

Abstract
AIM
To determine whether oral glutathione (GSH) administration can alleviate the effects of fasting-induced intestinal atrophy in the small intestinal mucosa.
METHODS
Rats were divided into eight groups. One group was fed ad libitum, another was fed ad libitum and received oral GSH, and six groups were administrated saline (SA) or GSH orally during fasting. Mucosal height, apoptosis, and cell proliferation in the jejunum were histologically evaluated. iNOS protein expression (by immunohistochemistry), nitrite levels (by high performance liquid chromatography, as a measure of NO production), 8-hydroxydeoxyguanosine formation (by ELISA, indicating ROS levels), glutathione/oxidized glutathione (GSH/GSSG) ratio (by enzymatic colorimetric detection), and γ-glutamyl transpeptidase (Ggt1) mRNA levels in the jejunum (by semi-quantitative RT-PCR) were also estimated.
RESULTS
Oral GSH administration was demonstrated to drastically reduce fasting-induced intestinal atrophy in the jejunum. In particular, jejunal mucosal height was enhanced in GSH-treated animals compared to SA-treated animals [527.2 ± 6.9 for 50 mg/kg GSH, 567.6 ± 5.4 for 500 mg/kg GSH vs 483.1 ± 4.9 (μm), P < 0.01 at 72 h]. This effect was consistent with decreasing changes in GSH-treated animals compared to SA-treated animals for iNOS protein staining [0.337 ± 0.016 for 50 mg/kg GSH, 0.317 ± 0.017 for 500 mg/kg GSH vs 0.430 ± 0.023 (area of staining part/area of tissue), P < 0.01 at 72 h] and NO [2.99 ± 0.29 for 50 mg/kg GSH, 2.88 ± 0.19 for 500 mg/kg GSH vs 5.34 ± 0.35 (nmol/g tissue), P < 0.01 at 72 h] and ROS [3.92 ± 0.46 for 50 mg/kg GSH, 4.58 ± 0.29 for 500 mg/kg GSH vs 6.42 ± 0.52 (8-OHdG pg/μg DNA), P < 0.01, P < 0.05 at 72 h, respectively] levels as apoptosis mediators in the jejunum. Furthermore, oral GSH administration attenuated cell proliferation decreases in the fasting jejunum [182.5 ± 1.9 for 500 mg/kg GSH vs 155.8 ± 3.4 (5-BrdU positive cells/10 crypts), P < 0.01 at 72 h]. Notably, both GSH concentration and Ggt1 mRNA expression in the jejunum were also attenuated in rats following oral administration of GSH during fasting as compared with fasting alone [0.45 ± 0.12 vs 0.97 ± 0.06 (nmol/mg tissue), P < 0.01; 1.01 ± 0.11 vs 2.79 ± 0.39 (Ggt1 mRNA/Gapdh mRNA), P < 0.01 for 500 mg/kg GSH at 48 h, respectively].
CONCLUSION
Oral GSH administration during fasting enhances jejunal regenerative potential to minimize intestinal mucosal atrophy by diminishing fasting-mediated ROS generation and enterocyte apoptosis and enhancing cell proliferation.
Keywords: Intestinal atrophy, Glutathione, Apoptosis, Cell proliferation, Inducible nitric oxide synthase
Core tip: We have previously demonstrated that the intestinal mucosal atrophy consequent to fasting is due in large part to increased apoptosis in jejunal villi and decreased cell proliferation in jejunal crypts, with concomitant increased reactive oxygen species (ROS) and NO production and decreased glutathione (GSH). Here we demonstrate protection against fasting-induced intestinal mucosal atrophy by minimizing ROS induction in the intestinal mucosa through supplemental oral administration of an antioxidant such as GSH during fasting in rats. In particular, oral GSH administration during fasting enhances jejunal regenerative potential by diminishing enterocyte apoptosis and enhancing cell proliferation.
INTRODUCTION
Fasting induces small intestinal mucosal atrophy including increased epithelial permeability and compromised tight junctions, which can lead to bacterial translocation, particularly in patients receiving a prolonged course of total parenteral nutrition (TPN)[1,2]. Many surgeons involved in nutritional support therapy desire the identification of bioactive substances that may be efficacious in protecting against intestinal injury during long-term TPN administration, particularly with respect to intestinal barrier function and subsequent septic complications. Fasting and other states of malnutrition are associated with increased reactive oxygen species (ROS) formation, which has been implicated in the loss of intestinal mucosal structure and function under conditions of inflammation, injury, and shock[3,4]. In addition, these fasting states are also accompanied by depletion of the critical antioxidant glutathione (GSH), which functions to eliminate induced ROS in the intestinal mucosa[5-7]. Therefore, GSH is the most prevalent and important low-molecular-weight thiol in mammalian tissues[8]. As GSH deficiency in tissues is associated with increased oxidative stress, aging, and increased risk of numerous chronic diseases as well as fasting[9], the maintenance of tissue levels of GSH is critical for maintaining health and preventing disease and age-related biological insults.
GSH sources in the intestinal mucosa include intracellular synthesis, biliary supply, and dietary intake. The intestinal lumen receives a large quantity of hepatic GSH from biliary secretion[10] and dietary GSH from fresh fruits, vegetables, and many types of meat[11]. As the only enzyme of the γ-glutamyl cycle located on the outer surface of the plasma membrane, γ-glutamyl transpeptidase (GGT) plays a key role in GSH homeostasis by breaking down extracellular GSH and providing cysteine, the rate-limiting substrate for intracellular de novo GSH synthesis[12]. The two intracellular enzymes, γ-glutamylcysteine synthetase and GSH synthetase, catalyze intracellular GSH synthesis from glutamate, cysteine, and glycine, which are translocated into cells by amino acid transporters on the surface of the plasma membrane following the resolution of extracellular GSH by GGT. Intracellular GSH plays an important role in antioxidant defense and the regulation of pathways essential for in vivo homeostasis through its catalysis by glutathione S-transferase (GST) and glutathione peroxidase[9]. Studies of oral GSH supplementation in humans and laboratory animals have shown that the enhancement of intestinal mucosal GSH levels by oral GSH supplementation under conditions in which intracellular GSH status is compromised (i.e., GSH deficiency-related biological insults) can restore tissue GSH and promote ROS metabolism[10,13,14]. Thus, it has been described that orally administered GSH acts as backup for GSH-deficient tissue.
As an alternative, the ability to control extracellular redox provides another possible role for intestinal luminal GSH. Recent studies have shown that the thiol/disulfide redox status in the extracellular compartment regulates the ROS that are generated primarily in the intracellular compartment and serve an important function in the activation of proteins and up-regulation of antioxidant and detoxification systems[15-17]. Furthermore, many proteins including transporters, receptors, and enzymes present on the surface of the plasma membrane and located in extracellular fluids contain cysteine and methionine residues that are subject to oxidation. Accordingly, these proteins respond to variations in the extracellular thiol/disulfide redox environment, which can alter their activity[18]. Notably, recent demonstrations that treatment with GSH leads to a significant increase in the number of free sulfhydryls on the cell surface have led to the conclusion that extracellular GSH modulates the interaction between bacteria and epithelial respiratory cells and inhibits bacterial invasion into these cells[19].
We previously suggested that fasting for 2-3 d causes intestinal mucosal atrophy resulting from increased apoptosis in jejunal villi, nitric oxide (NO) and ROS production following elevated inducible nitric oxide synthase (iNOS) expression, and decreased cell proliferation in jejunal crypts[20]. Conversely, these apoptosis mediators are all suppressed by treatment with aminoguanidine, a selective iNOS inhibitor, with consequent mucosal recovery, suggesting that iNOS, which induces the production of ROS, is likely to constitute a significant upstream factor promoting fasting-induced apoptosis in enterocytes. Furthermore, refeeding repaired fasting-induced jejunal atrophy by inhibiting these apoptosis mediators, including ROS, and also showed an association between ROS inhibition and increased cell proliferation for decreasing intestinal mucosal atrophy[21]. Therefore, we hypothesized that protection against intestinal mucosal atrophy might be provided through the elimination of induced ROS in the intestinal mucosa by introducing an antioxidant such as GSH during a fasting period in rats.
Therefore, the objective of the present study was to investigate the effects of oral GSH administration on the manifestation of fasting-induced intestinal atrophy through the reduction of oxidative stress, with a particular focus on the possible participation of intracellular GSH levels and GGT expression on the cell surface as parameters for de novo intracellular GSH synthesis.
MATERIALS AND METHODS
Animals and experimental design
The experimental protocol and design were approved by the Institutional Animal Care and Use Committee at the Life Science Center of Josai University. Male Wistar rats at 9 wk of age were purchased from SLC (Shizuoka, Japan) and housed in wire-bottomed cages. The rats were placed in a room illuminated from 7:00 am to 7:00 pm (12:12-h light:dark cycle). The animals were allowed free access to deionized water and standard rat chow (CE-2, CLEA Japan) until the study began. At 10 wk of age, 59 rats were randomly divided into eight groups (Figure 1). Each group received saline (SA) or GSH (50 or 500 mg/kg b.w./d) using oral gavage needles at 24, 48, and 72 h before fasting, including a normally fed control group and a normally fed control that received GSH, with the following fasting durations in each group: (1) SA + ad libitum (normal fed control); (2) SA + 48-h fast; (3) SA + 72-h fast; (4) 500 mg/kg GSH + ad libitum (normal fed control received GSH); (5) 50 mg/kg GSH + 48-h fast; (6) 50 mg/kg GSH + 72-h fast; (7) 500 mg/kg GSH + 48-h fast; and (8) 500 mg/kg GSH + 72-h fast. All rats received SA or GSH by oral gavage for 2, 26, and 50 h after fasting. The GSH (Setria® reduced glutathione) used in this work was provided by Kyowa Hakko Bio Co., Ltd. The rats were weighed daily.
