

Summary
While the physiological benefits of the fibroblast growth factor 21 (FGF21) hepatokine are documented in response to fasting, little information is available on Fgf21 regulation in a glucose-overload context. We report that peroxisome-proliferator-activated receptor α (PPARα), a nuclear receptor of the fasting response, is required with the carbohydrate-sensitive transcription factor carbohydrate-responsive element-binding protein (ChREBP) to balance FGF21 glucose response. Microarray analysis indicated that only a few hepatic genes respond to fasting and glucose similarly to Fgf21. Glucose-challenged Chrebp−/− mice exhibit a marked reduction in FGF21 production, a decrease that was rescued by re-expression of an active ChREBP isoform in the liver of Chrebp−/− mice. Unexpectedly, carbohydrate challenge of hepatic Pparα knockout mice also demonstrated a PPARα-dependent glucose response for Fgf21 that was associated with an increased sucrose preference. This blunted response was due to decreased Fgf21 promoter accessibility and diminished ChREBP binding onto Fgf21 carbohydrate-responsive element (ChoRE) in hepatocytes lacking PPARα. Our study reports that PPARα is required for the ChREBP-induced glucose response of FGF21.
Keywords: ChREBP, PPARα, FGF21, glucose intake, sucrose preference
Graphical Abstract
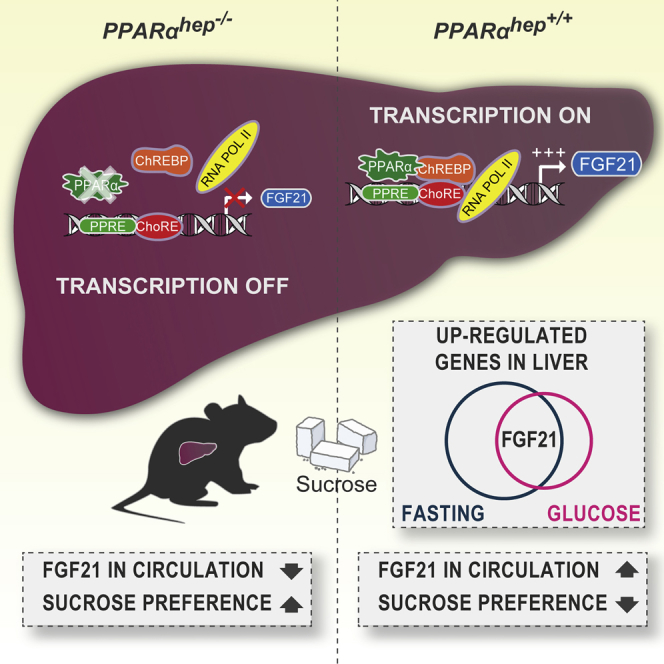
Highlights
-
•
Fgf21 is a unique hepatic gene inducible by both catabolic and anabolic signals
-
•
The ChREBP-mediated induction of Fgf21 in hepatocytes requires PPARα
-
•
Loss of PPARα impairs Fgf21 promoter accessibility at the ChoRE
-
•
PPARα is required for the control of sucrose preference in vivo
FGF21 is a hepatokine with beneficial metabolic effects, including control of sucrose preference. Iroz et al. demonstrate that Fgf21 is a unique hepatic gene inducible by both fasting and glucose signals and that the transcription factors PPARα and ChREBP both regulate the endocrine control of sugar intake by hepatic FGF21.
Introduction
The liver is central for the regulation of energy homeostasis, controlling several biochemical pathways important for metabolism of lipids, carbohydrates, proteins, bile synthesis, and detoxification of drugs and toxins. The liver also controls endocrine responses through the production of hepatokines. These proteins secreted by the hepatocytes act as hormones, and several hepatokines are considered promising leads for metabolic therapy development (Iroz et al., 2015). Among them, fibroblast growth factor 21 (FGF21) has emerged as an interesting target (Kharitonenkov and Adams, 2013). Originally targeted for its glucose-lowering properties in rodents and primates (Berglund et al., 2009, Fisher et al., 2011, Kharitonenkov et al., 2007), FGF21 is also able to improve insulin sensitivity and lipid homeostasis and induce weight loss (Coskun et al., 2008, Markan et al., 2014).
