparin inhibits Hepatocyte Growth Factor induced motility and invasion of hepatocellular carcinoma cells through early growth response protein 1. PLoS One. 2012;7:e42717. doi: 10.1371/journal.pone.0042717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.İşcan E, Güneş A, Korhan P, Yılmaz Y, Erdal E, Atabey N. The regulatory role of heparin on c-Met signaling in hepatocellular carcinoma cells. J Cell Commun Signal. 2017;11:155–166. doi: 10.1007/s12079-016-0368-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pacchiana G, Chiriaco C, Stella MC, Petronzelli F, De Santis R, Galluzzo M, Carminati P, Comoglio PM, Michieli P, Vigna E. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J Biol Chem. 2010;285:36149–36157. doi: 10.1074/jbc.M110.134031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knudsen BS, Zhao P, Resau J, Cottingham S, Gherardi E, Xu E, Berghuis B, Daugherty J, Grabinski T, Toro J, et al. A novel multipurpose monoclonal antibody for evaluating human c-Met expression in preclinical and clinical settings. Appl Immunohistochem Mol Morphol. 2009;17:57–67. doi: 10.1097/PAI.0b013e3181816ae2. [DOI] [PMC free article] [PubMed] [Google Scholar]