Figure 1.
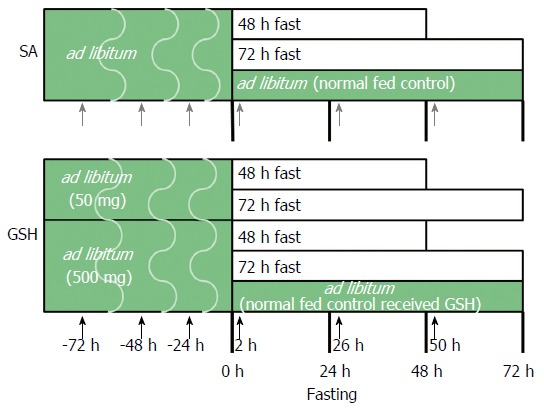
Experimental design. Rats were divided into eight groups, one of which was fed ad libitum, another of which was fed ad libitum and received oral glutathione (GSH), and six of which were administered saline (SA) or GSH orally prior to and after fasting. The large block arrows represent treatment with SA (closed gray arrow) and GSH (closed block arrow).
Collection of intestinal mucosa
After fasting, the rats were anesthetized and then euthanized by exsanguination. The entire small intestine was carefully removed and placed on ice. Then, 10 cm of tissue from the oral (duodenum) side was removed and the remainder of the intestine was divided into two segments: proximal (jejunum) and distal (ileum). The segments used in analyses comprised the jejunum from 3 to 5 cm distal to the duodenum[22]. Samples with a length of approximately 3 cm were fixed in 10% neutral buffered formalin for measurement of mucosal height and immunohistochemistry. The remaining segments were snap-frozen in liquid nitrogen and stored at -80 °C.
Histopathological analysis of mucosal height and apoptotic index
Fixed tissue samples were embedded in paraffin and sectioned (2-3 μm thickness) prior to being stained with hematoxylin and eosin (H&E). Mucosal height (villous height plus crypt depth) was measured using a microscope (BX41; Olympus, Tokyo, Japan) and a digital camera system (Penguin 150CL; Pixera, San Jose, CA, United States). Mucosal height was measured for at least 30 villi per animal.
To detect enterocyte apoptosis in the jejunum, apoptotic index (AI) using conventional light microscopy of H&E-stained specimens was used. We followed previously published methods[20,23]. In brief, jejunal sections as used above for histopathological analysis were examined in a blinded manner for the typical attributes of apoptotic cells. We assessed 50 villus-crypt columns per rat. For each column, the number and position of apoptotic cells and the total number of cells were recorded. To account for the effects of fasting and GSH treatment on apoptosis, the average number of apoptotic cells in villi and crypts was determined along with AI. To identify the locations of apoptosis along the villi and crypts, AI distribution curves were constructed on the basis of group means, in which cell position vs AI was plotted at each position. Here, AI was defined as the total number of apoptotic cells at each cell position and is expressed as the percentage of the total number of cells counted at that cell position. Cell position 1 was set as the cell at the crypt-villus junction and the cell at the base of the crypt column for villus and crypt data, respectively.
Immunohistochemical assessment of iNOS expression and 5-bromo-2’-deoxyuridine (5-BrdU)-positive cells
Immunohistochemical staining was performed using a rabbit anti-iNOS polyclonal antibody[24]. The specimens were dewaxed and treated for antigen retrieval by boiling in 10 mmol/L citrate buffer (pH 6.0)[25]. After being washed with PBS, the specimens were incubated in 6% hydrogen peroxide and nonspecific binding was blocked with a 20% goat serum solution in PBS. Specimens were subsequently incubated with an anti-iNOS primary antibody (1:100; BD Transduction Laboratories, Lexington, KY, United States), except for the control sections, for which no primary antibody was used. A biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA, United States) was used as a secondary antibody. The sections were then treated using the VECTASTAIN Elite ABC Kit (Vector Laboratories), and reaction products were detected using color development with diaminobenzidine. Finally, the sections were counterstained with hematoxylin and examined under a light microscope and a digital camera system. For each rat, 5 clearly dyed sections were chosen randomly and 5 random fields for each section were assessed (at 40 × magnification). The content of iNOS was quantitatively measured based on the average optical density using a digital camera and ImageJ software (National Institutes of Health, Bethesda, Maryland, United States)[26].
To assess cell proliferation in the crypt, the cell proliferation index was determined using conventional light microscopy of specimens immunohistochemically stained for 5-BrdU[27]. Rats were intraperitoneally injected with 100 mg/kg 5-BrdU prior to euthanasia. After paraffin embedding and sectioning, tissue sections were dewaxed and immersed in 3% hydrogen peroxide-methanol solution. The specimens were washed with PBS and denatured in 2 N hydrochloric acid. Following further PBS washing, the specimens were immersed in 0.1 mol/L boric acid buffer (pH 8.5), incubated with 20 μg/mL proteinase K at 37 °C, and then the reaction was terminated with PBS containing bovine serum albumin. The sections were incubated with a mouse anti-5-BrdU monoclonal antibody (1:50; Chemicon International, Billerica, MA, United States), with the exception of control samples, for which the primary antibody was omitted. A biotinylated goat anti-mouse IgG (1:200; Vector Laboratories) was used as a secondary antibody. The sections were then treated using the VECTASTAIN Elite ABC Kit (Vector Laboratories) and antibody binding was detected by color development after addition of diaminobenzidine. Finally, the sections were counterstained with hematoxylin and examined under a light microscope with a digital camera system. The number of labeled cells in at least 10 well-oriented longitudinal crypts was determined for each rat. Results are expressed as the number of 5-BrdU-labeled cells per crypt.
Jejunal nitrite concentrations
Nitrite concentrations in the jejunum were measured using a dedicated high-performance liquid chromatography (HPLC) system (ENO-20; EiCom, Kyoto, Japan)[28]. Jejunal segments were homogenized with an equal volume of methanol and centrifuged for deproteinization at 12000 g at 4 °C for 5 min. The samples were then applied to the HPLC system. The nitrites and nitrates were separated using a reverse-phase column (NO-PAK; EiCom), after which nitrate was reduced to nitrite in a reduction column packed with copperized cadmium (NO-RED; EiCom). These nitrites were then mixed with the Griess reagent in a reaction coil and the change in absorbance was monitored at 540 nm.
DNA oxidation analysis
Oxidative stress in the jejunum was evaluated by quantifying 8-hydroxydeoxyguanosine (8-OHdG) present in DNA[29]. 8-OHdG is a product of oxidative DNA damage following specific enzymatic cleavage after 8-hydroxylation of the guanine base and is thought to constitute a marker of oxidative DNA damage, reflecting the DNA repair rate[30]. Jejunal DNA was purified using the DNA Extractor TIS Kit (Wako Pure Chemical Industries Ltd., Osaka, Japan)[31]. DNA samples were hydrolyzed to nucleosides by sequential incubation with 6 U of nuclease P1 (Wako) followed by 2 U of alkaline phosphatase (Wako). Hydrolysates were filtered through a VIVASPIN 500 MWCO 10000 filter (Sartorius Stedim Biotech, Gottingen, Germany). The levels of 8-OHdG in the filtered samples were determined using an ELISA kit (Japan Institute for the Control of Aging, Shizuoka, Japan).
Jejunal GSH and GSSG concentrations
GSH and GSSG concentrations were measured by using a GSSG/GSH Quantification Kit (Dojindo Molecular Technologies, Inc., Rockville, MD, United States). Jejunal segments were homogenized with 5% sodium sulfosalicylate (Wako) and centrifuged to remove proteins. For each sample, the supernatant was added to a well to which coenzyme working solution and enzyme working solution had been previously added, and incubated at room temperature. Then, the substrate working solution was added to the well and it was incubated at room temperature. The absorbance of samples and the GSH standard was measured at 405 nm using a microplate reader (TECAN GENios, Grodig, Austria). For measurement of GSSG, the supernatant was treated with masking solution and then added to a well as described above. The absorbance of samples and standard GSSG was measured at 405 nm.