Fgf21 is a direct target of the nuclear receptor peroxisome-proliferator-activated receptor α (PPARα) in response to fasting (Badman et al., 2007, Lundåsen et al., 2007). Activated by free fatty acids derived from lipolysis (Jaeger et al., 2015, Montagner et al., 2016), PPARα is essential to liver health, as its deletion promotes the development of non-alcoholic fatty liver disease (NAFLD) and hypercholesterolemia during aging (Montagner et al., 2016). The beneficial role of PPARα in response to dyslipidemia is thought to be mediated, at least in part, through FGF21 (Ong et al., 2012). Indeed, anti-diabetic therapies and PPARα agonists (fenofibrate or Wy-14653) significantly induce liver-derived FGF21 in both mouse and human plasma (Christodoulides et al., 2009, Gälman et al., 2008, Lundåsen et al., 2007).
Hepatocyte PPARα plays an essential role during fasting, which triggers transcriptional regulation for the maintenance of glycemia and ketogenesis through fatty acid catabolism for use as an alternative energy source (Goldstein and Hager, 2015). In agreement, PPARα-deficient mice exhibit impaired fatty acid oxidation and ketogenesis that promotes hepatic steatosis during fasting (Kersten et al., 1999, Kroetz et al., 1998, Leone et al., 1999, Montagner et al., 2016).
Recent work reported that FGF21 is also activated in response to glucose and fructose in rodents and humans (Herman et al., 2012, Iizuka et al., 2009, Sánchez et al., 2009, Uebanso et al., 2011). Enriched in liver, the transcription factor carbohydrate-responsive element-binding protein (ChREBP) mediates the response to dietary carbohydrates (Abdul-Wahed et al., 2017). A physiological role for the ChREBP-FGF21 axis was revealed in experiments showing that in response to sugar consumption, ChREBP-enhanced FGF21 secretion from the liver blocked sugar-seeking behavior in mice and primates by targeting the paraventricular nucleus of the hypothalamus (Talukdar et al., 2016, von Holstein-Rathlou et al., 2016). The ChREBP protein contains a low-glucose inhibitory domain (LID) and a glucose responsive activation conserved element (GRACE) located in its N terminus (Li et al., 2006). Activation of GRACE by glucose promotes ChREBP transcriptional activity and binding to the carbohydrate-responsive element (ChoRE) of its target genes, including L-pyruvate kinase (Lpk), a rate-limiting enzyme in glycolysis, fatty acid synthase (Fas), and steroyl CoA desaturase (Scd1), key enzymes of de novo lipogenesis (Kawaguchi et al., 2001). Another isoform of Chrebp, Chrebpβ, originating from an alternative promoter, was identified in adipose tissue and liver (Herman et al., 2012). This alternative splicing results in a constitutively active ChREBP isoform lacking the LID, a domain associated with inhibition of ChREBP activity (Herman et al., 2012).
Understanding the regulation of FGF21 is currently a research focal point. Through the use of Chrebp knockout mice, we report here that ChREBP is required for the expression and secretion of hepatic FGF21 in response to carbohydrate intake. Unexpectedly, studies in hepatocyte-specific Pparα knockout mice reveal a physiological role for PPARα in the context of glucose challenge, as ChREBP is unable to induce Fgf21 in the absence of hepatic PPARα. Altogether, our results suggest that FGF21’s glucose-mediated response is dependent on both ChREBP and PPARα.
Results
FGF21 Is Induced by Both Fasting and Glucose Challenge
To characterize gene expression during fasting and a glucose challenge, a microarray analysis was conducted using liver samples from wild-type mice (Figure 1). Genes sensitive to glucose or fasting that were markedly different from the fed group were incorporated into a heatmap (Figure 1A). 67 genes were significantly induced by glucose in comparison to fed conditions (cluster 1), and 675 genes were significantly upregulated between fed and fasted groups (cluster 6). Gene ontology analysis revealed that pathways identified as specifically impacted by glucose and not by fasting are involved in pyruvate and insulin-sensitive metabolism (Figure 1B). Gene ontology revealed that pathways specifically sensitive to fasting, but not to glucose, are involved in PPAR signaling (Figure 1C). Interestingly, among the top genes upregulated by glucose and fasting (Figure S1A), only 3 genes (Fgf21, Fut1, and Atf5) were significantly upregulated (log fold change [FC] > 1; p ≤ 0.01) as compared to fed conditions (Figure 1D). When the stringency of the selection was increased to a log FC > 2 (Figure 1E), Fgf21 was left to be the sole gene upregulated by both fasting and glucose challenge (log FC = 3.9 and log FC = 3.7, respectively (p ≤ 0.01) (Figure 1E). qPCR analysis confirmed that Fgf21 expression was significantly upregulated by fasting and glucose compared to fed conditions (Figure 1F). The glucose effect was validated through analysis of Chrebp, Chrebpβ, and Lpk gene expression, while the effect of fasting was assessed by measuring the expression of two typical PPARα targets, Cyp4a10 and Vnn1 (Figure 1F).
Figure 1.

FGF21 Is Highly Induced by Both Fasting and Glucose Challenge
Wild-type C57BL/6J 10-week-old male mice were fed ad libitum, fasted for 24 hr, or fed for 24 hr a standard diet with addition of 20% glucose in drinking water (glucose challenge). Mice were killed at ZT14 (14 hr after the start of light period in the animal housing unit). Transcriptomic analysis was performed on livers (n = 6 per condition) using gene expression microarray.
(A) Heatmap of differentially expressed probes (false discovery rate [FDR] < 5%).
(B and C) KEGG categories corresponding to functions impacted by glucose (B) and fasting (C).
(D) Venn diagram presenting the overlap between glucose- and fasting-induced gene expression (log FC > 1; adjusted p < 0.01).
(E) Venn diagram presenting the overlap between glucose- and fasting-induced gene expression (log FC > 2; adjusted p < 0.01).
(F) Gene expression determined by qPCR. Data are expressed as means ± SEM, n = 6 individual mice per group. Significance is based on two-way ANOVA followed by a Bonferroni post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
ChREBP Is Necessary for Glucose-Mediated Expression and Secretion of Hepatic FGF21
To address the importance of ChREBP in the Fgf21 glucose response, experiments were first performed in mouse hepatocytes from wild-type mice (Figure 2). We observed that Fgf21 expression was induced by elevated glucose concentrations and paralleled with Chrebp, Chrebpβ, and target gene expression (Figure 2A). ChREBP recruitment onto the Fgf21 and Lpk promoters significantly increased under high glucose concentrations (25 mM) (Figure 2B). This stimulatory effect of glucose was specific and not linked to an osmotic shock, since Fgf21 expression was not induced in response to mannitol (Figure S2). To determine whether ChREBP is mandatory for upregulation of FGF21 in response to glucose, experiments were completed in hepatocytes lacking ChREBP (Chrebp−/−) (Figures 2C–2E). Mice lacking exons 9–15 of the Chrebp gene were generated through homologous recombination (Figure S3). The absence of the ChREBP protein (α isoform, 94 kDa) was validated in Chrebp−/− hepatocytes by western blot analysis (Figure 2C). Under these conditions, the ChREBPβ protein (72 kDa) could not be detected (data not shown). Similarly to Chrebp, Chrebpβ, and the ChREBP and ChREBPβ target genes Lpk and Scd1, Fgf21 robustly responded to 25 mM glucose stimulation in wild-type hepatocytes, but this response was blunted in Chrebp−/− hepatocytes (Figure 2D). In addition, no increase in FGF21 in culture medium was detected when Chrebp−/− hepatocytes were cultured in 25 mM glucose (Figure 2E). Of note, basal FGF21 production (5 mM glucose) was also significantly decreased in culture medium from Chrebp−/− hepatocytes (Figure 2E). These findings indicate that ChREBP is mandatory for the glucose-mediated expression and secretion of FGF21 by hepatocytes.
Figure 2.