Analysis of Ggt1 mRNA expression by semi-quantitative reverse transcription-PCR
Total RNA was purified using the TaKaRa RNAiso Reagent (TaKaRa Bio, Kusatsu, Japan). Reverse transcription (RT)-PCR was performed with total RNA using the RNA PCR kit (AMV) Version 3.0 (TaKaRa Bio): 1 cycle at 42 °C for 30 min, 99 °C for 5 min, and 5 °C for 5 min for reverse transcription, and 30 cycles at 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min for PCR. The following primer pairs (synthesized by TaKaRa Bio) were used: Ggt1 forward primer, 5’-ACCACTCACCCAACCGCCTAC-3’; Ggt1 reverse, 5’-ATCCGAACCTTGCCGTCCTT-3’ (product size: 317 bp)[12]. Expression of target mRNAs was measured relative to that of glyceraldehyde 3-phosphate dehydrogenase (Gapdh), which was determined using the following primers: Gapdh forward, 5’-GGCACAGTCAAGGCTGAGAATG-3’; Gapdh reverse, 5’-ATGGTGGTGAAGACGCCAGTA-3’ (143 bp, TaKaRa Bio ID: RA015380). A portion of each PCR mixture was electrophoresed on a 2% agarose gel in Tris-borate-EDTA buffer and DNA bands were visualized using ethidium bromide staining. PCR product intensity was measured using the Gene Genius Bioimaging System (Syngene, Cambridge, United Kingdom).
Statistical analysis
Statistical analyses were performed using SPSS ver. 22 for Windows. All values are expressed as the mean ± SE. One-way analysis of variance (ANOVA) followed by a Bonferroni multiple-comparison test was used for analyzing the statistical difference between the fasting periods in SA- or GSH-treated groups. Two-way ANOVA followed by a Bonferroni multiple-comparison test was used for analyzing the statistical difference between the SA- and GSH-treated groups for the fasting periods. Statistical significance was accepted at P < 0.05. The statistical analysis of this study was performed by Uchida H, who acquired biostatistics expertise during his training in public health.
RESULTS
Body weight changes
Figure 2 shows body weight changes in SA-treated and GSH-treated rats. Decreases in body weight in both groups were observed along with fasting. Rats fasted for 48 and 72 h with SA treatment showed approximately 15% (P < 0.01) and 19% (P < 0.01) body weight loss, respectively, compared with normally fed controls. There was no difference in weight loss between the GSH treatments and the respective SA-treated groups at each fasting period.
Figure 2.
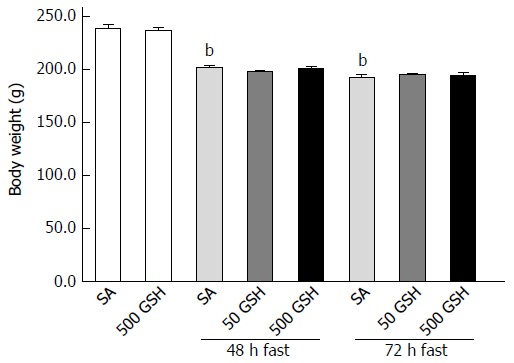
Effects of fasting and glutathione treatment on body weight. Fasting caused gradual decreases in body weight in both SA- and GSH-treated groups. There was no difference in weight loss between the GSH treatments and the respective SA-treated groups in each fasting period. Values represent the mean ± SE. bP < 0.01 vs the normally fed control. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline.
Histological characterization of jejunal mucosal atrophy
Jejunal mucosal atrophy was assessed by jejunal mucosal height. Decreased jejunal mucosal height in both SA- and GSH-treated rats was observed along with fasting (Figure 3). Rats fasted for 48 and 72 h with SA treatment showed a significant decrease (P < 0.01) in mucosal height compared with normally fed controls. However, significant differences (P < 0.01) in jejunal mucosal height were found between the SA- and GSH-treated rats with 48 and 72 h of fasting.
Figure 3.
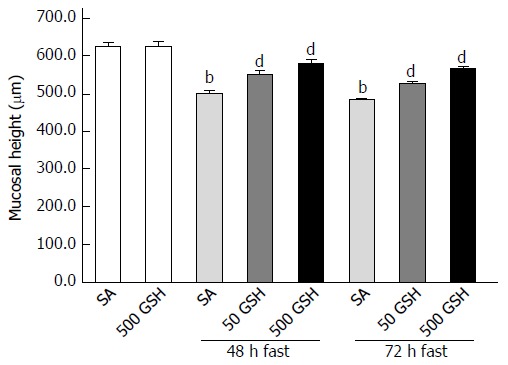
Effects of fasting and glutathione treatment on jejunal mucosal atrophy. Jejunal mucosal atrophy was assessed by jejunal mucosal height. Values represent the mean ± SE. bP < 0.01 vs the normally fed control. dP < 0.01 vs the respective SA-treated group in each fasting period. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline.
iNOS protein expression in the jejunum
As shown in Figure 4A, iNOS protein staining was localized almost exclusively in the mucosal epithelial monolayer in the 48- and 72-h fasting periods compared with the normally fed controls, and a decreased level of staining occurred with GSH treatment. Quantitative measurement using the average optical density revealed that this expression significantly increased following fasting for 48 and 72 h with SA treatment (P < 0.01), whereas GSH treatments significantly decreased the fasting-induced enhancement of jejunal iNOS protein expression (P < 0.05 vs SA at 48 h fasting, P < 0.01 vs SA at 72 h fasting; Figure 4B).
Figure 4.
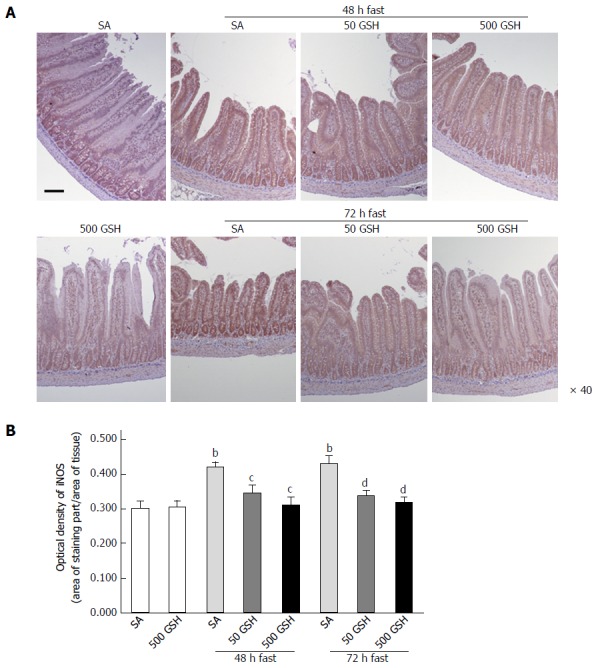
Effects of fasting and glutathione treatment on inducible nitric oxide synthase protein expression in the jejunum. A: Light micrographs of immunohistochemical staining for iNOS. In SA-treated groups, iNOS protein staining was localized almost exclusively in the mucosal epithelial monolayer with 48 and 72 h of fasting compared with the normally fed control. Decreased staining occurred with GSH treatment. Bar = 100 μm. B: Optical density of iNOS protein. The content of iNOS protein was quantitatively measured by averaging the optical density. Values represent the mean ± SE. bP < 0.01 vs normally fed controls. dP < 0.01, cP < 0.05 vs the respective SA group in each fasting period. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline; iNOS: Inducible nitric oxide synthase.
Nitrite level in the jejunum
We next measured the levels of nitrite, which is a stable oxidation product of endogenous NO that is induced by iNOS. In SA-treated groups, fasting significantly increased the jejunal nitrite concentration with 48 and 72 h of fasting compared with the normally fed controls (P < 0.01; Figure 5). Conversely, GSH treatments significantly decreased the fasting-induced enhancement of the jejunal nitrite concentration compared with the respective SA-treated group for each fasting period (P < 0.01).
Figure 5.
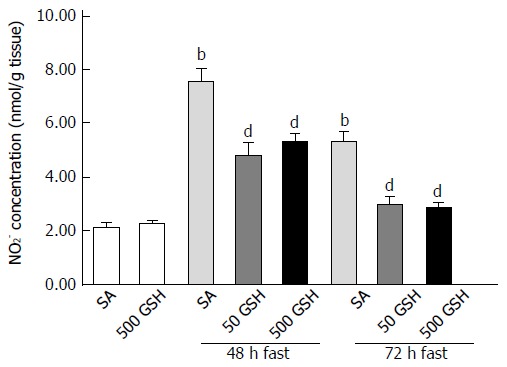
Effects of fasting and glutathione treatment on nitrite concentration in the jejunum. Nitrite concentrations were measured by HPLC using postcolumn derivatization with Griess reagent. Values represent the mean ± SE. bP < 0.01 vs the SA-treated group. dP < 0.01 vs the respective SA-treated group in each fasting period. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline.
8-OHdG level in the jejunum
As we previously determined that fasting causes jejunal apoptosis via ROS production and induction of NO following increased iNOS expression, in the present study we measured levels of iNOS expression, nitrite (indicating NO production), and 8-OHdG (as a marker of ROS presence). Consistent with the abovementioned changes in iNOS expression, fasting increased intestinal nitrite levels, whereas the degree of increase was significantly reduced by GSH treatment. Furthermore, the elevated jejunal 8-OHdG levels observed after fasting were significantly diminished by both 48 h and 72 h of GSH treatment (P < 0.01 for 50 mg/kg GSH vs SA with 72 h of fasting, P < 0.05 for 500 mg/kg GSH vs SA with 72 h of fasting; Figure 6).
Figure 6.
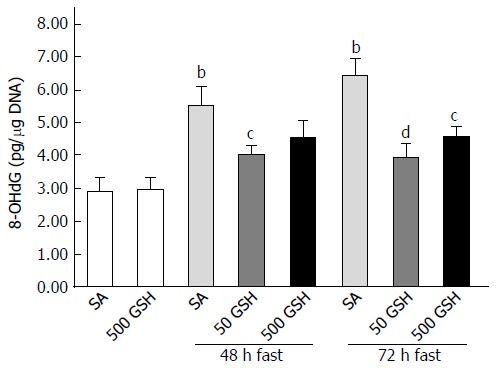
Effects of fasting and glutathione treatment on DNA oxidative damage in the jejunum. DNA oxidative damage was assessed by the level of 8-hydroxydeoxyguanosine (8-OHdG) in the jejunum. Values represent the mean ± SE. bP < 0.01 vs the normally fed control. dP < 0.01, cP < 0.05 vs the SA-treated group with 48 and 72 h of fasting. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline.
Evaluation of enterocyte apoptosis
Apoptosis was determined by histomorphometry, which is preferable to terminal deoxynucleotidyl transferase dUTP nick-end labeling for quantitative assessment. Using histomorphometric assessment of jejunal cells, we evaluated the contribution of reduced apoptosis to the recovery from fasting-induced mucosal atrophy mediated by oral GSH treatment. The representative apoptosis changes as visualized by conventional light microscopy of H&E-stained specimens are shown in Figure 7A. AI distribution profiles in the villi and the crypt are also shown in Figure 7B. From the AI distribution profiles, increased apoptosis in the lower half of the villus (cell positions 1 to 40) was evident with 48 and 72 h of fasting in the SA-treated groups, and this increase was diminished by GSH treatment. In the SA-treated groups, fasting significantly increased jejunal villus AI with 48 and 72 h of fasting compared with that in the normally fed controls (P < 0.01; Table 1). GSH treatment significantly decreased the fasting-induced enhancement of villus AI compared with that in the respective SA-treated group for each fasting period (P < 0.05 for 50 mg/kg GSH vs SA with 48 or 72 h of fasting, P < 0.01 for 500 mg/kg GSH vs SA with 48 or 72 h of fasting). From the AI distribution profiles, increased apoptosis in the lower two-thirds of crypts (cell positions 1 to 20) was evident with 48 and 72 h of fasting in the SA-treated groups (Figure 7B), and this effect was diminished by GSH treatment. In the SA-treated groups, fasting significantly increased jejunal crypt AIs with 48 and 72 h of fasting compared with the normally fed controls (P < 0.01; Table 1). GSH treatments significantly ameliorated the fasting-induced increase in crypt AI compared with the respective SA-treated group for each fasting period (P < 0.01).
Figure 7.
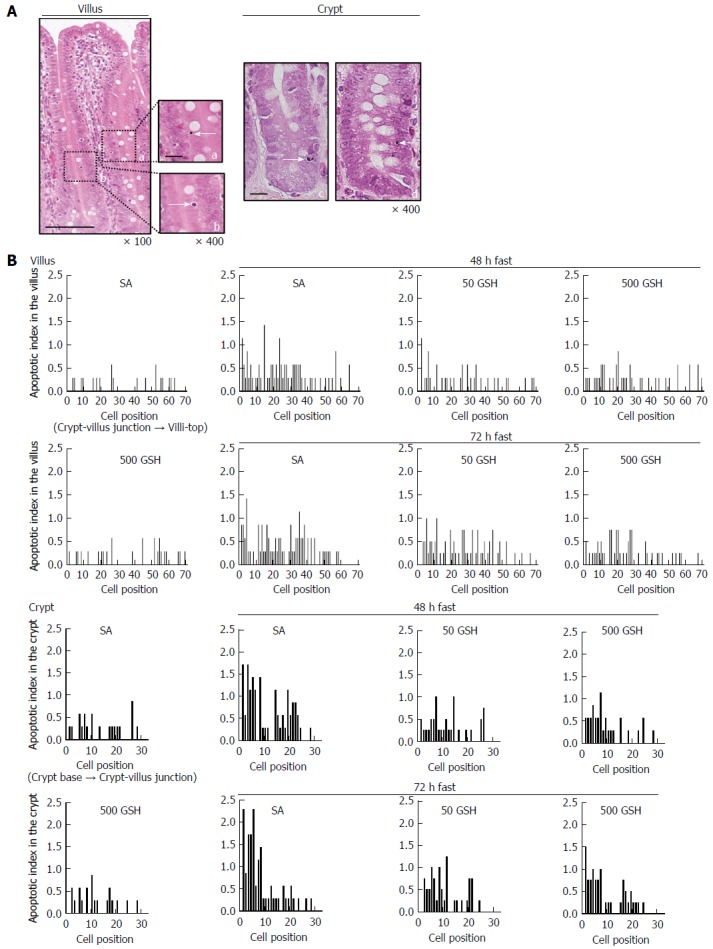
Effects of fasting and glutathione treatment on apoptotic index in the jejunal villus and crypt. Representative apoptotic changes determined by conventional light microscopy of hematoxylin and eosin (HE)-stained sections of the jejunal mucosa are shown in A. Left side: A jejunal villus from a 72-h fasted rat with 500 mg/kg GSH treatment. Apoptotic cells in the villus are indicated by an arrow showing an apoptotic corpuscle (a) and condensed chromatin (b). Bar = 100 μm (low magnification). Bar = 20 μm (high magnification). Right side: Jejunal crypts from 48- and 72-h fasted rats. Apoptotic cells in the crypt are indicated by an arrow showing an intensely eosinophilic cytoplasm and nuclear fragmentation (c) and condensed chromatin (d). Bar = 20 μm. AI distribution curves in the villus and the crypt are shown in B. AI is defined as the total number of apoptotic cells at each cell position and is expressed as a percentage of the total number of cells counted at that cell position. Cell position 1 is defined as the cell at the crypt-villus junction and the cell at the base of the crypt column for the villus and crypt data, respectively. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline.
Table 1.
Enterocyte apoptosis of the jejunal villus and crypt evaluated by conventional light microscopy of hematoxylin and eosin-stained specimens
| SA | 500 GSH |
48 h fast |
72 h fast |
|||||
| SA | 50 GSH | 500 GSH | SA | 50 GSH | 500 GSH | |||
| Villus | ||||||||
| Cells per villus column, n | 70 ± 1 | 69 ± 1 | 56 ± 2b | 64 ± 1d | 67 ± 2d | 56 ± 2b | 63 ± 1d | 65 ± 2d |
| Apoptotic cells per villus column, n | 0.06 ± 0.01 | 0.09 ± 0.01 | 0.20 ± 0.03b | 0.12 ± 0.01d | 0.11 ± 0.01d | 0.23 ± 0.03b | 0.18 ± 0.01 | 0.14 ± 0.02c |
| Apoptotic index, % | 0.09 ± 0.01 | 0.12 ± 0.01 | 0.38 ± 0.06b | 0.19 ± 0.02c | 0.18 ± 0.02d | 0.42 ± 0.06b | 0.29 ± 0.02c | 0.21 ± 0.02d |
| Crypt | ||||||||
| Cells per villus column, n | 28 ± 0.2 | 27 ± 0.4 | 22 ± 0.4b | 25 ± 0.3d | 27 ± 0.4d | 22 ± 0.3b | 23 ± 0.4c | 26 ± 0.3d |
| Apoptotic cells per villus column, n | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.18 ± 0.01b | 0.08 ± 0.01d | 0.09 ± 0.01d | 0.17 ± 0.02b | 0.10 ± 0.01d | 0.11 ± 0.01d |
| Apoptotic index, % | 0.21 ± 0.04 | 0.22 ± 0.02 | 0.81 ± 0.05b | 0.32 ± 0.03d | 0.33 ± 0.03d | 0.78 ± 0.10b | 0.41 ± 0.06d | 0.40 ± 0.05d |
Apoptosis was measured as in Figure 7A. 7-8 rats were tested in each group. Values represent the mean ± SE.
P < 0.01 vs the normally fed control;
P < 0.01 or
P < 0.05 vs the SA-treated group in each fasting period. GSH: Glutathione; SA: Saline.
Evaluation of cell proliferation
GSH is known to exhibit an important function related to the regulation of cell proliferation by protecting against the damaging effects of ROS[32]. To evaluate the effect of oral GSH administration on the reduced cell proliferation in fasting-induced jejunal mucosal atrophy, we assessed 5-BrdU incorporation, a proliferation indicator, in the jejunum. Although cell proliferation decreased with 48 and 72 h of fasting compared with that in the normally fed controls (P < 0.01; Figure 8), animals treated with GSH exhibited significantly higher levels of jejunal cell proliferation compared with those in the respective SA-treated group for each fasting period (P < 0.01 for 50 mg/kg GSH vs SA at 48 h fasting, and for 500 mg/kg GSH vs SA at 48 and 72 h fasting).
Figure 8.