ChREBP Is Necessary for Glucose-Mediated Expression and Secretion of FGF21 In Vitro
(A and B) Hepatocytes prepared from male C57BL/6J mouse livers were stimulated 1 day after platting for 24 hr with medium containing 5, 10, 15, 20, 25, or 30 mM glucose. (A) qPCR analysis of Fgf21, Chrebp, Chrebpβ, Lpk, and Scd1 gene expression. Data are presented as means ± SEM from 4 independent cultures done in triplicate. Significance is based on two-way ANOVA followed by a Bonferroni post hoc test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). (B) ChIP analysis for ChREBP binding on the Fgf21 and Lpk ChoRE followed by qPCR in mouse hepatocytes challenged with 5 or 25 mM glucose for 24 hr. Data are expressed as means ± SEM (n = 3). Significance is based on Student’s t test followed by Mann-Whitney post hoc test (∗p < 0.05).
(C–E) Primary hepatocytes from female Chrebp−/− and Chrebp+/+ littermates were stimulated 1 day after platting for 24 hr with cell culture medium containing 5 or 25 mM glucose. (C) Western blot analysis of protein from whole hepatocyte lysate. Two representative samples are presented. β actin was used as loading control. (D) Gene expression determined by qPCR. (E) ELISA quantification of FGF21 protein in medium collected at the end of glucose stimulation.
Data are presented as means ± SEM from 4 independent cultures in triplicate. Significance is based on two-way ANOVA followed by a Bonferroni post hoc test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
Liver-Specific ChREBP Expression Rescues FGF21 Plasma Concentrations in ChREBP Knockout Mice
We next performed glucose challenge experiments in vivo. 10- to 12-week-old Chrebp+/+ and Chrebp−/− male mice were given 24-hr access to a bottle of glucose-free water (fed) or a bottle containing 20% glucose (glucose challenge) (Figure 3). A significant elevation in blood glucose was observed in glucose-challenged Chrebp−/− mice compared to Chrebp+/+ mice under the same conditions (Figure 3A). A trend toward higher insulin concentrations was observed in Chrebp−/− mice compared to controls (under fed and glucose conditions), but this difference did not reach statistical significance (Figure 3B). Glucose challenge raised ChREBP protein content in liver of Chrebp+/+ mice (Figure 3C). A significant stimulation in hepatic triglyceride (TG) concentrations was also observed in glucose-challenged Chrebp+/+ mice, but not Chrebp−/− mice (Figure 3D). A significant increase in Chrebp, Chrebpβ, Lpk, and Scd1 mRNA levels was observed in liver of glucose-challenged Chrebp+/+ mice compared to fed mice from the same genotype (Figure 3E). Fgf21 mRNA and plasmatic FGF21 protein concentrations markedly increased in glucose-challenged Chrebp+/+ mice. While a residual (nonsignificant) glucose effect was observed in Chrebp−/− mice, this response was significantly reduced compared to Chrebp+/+ mice (Figures 3F and 3G), despite elevated blood glucose levels (Figure 3A). These results show that ChREBP is required for the in vivo glucose-mediated induction of Fgf21.
Figure 3.

FGF21 Is Unable to Respond to a Glucose Challenge without ChREBP
(A–G) Adult male Chrebp−/− and Chrebp+/+ littermates were allowed access to a 20% glucose drinking water solution and standard chow diet ad libitum for 18 hr. Fed mice received drinking water from the same water source used to make the glucose solution. (A) Blood glucose (mg/dl) recovered at the time of harvest from tail snip. (B) Insulin concentrations (ng/ml). (C) Western blot analysis of protein from whole liver lysate. β actin was used as loading control. Three representative samples are presented. (D) Hepatic triglyceride (TG) concentrations. Relative Chrebp, Chrebpβ, Lpk, and Scd1 gene expression (E) and Fgf21 (F) gene expression determined by qPCR. (G) ELISA quantification of FGF21 protein (ng/mL) in plasma. Data are presented as means ± SEM from 6 to 8 individual mice. Significance is based on two-way ANOVA followed by a Bonferroni post hoc test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; nd, not detectable).
(H–J) Adult male Chrebp+/+ and Chrebp−/− mice were injected intravenously with a single dose of 3 × 109 pfu GFP or ChREBPCA adenovirus on day 1. Four days later, analyses were performed. (H) Blood glucose (mg/dl) recovered at the time of harvest from tail snip. (I) Western blot analysis of protein extracted from whole liver lysate. β actin was used as loading control. Three representative samples are presented. (J) ELISA quantification of FGF21 protein (ng/ml) in plasma.
(K) Relative expression of hepatic genes determined by qPCR.
Data are presented as means ± SEM from 8 to 12 individual mice. Significance is based on two-way ANOVA followed by a Bonferroni post hoc test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; nd, not detectable).
We next addressed whether liver-specific re-expression of Chrebp in a context of global ChREBP deficiency could rescue FGF21 production. Chrebp+/+ and Chrebp−/− adult mice were injected with an adenovirus vehicle containing the GFP protein or a truncated isoform of ChREBP lacking the LID domain (Li et al., 2006) corresponding to a constitutively active ChREBP isoform (ChREBPCA). Mice received a 20% glucose solution for 24 hr before sacrifice (Figures 3H–3K). Blood glucose concentrations were higher in GFP-ChREBP−/− mice than in GFP-ChREBP+/+ mice and were rescued to basal values when ChREBPCA was injected into Chrebp−/− mice (Figure 3H). Western blot analysis confirmed the absence of native ChREBP protein (94 kDa) but the presence of ChREBPCA (72 kDa) in Chrebp−/− mice injected with ChREBPCA (Figure 3I). Importantly, ChREBPCA rescued the circulating level of FGF21 (Figure 3J). This correlates with the effect of ChREBPCA on the hepatic expression of Chrebp, Chrebpβ, and their targets, Lpk, Scd1, and Fgf21 mRNA (Figure 3K). Altogether, we report that ChREBPCA administration rescued ChREBP activity in the liver of Chrebp−/− mice and was sufficient to restore FGF21 gene expression and production.
Glucose Stimulation or ChREBP Overexpression Is Not Efficient to Induce FGF21 Expression or Secretion in the Absence of Hepatic PPARα
Since the Fgf21 promoter contains overlapping peroxisome proliferator response element (PPRE) and ChoRE units (−88 to −54 bp) (Girer et al., 2016, Uebanso et al., 2011), we investigated whether PPARα could impact the glucose response of Fgf21 mediated by Chrebp. Primary hepatocytes from liver-specific Pparα knockout mice (Pparαhep−/−) and their littermates, Pparαhep +/+ mice (Montagner et al., 2016), were stimulated by glucose in a dose-dependent manner (Figure 4). A similar glucose-mediated induction of Chrebp, Chrebpβ, Lpk, and Scd1 mRNA was observed in hepatocytes from Pparαhep +/+ and Pparαhep −/− mice (Figure 4A). Although a residual glucose effect was observed for Fgf21 expression in Pppaαhep −/− mice hepatocytes, this response was reduced compared to Pparαhep+/+ mice (Figure 4B). Importantly, FGF21 production in response to glucose was reduced by 60% in culture medium from Pppaαhep−/− hepatocytes compared to controls (Figure 4C). This profile was specific to Fgf21, since the expression of other typical PPARα targets (Cyp4a10, Cyp4a14, or Vnn1) was not induced by glucose (Figure S4). We next addressed whether Fgf21 could be rescued by Chrebp overexpression in the context of Pparα deficiency. Hepatocytes from Pparαhep+/+ and Pppaαhep −/− mice were infected with the constitutive active form of ChREBP (ChREBPCA) for 24 hr. The expression of Chrebp and Chrebpβ was significantly increased in response to ChREBPCA in both Pparα hep+/+ and Pppaαhep −/− hepatocytes, and as a result, Lpk and Scd1 mRNA expression was stimulated compared to GFP conditions (Figure 4D). However, while Chrebp overexpression led to a 50-fold increase in Fgf21 expression in Pparαhep+/+ hepatocytes, it failed to significantly induce Fgf21 expression in Pppaαhep −/− hepatocytes (Figure 4E). FGF21 measured in the medium of cell culture confirmed the Fgf21 mRNA expression profile (Figure 4F). These results suggest that neither glucose nor ChREBP overexpression is efficient in inducing Fgf21 gene expression or protein production in the absence of PPARα in hepatocytes.