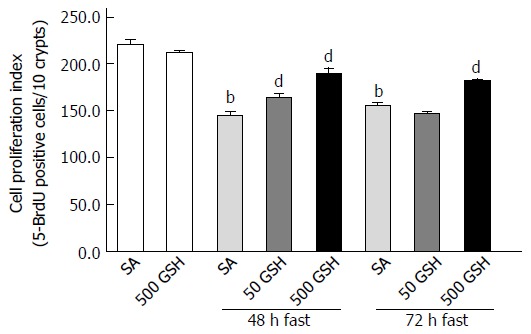
Effects of fasting and glutathione treatment on cell proliferation in the jejunum. Cell proliferation in the jejunal crypt was histologically assessed by 5-bromo-2’-deoxyuridine (5-BrdU) incorporation. The fraction of 5-BrdU-positive cells was expressed as the cell proliferation index (5-BrdU-positive cells/10 crypts). Values represent the mean ± SE. bP < 0.01 vs the normally fed control. dP < 0.01 vs the respective SA-treated group in each fasting period. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline.
GSH and GSSG levels in the jejunum
GSH is one of the most important scavengers of ROS; additionally, GSH and oxidized GSH (i.e., GSSG) are used as markers of oxidative stress[33]. To evaluate GSH redox balance and GSH levels in the jejunum consequent to oral GSH administration during fasting, we assessed GSH and GSSG levels in the jejunum. Fasting significantly decreased the GSH concentration in the jejunum (P < 0.01; Figure 9A). GSH treatment significantly further decreased the fasting-induced decreases in the jejunal GSH concentration for each fasting period (P < 0.01). Conversely, GSSG concentration in the jejunum was similar to that of normally fed controls except for the 500 mg/kg GSH treatment group with ad libitum feeding (Figure 9B).
Figure 9.
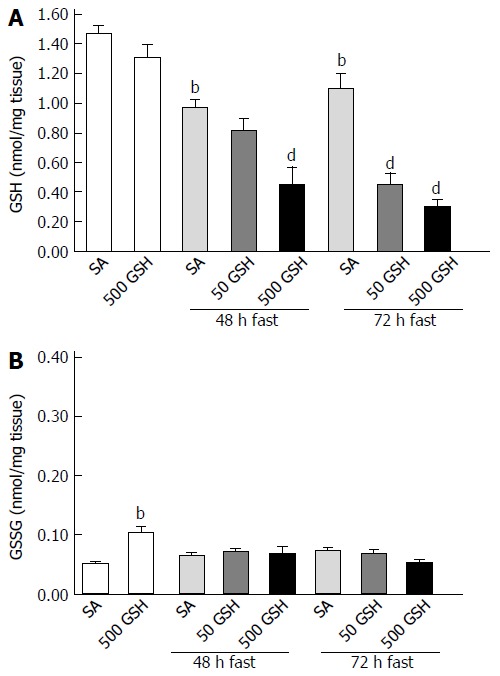
Effects of fasting and glutathione treatment on glutathione and oxidized glutathione concentration in the jejunum. GSH and GSSG concentration in the jejunum was determined by measuring the absorption derived from a colorimetric reaction with DTNB [5, 5’-dithiobis (2-nitrobenzoic acid)] coupled with the enzymatic recycling system. A: GSH concentration in the jejunum; B: GSSG concentration in the jejunum. Values are the mean ± SE. bP < 0.01 vs the normally fed control. dP < 0.01 vs the respective SA-treated group in each fasting period. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline; GSSG: Oxidized glutathione.
Ggt1 mRNA expression in the jejunum
Fasting significantly increased Ggt1 expression in the jejunum at 48 h of fasting (P < 0.05) (Figure 10) whereas GSH treatment significantly reduced Ggt1 elevation at 48 h of fasting (P < 0.05, P < 0.01 for 50 mg/kg GSH, 500 mg/kg GSH vs SA at 48 h of fasting, respectively). A similar tendency was obtained for the 72-h fasting group, although this difference did not reach statistical significance.
Figure 10.
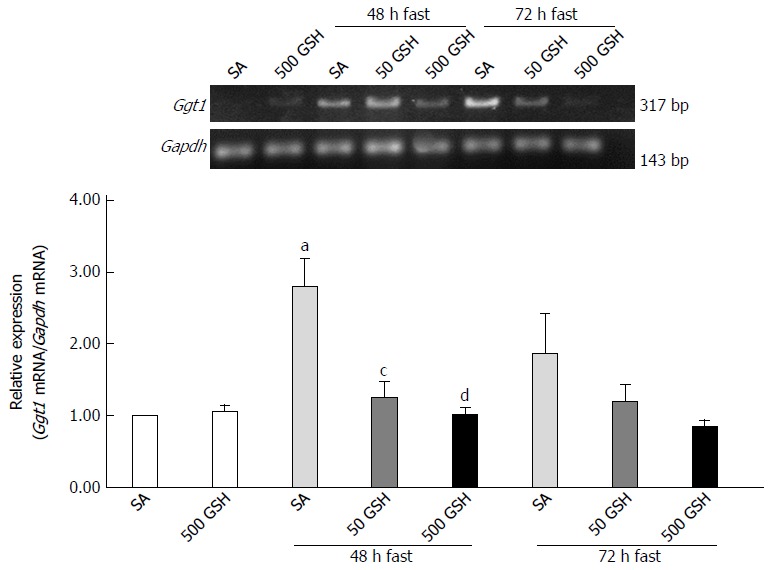
Effects of fasting and glutathione treatment on Ggt1 mRNA expression in the jejunum. GGT is the only enzyme of the gamma-glutamyl cycle located on the outer surface of plasma membrane and plays key roles in GSH homeostasis by breaking down extracellular GSH. Values are the mean ± SE. aP < 0.05 vs the normally fed control. dP < 0.01, cP < 0.05 vs the SA-treated group with 48 h of fasting. 7-8 rats were tested in each group. GSH: Glutathione; SA: Saline; GSSG: Oxidized glutathione.
DISCUSSION
Previous reports have suggested the involvement of GSH in fasting-induced intestinal mucosal atrophy; in addition, fasting has been associated with increased ROS formation and depletion of the critical antioxidant GSH in the intestine[4,6,34-36]. However, the roles of GSH in intestinal mucosal recovery from this condition as mediated by oral GSH administration have not yet been described. The objective of the present study was to investigate the effects of oral GSH administration on the development of fasting-induced intestinal atrophy, with a particular focus on the possible participation of intracellular GSH level and GGT expression on the cell surface as potential parameters of intracellular de novo GSH synthesis.
During fasting, the intestinal lumen receives very little or no GSH from two of the three primary sources, i.e., biliary secretion[10,37] and dietary intake[11]. Therefore, the overall GSH levels of the intestinal mucosa decrease during fasting because the remaining GSH source, intracellular GSH, is primarily synthesized from GSH in the intestinal lumen.
There is growing evidence that dysfunctional GSH homeostasis is involved in the etiology of several diseases. The previously reported conditions associated with GSH depletion include liver disease[38], immune disorders[39], neurodegenerative disease[40], cardiovascular disease[41], pulmonary disease[42], arthritis, diabetes[43], and the aging process itself[44]. Thus, oral supplementation with GSH to increase the depressed GSH level has been studied extensively as a potential means to prevent these diseases by countering the negative effects of oxidative stress, which is one of their underlying causes. Several studies have shown that GSH supplementation in laboratory animals is effective, with benefits including enhancement of immune function[45] and protection against carcinogenesis[46]. In addition, daily consumption of GSH supplements in humans was effective at increasing body compartment stores of GSH, which may be of importance as the maintenance of tissue levels of GSH might be critical for maintaining health and preventing disease as well as age-related biological insults[47]. Thus, it has been suggested that orally administered GSH may act as a backup for intracellular GSH for GSH-deficient tissue. Therefore, the enhancement of intestinal mucosal GSH levels by oral GSH supplementation under conditions of intracellular GSH deficiency-related biological insults such as fasting may restore tissue GSH and promote ROS metabolism, thus facilitating the recovery from intestinal mucosal atrophy.
Our previous studies suggested that fasting-induced intestinal mucosal atrophy could be alleviated by directly inhibiting the fasting-mediated NO production by iNOS and subsequent ROS enhancement associated with increased jejunal apoptosis via, e.g., iNOS inhibitor treatment[20] or refeeding[21]. Notably, an association was also observed between inhibition of ROS and increasing cell proliferation[21]. The intestinal epithelial layer is uniquely organized for rapid self-renewal, which is achieved by the well-regulated processes of crypt-to-villus apoptosis and crypt stem cell proliferation. The intestinal epithelium sits at the interface between the intestinal mucosa and the intestinal lumen, and as such is prone to oxidative damage induced by luminal and intracellular oxidants. A previous study[16] reviewed the GSH/GSSG redox mechanism, which modulates intestinal cell transition through cell proliferation, differentiation, or apoptosis and can govern the regenerative potential of the intestinal mucosa. Therefore, we hypothesized that protection against intestinal mucosal atrophy could be provided by controlling apoptosis and cell proliferation through the elimination of induced ROS in the intestinal mucosa by orally administering an antioxidant such as GSH during fasting in rats.
Here, we found that there was no difference in weight loss between fasting alone and oral GSH treatment during fasting (Figure 2) with the GSH levels used for this study. Conversely, jejunal mucosal atrophy was significantly increased by fasting but was remedied by oral GSH treatment during fasting (Figure 3), with significantly greater improvement resulting from the high vs low GSH dose. Furthermore, because the etiology of fasting-induced intestinal atrophy was shown to comprise ROS genesis from the NO produced by iNOS[20,21], we measured iNOS protein expression in the jejunum. We identified an inverse relationship between intestinal mucosal atrophy and iNOS protein expression (primarily in the mucosal epithelial monolayer) associated with oral GSH treatment during fasting, most particularly with high levels of GSH administration (Figure 4). This is supported by our previous observations that iNOS inhibitor treatment during fasting and refeeding after fasting decreases fasting-induced atrophy and suppresses iNOS expression in the jejunum.