Figure 4.

Fgf21 Is Not Efficiently Induced by Glucose or by ChREBP Overexpression in the Absence of Liver PPARα
(A–C) Primary hepatocytes derived from adult male Pparαhep+/+ or Pparαhep−/− mice were incubated 1 day after platting for 24 hr with medium containing 5, 10, 15, 20, 25, or 30 mM glucose. Relative Chrebp, Chrebpβ, Lpk, and Scd1 gene expression (A) and Fgf21 gene expression (B) was determined by qPCR. (C) ELISA quantification of FGF21 protein (ng/ml) in medium collected at end of glucose stimulation. Figures are presented as means ± SEM from 4 independent cultures completed in duplicate. Significance is based on two-way ANOVA followed by a Bonferroni post hoc test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; nd, not detectable).
(D–F) Primary hepatocytes derived from adult male mice Pparαhep+/+ or Pparαhep−/− were incubated 6 hr after platting with 3 × 109 pfu GFP or ChREBPCA adenovirus at a glucose concentration of 5 mM for 24 hr. Relative Chrebp, Chrebpβ, Lpk, and Scd1 expression (D) and of Fgf21 expression (E) was determined by qPCR. (F) ELISA quantification of FGF21 protein (in ng/ml) in medium collected at end of adenoviral treatment.
Data are presented as means ± SEM from 4 independent cultures completed in duplicate. Significance is based on two-way ANOVA followed by a Bonferroni post hoc test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
FGF21 Synergistically Responds to Glucose and a Pharmacological PPARα Activator in Both Mouse and Human Hepatocytes
To determine whether ChREBP and PPARα act in synergy to regulate Fgf21 gene expression, mouse and human hepatocytes were stimulated for 24 hr with low (5 mM) or high glucose concentrations (25 mM) in the presence of the PPARα agonist Wy-14643 (Figure S5). Expression of Chrebp and Chrebpβ mRNA confirmed a positive glucose response in both mouse and human hepatocytes (Figures S5A and S5B). Activation of PPARα by Wy-14643 was validated by a significant upregulation in Acox1 expression, a PPARα target gene (Figures S5C and S5D). Fgf21 mRNA levels were drastically increased when hepatocytes (mouse and human) were incubated in the combined presence of high glucose (25 mM) and Wy-14643 (Figures S5E and S5F). Altogether, these results show that ChREBP and PPARα can act synergistically to induce Fgf21 in mouse and in human liver cells.
Hepatocyte PPARα Is Required for the Effect of Glucose on FGF21 In Vivo
To investigate whether PPARα is involved in the glucose-mediated induction of Fgf21 in vivo, glucose challenge experiments were performed in 10- to 12-week-old Pparαhep+/+ and Pparα hep−/− mice. Three nutritional conditions were assigned to both genotypes: (1) mice fasted 24 hr with free access to water (fasted), (2) mice fed ad libitum with access to standard chow diet and free access to water (fed), and (3) mice fed ad libitum with access to standard chow diet and a 20% glucose solution in water (glucose challenge) (Figure 5). Chrebp, Chrebpβ, and their target genes (Lpk and Scd1) were induced in a similar manner in the liver of Pparα hep+/+ and Pparαhep−/− mice when challenged by glucose (Figure 5A). In contrast, the glucose-mediated induction of Fgf21 (compared to the fed state) was significantly reduced in the absence of PPARα (Figure 5A). FGF21 in the circulation matched hepatic mRNA levels, revealing an upregulation of FGF21 in the plasma of Pparα hep+/+ that was significantly reduced in glucose-challenged Pparαhep−/− mice (Figure 5B). As expected, loss of PPARα markedly impacted the fasting response of FGF21 (Figure 5A-B). Because the Fgf21 promoter contains overlapping PPRE and ChoRE units (Figure 5C), we investigated whether lack of PPARα could impair ChREBP binding in response to glucose in vivo. When primers amplifying both ChoRE and PPRE were used (Figure 5C), a significant enrichment in PPARα binding within the proximal region of the Fgf21 promoter in liver of Pparα hep+/+ mice was observed (Figure 5D). While PPARα binding was elevated under fasting conditions, a significant PPARα recruitment onto the proximal region of the Fgf21 promoter was