Oral GSH treatment during fasting inhibited the fasting-mediated generation of the apoptosis mediators NO and ROS concomitant with decreased iNOS expression (Figures 5 and 6). To determine the importance of apoptotic changes in the jejunum for intestinal recovery by oral GSH treatment (Figure 7A), we performed a histomorphometric assessment including AI score (Table 1) and distribution profiles (Figure 7B). The increased AI recorded in the villi and crypts after fasting decreased with oral GSH treatment (Table 1). Specifically, the lower half of the apoptotic enterocytes distributed across the entire jejunal villi and the apoptotic cells primarily located in the lower two-thirds of the crypt (Figure 7B) were particularly affected by oral GSH treatment during fasting. These results were broadly consistent with changes in iNOS, NO, and ROS as apoptosis mediators, suggesting that oral GSH administration during fasting may inhibit enterocyte apoptosis by diminishing the ROS in the jejunum generated by fasting.
GSH treatment also was found to alleviate the decreased jejunal cell proliferation mediated by fasting (Figure 8), with significantly greater improvement in fasting-induced jejunal mucosal atrophy resulting from high vs low dose GSH. These observations are consistent with the role of GSH in cell proliferation through protection against damage from ROS[16,32]. Together, these findings indicate that oral GSH administration leads to less fasting-mediated enhancement and therefore decreased overall ROS levels in the jejunal mucosa, thereby functioning to increase cell proliferation. This suggests that oral GSH administration may provide regenerative potential to the mucosa during fasting.
Notably, although our findings replicated prior results[6,34-36] that fasting significantly decreased jejunal GSH concentrations (Figure 9A), we found that the concentrations were further significantly decreased by oral GSH supplementation during fasting, with a greater effect from high vs low GSH dose. Conversely, the jejunal concentrations of the GSH oxidation product GSSG were not changed by any feeding regime (Figure 9B). Furthermore, oral GSH treatment during fasting significantly decreased the fasting-induced ROS levels in the jejunum (Figure 6), but we consider it unlikely that this effect depends on antioxidant activity given the depressed GSH concentration in the jejunum. These results demonstrate that oral GSH administration during fasting did not contribute to the supply of GSH in the jejunum, and an increase in GSH oxidation was not observed, suggesting that oral GSH administration during fasting does not provide a backup for the jejunum-depleted GSH.
The synthesis of intracellular de novo GSH, which is critical for ROS removal and maintenance of GSH homeostasis, relies upon the enzyme activity of the plasma membrane protein GGT to break down extracellular GSH[12]. In the present study, we demonstrated that oral GSH treatment significantly ameliorated the increased jejunal Ggt1 mRNA expression observed following 48 h of fasting (Figure 10). In comparison, Jonas et al[4] indicated that fasting for 72 h did not change Ggt1 mRNA levels in the ileum, although this was determined via ribonuclease protection assay rather than by semi-quantitative RT-PCR as used in the present study. The different findings may also be a consequence of the disparity in fasting duration. Notably, Ogasawara et al[34] showed that starvation causes a striking decrease of GGT activity in the jejunum, which is consistent with our finding of fasting-induced increases in Ggt1 mRNA levels, as the mRNA expression of GGT might be upregulated by the decline of GGT activity[12]. Furthermore, Zhang et al[12] described that Ggt1 mRNA expression is increased during oxidative stress, which was considered to constitute an adaptation to such adverse conditions. Similarly, we found that fasting increased jejunal ROS levels and up-regulated Ggt1 mRNA expression in the jejunum in this study. However, considering that oral GSH treatment during fasting significantly decreased both jejunal GSH concentration and Ggt1 mRNA expression, the supplemented GSH in the intestinal lumen may not be involved in intracellular de novo GSH synthesis.
The ability of GSH to control extracellular redox provides another possible role for intestinal luminal GSH during fasting, as the present study demonstrated that intracellular GSH levels of the jejunum mucosa were not increased by intestinal luminal GSH administration during fasting, although the GSH administration attenuated the fasting-induced ROS in the intestinal mucosa. Reversible redox reactions of intracellular thiol/disulfide pairs such as GSH/GSSG regulate diverse biological processes in vivo, including signaling for apoptosis and cell proliferation, enzyme catalysis, gene expression, and molecular folding and trafficking[16-18,48,49]. Most studies have focused on the major intracellular thiols such as GSH[50,51]; however, relatively little is known regarding the effects of changes in extracellular thiol/disulfide pairs. Biliary GSH is an important intestinal luminal source of GSH and attenuates ROS of the intestinal mucosa by increasing the GSH levels of the intestinal mucosa. However, it has been suggested that biliary GSH controls the status of the intestinal lumen and contributes to protection of the intestinal mucosa[10]. The GSH of the intestinal lumen may act on the intestinal mucosa via both of these two pathways, and it is important to consider the control of the redox status of the intestinal lumen. In addition, the roles of extracellular (i.e., intestinal luminal) GSH in the recovery from fasting-induced intestinal mucosal atrophy enabled by oral GSH treatment have not yet been described.
A few studies have examined the ability of the extracellular thiol/disulfide redox state to regulate apoptosis and cell proliferation. Circu et al[16] demonstrated that the luminal/extracellular redox environment is determined by the cysteine/cystine (Cys/CySS) redox pair with contributions from the GSH system, with the majority of Cys in the intestinal lumen originating from GGT enzymatic hydrolysis of GSH obtained from dietary intake and biliary supply. Intestinal cell proliferation and apoptosis are associated with quantitative changes in the redox potential (Eh) of the extracellular GSH/GSSG or Cys/CySS redox pair. In the present study, the changes in apoptosis and cell proliferation in the intestinal mucosa resulting from oral GSH administration during fasting may derive from Eh changes mediated by GSH and Cys in the intestinal lumen, which in turn may ultimately control signaling proteins, enzyme catalysis, and gene expression. For example, Devadas et al[52] linked ROS formation with the progression of Fas-induced apoptosis via the protective effects of extracellular GSH, which are ascribed to its known capacity as an antioxidant. Specifically, extracellular GSH was deduced to inhibit Fas-mediated apoptosis following ROS reduction in cells because Fas crosslinking induced rapid generation of ROS well before the appearance of characteristic apoptotic changes. Furthermore, Fujise et al[22] suggested that fasting-induced apoptosis in the rat small intestine occurred via a receptor-mediated type I apoptotic pathway including induced expression of FasL, Fas, and TNFR1. Selleri et al[53] and Viard-Leveugle et al[54] provided evidence that iNOS expression and NO production are involved in the mechanism of FasL upregulation and Fas-mediated apoptosis. The iNOS-NO-ROS-FasL pathway represents a potential link between the apoptosis and intestinal atrophy observed in the fasting.
In the present study, fasting-induced intestinal mucosal atrophy resulting from increased apoptosis was caused by increased production of NO and ROS as apoptosis mediators following elevation of iNOS expression. Because the iNOS-NO-ROS-FasL pathway is part of the apoptotic mechanism in the intestinal mucosa atrophy occurring after fasting[22,52-54], oral GSH treatment during fasting might inhibit Fas-mediated apoptosis following reduction of ROS levels in the jejunal mucosa resulting from Eh changes mediated by GSH and Cys in the intestinal lumen. Additionally, because iNOS protein expression was also induced by ROS produced from NO in fasting-induced intestinal mucosal atrophy in a previous study[20], the decrease in ROS levels in the intestinal mucosa reduces iNOS protein expression and NO production. Moreover, Jonas et al[55] showed that the extracellular thiol/disulfide redox state modulates cell proliferation and that this system interacted with growth factor signaling in a human colon carcinoma cell line. Thus, the change in extracellular thiol/disulfide redox state in response to peptide growth factors indicated an interaction of growth factor-activated pathways and thiol/disulfide metabolism during intestinal cell proliferation.
Therefore, oral GSH administration during fasting may regulate the redox state of the intestinal lumen and consequently may both relieve Fas-mediated apoptosis and increase growth factor-mediated cell proliferation by ROS removal in jejunal epithelial cells (Figure 11). However, because our present study focused on intracellular GSH, further studies focusing on extracellular GSH are required to confirm this model. In particular, a study utilizing measurements of GSH/GSSG and Cys/CySS levels in the intestinal lumen along with Fas and growth factor signaling in the jejunum may be necessary to identify the mechanism underlying the recovery of the intestinal mucosal atrophy mediated by oral GSH administration during fasting.
Figure 11.
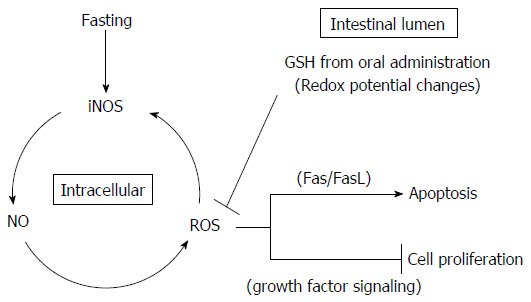
Schematic diagram of protective effects of glutathione in the intestinal lumen against fasting-induced intestinal atrophy, mediated through oxidative stress. The schematic diagram depicts a possible role of intestinal lumen redox status in the regulation of jejunal mucosa apoptosis and cell proliferation in fasting-induced intestinal atrophy, mediated through oxidative stress. Fasting causes increased production of NO and ROS as apoptosis mediators following elevation of iNOS expression. The changes in apoptosis and cell proliferation in the intestinal mucosa resulting from oral GSH administration during fasting may derive from intracellular ROS removal by redox potential changes mediated by GSH and Cys (originating from enzymatic hydrolysis of GSH) in the intestinal lumen. Intracellular ROS removal is considered to inhibit Fas-mediated apoptosis and increase growth factor-mediated cell proliferation. GSH: Glutathione; ROS: Reactive oxygen species.
In conclusion, oral GSH administration during fasting may provide regenerative potential to the jejunal mucosa through control of enterocyte apoptosis and cell proliferation. The GSH in the intestinal lumen provided through oral administration during fasting may not be involved in intracellular de novo GSH but may be useful to prevent intestinal mucosal atrophy by diminishing the levels of ROS in the jejunum generated by fasting.
COMMENTS
Background
Fasting induces small intestinal mucosal atrophy, which can lead to bacterial translocation, particularly in patients receiving a prolonged course of total parenteral nutrition (TPN), and is associated with increased reactive oxygen species (ROS) formation. This fasting state is also accompanied by depletion of the critical antioxidant glutathione (GSH), which functions to eliminate induced ROS in the intestinal mucosa. As GSH deficiency in tissues is associated with increased oxidative stress and fasting, the maintenance of tissue levels of GSH is critical for protection of small intestinal mucosa.
Research frontiers
The roles of GSH in intestinal mucosal recovery from fasting as mediated by oral GSH administration have not been investigated.
Innovations and breakthroughs
GSH treatment in the intestinal lumen could be a promising strategy to improve clinical outcome in patients receiving a prolonged course of TPN.
Applications
Oral GSH administration during fasting enhances jejunal regenerative potential to minimize intestinal mucosal atrophy by diminishing fasting-mediated ROS generation and enterocyte apoptosis and enhancing cell proliferation.
Peer-review
The manuscript by Uchida et al describes effects of oral GSH administration on fasting-induced intestinal atrophy in the small intestinal mucosa. The authors found that oral GSH administration of rats during fasting significantly enhanced jejunal regenerative potential to minimize mucosal atrophy. This reduction of intestinal atrophy correlated with correction of histopathological outcomes and many biochemical parameters. The authors’ conclusion that oral GSH improves intestinal atrophy is supported by convincing results.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee at the Life Science Center of Josai University (IACUC Protocol Number [H25057], [H26080]).
Conflict-of-interest statement: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data sharing statement: No additional data are available.
Peer-review started: March 23, 2017
First decision: April 28, 2017
Article in press: July 22, 2017
P- Reviewer: Hernandez C S- Editor: Gong ZM L- Editor: A E- Editor: Zhang FF
Contributor Information
Hiroyuki Uchida, Division of Pathophysiology, Department of Clinical Dietetics and Human Nutrition, Faculty of Pharmaceutical Science, Josai University, Sakado, Saitama 350-0295, Japan. mrhiro@josai.ac.jp.
Yukari Nakajima, Division of Pathophysiology, Department of Clinical Dietetics and Human Nutrition, Faculty of Pharmaceutical Science, Josai University, Sakado, Saitama 350-0295, Japan.
Kazuo Ohtake, Division of Pathophysiology, Department of Clinical Dietetics and Human Nutrition, Faculty of Pharmaceutical Science, Josai University, Sakado, Saitama 350-0295, Japan.
Junta Ito, Division of Pathophysiology, Department of Clinical Dietetics and Human Nutrition, Faculty of Pharmaceutical Science, Josai University, Sakado, Saitama 350-0295, Japan.
Masahiko Morita, Kyowa Hakko Bio Co., Ltd. Healthcare Products Development Center, Tsukuba-shi, Ibaraki 305-0841, Japan.
Ayako Kamimura, Kyowa Hakko Bio Co., Ltd. Healthcare Products Development Center, Tsukuba-shi, Ibaraki 305-0841, Japan.
Jun Kobayashi, Division of Pathophysiology, Department of Clinical Dietetics and Human Nutrition, Faculty of Pharmaceutical Science, Josai University, Sakado, Saitama 350-0295, Japan.
References
- 1.Ziegler TR. Molecular mechanisms of intestinal injury, repair, and growth. In: Takala J, Rombeau JL (eds), editors. Gut Dysfunction in Critical Illness. New York: Springer-Verlag; 1996. pp. 25–52. [Google Scholar]
- 2.Shaw D, Gohil K, Basson MD. Intestinal mucosal atrophy and adaptation. World J Gastroenterol. 2012;18:6357–6375. doi: 10.3748/wjg.v18.i44.6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darmon N, Pélissier MA, Heyman M, Albrecht R, Desjeux JF. Oxidative stress may contribute to the intestinal dysfunction of weanling rats fed a low protein diet. J Nutr. 1993;123:1068–1075. doi: 10.1093/jn/123.6.1068. [DOI] [PubMed] [Google Scholar]
- 4.Jonas CR, Farrell CL, Scully S, Eli A, Estívariz CF, Gu LH, Jones DP, Ziegler TR. Enteral nutrition and keratinocyte growth factor regulate expression of glutathione-related enzyme messenger RNAs in rat intestine. JPEN J Parenter Enteral Nutr. 2000;24:67–75. doi: 10.1177/014860710002400267. [DOI] [PubMed] [Google Scholar]
- 5.Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol Rev. 1994;74:139–162. doi: 10.1152/physrev.1994.74.1.139. [DOI] [PubMed] [Google Scholar]
- 6.Jonas CR, Estívariz CF, Jones DP, Gu LH, Wallace TM, Diaz EE, Pascal RR, Cotsonis GA, Ziegler TR. Keratinocyte growth factor enhances glutathione redox state in rat intestinal mucosa during nutritional repletion. J Nutr. 1999;129:1278–1284. doi: 10.1093/jn/129.7.1278. [DOI] [PubMed] [Google Scholar]
- 7.Aw TY. Intestinal glutathione: determinant of mucosal peroxide transport, metabolism, and oxidative susceptibility. Toxicol Appl Pharmacol. 2005;204:320–328. doi: 10.1016/j.taap.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI. The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem. 2007;113:234–258. doi: 10.1080/13813450701661198. [DOI] [PubMed] [Google Scholar]
- 9.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 10.Aw TY. Biliary glutathione promotes the mucosal metabolism of luminal peroxidized lipids by rat small intestine in vivo. J Clin Invest. 1994;94:1218–1225. doi: 10.1172/JCI117439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones DP, Coates RJ, Flagg EW, Eley JW, Block G, Greenberg RS, Gunter EW, Jackson B. Glutathione in foods listed in the National Cancer Institute’s Health Habits and History Food Frequency Questionnaire. Nutr Cancer. 1992;17:57–75. doi: 10.1080/01635589209514173. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Forman HJ, Choi J. Gamma-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005;401:468–483. doi: 10.1016/S0076-6879(05)01028-1. [DOI] [PubMed] [Google Scholar]
- 13.Aw TY, Williams MW. Intestinal absorption and lymphatic transport of peroxidized lipids in rats: effect of exogenous GSH. Am J Physiol. 1992;263:G665–G672. doi: 10.1152/ajpgi.1992.263.5.G665. [DOI] [PubMed] [Google Scholar]
- 14.Schmitt B, Vicenzi M, Garrel C, Denis FM. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015;6:198–205. doi: 10.1016/j.redox.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imhoff BR, Hansen JM. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem J. 2009;424:491–500. doi: 10.1042/BJ20091286. [DOI] [PubMed] [Google Scholar]
- 16.Circu ML, Aw TY. Intestinal redox biology and oxidative stress. Semin Cell Dev Biol. 2012;23:729–737. doi: 10.1016/j.semcdb.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pérez S, Taléns-Visconti R, Rius-Pérez S, Finamor I, Sastre J. Redox signaling in the gastrointestinal tract. Free Radic Biol Med. 2017;104:75–103. doi: 10.1016/j.freeradbiomed.2016.12.048. [DOI] [PubMed] [Google Scholar]
- 18.Moriarty-Craige SE, Jones DP. Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr. 2004;24:481–509. doi: 10.1146/annurev.nutr.24.012003.132208. [DOI] [PubMed] [Google Scholar]
- 19.D'Orazio M, Pacello F, Battistoni A. Extracellular glutathione decreases the ability of Burkholderia cenocepacia to penetrate into epithelial cells and to induce an inflammatory response. PLoS One. 2012;7:e47550. doi: 10.1371/journal.pone.0047550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito J, Uchida H, Yokote T, Ohtake K, Kobayashi J. Fasting-induced intestinal apoptosis is mediated by inducible nitric oxide synthase and interferon-{gamma} in rat. Am J Physiol Gastrointest Liver Physiol. 2010;298:G916–G926. doi: 10.1152/ajpgi.00429.2009. [DOI] [PubMed] [Google Scholar]
- 21.Ito J, Uchida H, Machida N, Ohtake K, Saito Y, Kobayashi J. Inducible and neuronal nitric oxide synthases exert contrasting effects during rat intestinal recovery following fasting. Exp Biol Med (Maywood) 2017;242:762–772. doi: 10.1177/1535370217694434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujise T, Iwakiri R, Wu B, Amemori S, Kakimoto T, Yokoyama F, Sakata Y, Tsunada S, Fujimoto K. Apoptotic pathway in the rat small intestinal mucosa is different between fasting and ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2006;291:G110–G116. doi: 10.1152/ajpgi.00393.2005. [DOI] [PubMed] [Google Scholar]
- 23.Dahly EM, Guo Z, Ney DM. Alterations in enterocyte proliferation and apoptosis accompany TPN-induced mucosal hypoplasia and IGF-I-induced hyperplasia in rats. J Nutr. 2002;132:2010–2014. doi: 10.1093/jn/132.7.2010. [DOI] [PubMed] [Google Scholar]
- 24.Morin MJ, Karr SM, Faris RA, Gruppuso PA. Developmental variability in expression and regulation of inducible nitric oxide synthase in rat intestine. Am J Physiol Gastrointest Liver Physiol. 2001;281:G552–G559. doi: 10.1152/ajpgi.2001.281.2.G552. [DOI] [PubMed] [Google Scholar]
- 25.Shi SR, Chaiwun B, Young L, Cote RJ, Taylor CR. Antigen retrieval technique utilizing citrate buffer or urea solution for immunohistochemical demonstration of androgen receptor in formalin-fixed paraffin sections. J Histochem Cytochem. 1993;41:1599–1604. doi: 10.1177/41.11.7691930. [DOI] [PubMed] [Google Scholar]
- 26.Lu H, Zhu B, Xue XD. Role of neuronal nitric oxide synthase and inducible nitric oxide synthase in intestinal injury in neonatal rats. World J Gastroenterol. 2006;12:4364–4368. doi: 10.3748/wjg.v12.i27.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang Y, Swartz-Basile DA, Swietlicki EA, Yi L, Rubin DC, Levin MS. Bax is required for resection-induced changes in apoptosis, proliferation, and members of the extrinsic cell death pathways. Gastroenterology. 2004;126:220–230. doi: 10.1053/j.gastro.2003.10.077. [DOI] [PubMed] [Google Scholar]
- 28.Ohtake K, Ishiyama Y, Uchida H, Muraki E, Kobayashi J. Dietary nitrite inhibits early glomerular injury in streptozotocin-induced diabetic nephropathy in rats. Nitric Oxide. 2007;17:75–81. doi: 10.1016/j.niox.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Inoue S, Kawanishi S. Oxidative DNA damage induced by simultaneous generation of nitric oxide and superoxide. FEBS Lett. 1995;371:86–88. doi: 10.1016/0014-5793(95)00873-8. [DOI] [PubMed] [Google Scholar]
- 30.Shigenaga MK, Gimeno CJ, Ames BN. Urinary 8-hydroxy-2’-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc Natl Acad Sci USA. 1989;86:9697–9701. doi: 10.1073/pnas.86.24.9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saito S, Yamauchi H, Hasui Y, Kurashige J, Ochi H, Yoshida K. Quantitative determination of urinary 8-hydroxydeoxyguanosine (8-OH-dg) by using ELISA. Res Commun Mol Pathol Pharmacol. 2000;107:39–44. [PubMed] [Google Scholar]
- 32.Filomeni G, Rotilio G, Ciriolo MR. Cell signalling and the glutathione redox system. Biochem Pharmacol. 2002;64:1057–1064. doi: 10.1016/s0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- 33.Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J, Trnkova L, Kruseova J, Eckschlager T, Kizek R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012;4:1247–1253. doi: 10.3892/ol.2012.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogasawara T, Ohnhaus EE, Hoensch HP. Glutathione and its related enzymes in the small intestinal mucosa of rats: effects of starvation and diet. Res Exp Med (Berl) 1989;189:195–204. doi: 10.1007/BF01852168. [DOI] [PubMed] [Google Scholar]
- 35.Viña J, Perez C, Furukawa T, Palacin M, Viña JR. Effect of oral glutathione on hepatic glutathione levels in rats and mice. Br J Nutr. 1989;62:683–691. doi: 10.1079/bjn19890068. [DOI] [PubMed] [Google Scholar]
- 36.Kelly FJ. Glutathione content of the small intestine: regulation and function. Br J Nutr. 1993;69:589–596. doi: 10.1079/bjn19930058. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi I, Kern MK, Dodds WJ, Hogan WJ, Layman RD, Ammon HV. Fasting and postprandial hepatic bile flow in unanesthetized opossums. Am J Physiol. 1990;259:G745–G752. doi: 10.1152/ajpgi.1990.259.5.G745. [DOI] [PubMed] [Google Scholar]
- 38.Loguercio C, Taranto D, Vitale LM, Beneduce F, Del Vecchio Blanco C. Effect of liver cirrhosis and age on the glutathione concentration in the plasma, erythrocytes, and gastric mucosa of man. Free Radic Biol Med. 1996;20:483–488. doi: 10.1016/0891-5849(96)02057-6. [DOI] [PubMed] [Google Scholar]
- 39.Herzenberg LA, De Rosa SC, Dubs JG, Roederer M, Anderson MT, Ela SW, Deresinski SC, Herzenberg LA. Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci USA. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smeyne M, Smeyne RJ. Glutathione metabolism and Parkinson’s disease. Free Radic Biol Med. 2013;62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Usal A, Acartürk E, Yüregir GT, Unlükurt I, Demirci C, Kurt HI, Birand A. Decreased glutathione levels in acute myocardial infarction. Jpn Heart J. 1996;37:177–182. doi: 10.1536/ihj.37.177. [DOI] [PubMed] [Google Scholar]
- 42.Gul M, Kutay FZ, Temocin S, Hanninen O. Cellular and clinical implications of glutathione. Indian J Exp Biol. 2000;38:625–634. [PubMed] [Google Scholar]
- 43.Nuttall SL, Martin U, Sinclair AJ, Kendall MJ. Glutathione: in sickness and in health. Lancet. 1998;351:645–646. doi: 10.1016/s0140-6736(05)78428-2. [DOI] [PubMed] [Google Scholar]
- 44.Viña J, Sastre J, Anton V, Bruseghini L, Esteras A, Asensi M. Effect of aging on glutathione metabolism. Protection by antioxidants. EXS. 1992;62:136–144. doi: 10.1007/978-3-0348-7460-1_14. [DOI] [PubMed] [Google Scholar]
- 45.Furukawa T, Meydani SN, Blumberg JB. Reversal of age-associated decline in immune responsiveness by dietary glutathione supplementation in mice. Mech Ageing Dev. 1987;38:107–117. doi: 10.1016/0047-6374(87)90071-6. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz JL, Shklar G. Glutathione inhibits experimental oral carcinogenesis, p53 expression, and angiogenesis. Nutr Cancer. 1996;26:229–236. doi: 10.1080/01635589609514479. [DOI] [PubMed] [Google Scholar]
- 47.Richie JP Jr, Nichenametla S, Neidig W, Calcagnotto A, Haley JS, Schell TD, Muscat JE. Randomized controlled trial of oral glutathione supplementation on body stores of glutathione. Eur J Nutr. 2015;54:251–263. doi: 10.1007/s00394-014-0706-z. [DOI] [PubMed] [Google Scholar]
- 48.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 49.Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 50.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 51.Sato N, Iwata S, Nakamura K, Hori T, Mori K, Yodoi J. Thiol-mediated redox regulation of apoptosis. Possible roles of cellular thiols other than glutathione in T cell apoptosis. J Immunol. 1995;154:3194–3203. [PubMed] [Google Scholar]
- 52.Devadas S, Hinshaw JA, Zaritskaya L, Williams MS. Fas-stimulated generation of reactive oxygen species or exogenous oxidative stress sensitize cells to Fas-mediated apoptosis. Free Radic Biol Med. 2003;35:648–661. doi: 10.1016/s0891-5849(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 53.Selleri C, Sato T, Raiola AM, Rotoli B, Young NS, Maciejewski JP. Induction of nitric oxide synthase is involved in the mechanism of Fas-mediated apoptosis in haemopoietic cells. Br J Haematol. 1997;99:481–489. doi: 10.1046/j.1365-2141.1996.4323240.x. [DOI] [PubMed] [Google Scholar]
- 54.Viard-Leveugle I, Gaide O, Jankovic D, Feldmeyer L, Kerl K, Pickard C, Roques S, Friedmann PS, Contassot E, French LE. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489–498. doi: 10.1038/jid.2012.330. [DOI] [PubMed] [Google Scholar]
- 55.Jonas CR, Ziegler TR, Gu LH, Jones DP. Extracellular thiol/disulfide redox state affects proliferation rate in a human colon carcinoma (Caco2) cell line. Free Radic Biol Med. 2002;33:1499–1506. doi: 10.1016/s0891-5849(02)01081-x. [DOI] [PubMed] [Google Scholar]