detected under both fed and glucose-challenge conditions (Figure 5D). Surprisingly, in liver of glucose challenged Pparα hep−/− mice, recruitment of ChREBP on the Fgf21 promoter ChoRE was significantly reduced (Figure 5D). Interestingly, a significant decrease in RNA polymerase II (Pol II) recruitment and histone H3 lysine 9 acetylation (H3K9ac), a histone mark related to transcriptional activation, was observed in parallel in the liver of these mice (Figure 5D). In contrast, ChREBP recruitment on the Lpk ChoRE was similar in glucose-challenged Pparαhep+/+ and Pparαhep −/− mice (Figure 5E). The presence of Pol II on the Lpk promoter (Figure 5E) was significantly elevated in response to glucose in liver of mice from both genotypes and correlated with Lpk mRNA expression (Figure 5A). Altogether, our results show that the absence of PPARα impairs the binding of ChREBP to its ChoRE and the subsequent glucose-mediated induction of Fgf21. Surprisingly, significant ChREBP recruitment was also detected on the Fgf21 ChoRE under fasting conditions (Figure 5D). Further analysis will be required to determine whether ChREBP interferes with the fasting-mediated induction of Fgf21 expression in the liver, as suggested in a recent report showing that liver-specific Chrebp knockout mice have decreased Fgf21 expression compared to controls (Jois et al., 2017).
Figure 5.

PPARα Is Required for the Glucose-Dependent Expression and Secretion of FGF21 by the Liver
Male Pparαhep+/+ or Pparαhep−/− mice were assigned one of three treatment groups: (1) fasted 24 hr with free access to drinking water (fasted), (2) fed ad libitum with free access to drinking water (fed), or (3) fed ad libitum with access to a 20% glucose drinking water solution for 24 hr (glucose challenge).
(A) Relative gene expression of Chrebp, Chrebpβ, Lpk, Scd1, and Fgf21 was determined by qPCR. Data are expressed as means ± SEM (n = 6 individual mice per group). Significance is based on two-way ANOVA followed by Bonferroni post hoc test (fasting or glucose versus fed; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001). Significance of the effect of genotype is based two-way ANOVA followed by Bonferroni post hoc test (Pparαhep−/− versus Pparαhep +/+; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001).
(B) ELISA quantification of FGF21 in plasma. Data are expressed as means ± SEM from n = 6 individual mice per group. Significance of the effect of fasting or glucose challenge is based on two-way ANOVA followed by post hoc test (fasting or glucose versus fed; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001). Significance of the effect of genotype is based on two-way ANOVA followed by Bonferroni post hoc test (Pparαhep−/− versus Pparαhep +/+; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001).
(C) Fgf21 promoter sequence from the transcription start site to −184 bp. The PPARα binding site (PPRE) is indicated in gray. The ChREBP binding site (ChoRE) is indicated in yellow. Primers used for ChIP analysis are indicated on the sequence.
(D) ChIP analysis followed by qPCR of whole mouse liver tissue. Immunoprecipitation (IP) was conducted with PPARα, ChREBP, H3K9ac, and RNA Pol II antibodies. The DNA region of the Fgf21 promoter was amplified using the primers indicated in (C).
(E) ChIP analysis followed by qPCR of whole mouse liver tissue. IP were conducted with ChREBP and RNA Pol II antibodies. Data are expressed as means ± SEM from n = 3 individual mice per group. Significance is based on two-way ANOVA followed by Bonferroni post hoc test (∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001).
Fgf21 Promoter Accessibility Is Reduced in the Absence of PPARα
To provide insights into the mechanisms by which PPARα affects ChREBP binding onto the Fgf21 promoter in response to glucose, a series of experiments were conducted in vitro (Figure 6). First, we performed formaldehyde-assisted isolation of regulatory elements (FAIRE)-qPCR analysis (Simon et al., 2012) to determine whether ChREBP accessibility to the Fgf21 promoter could be altered in the absence of PPARα (Figure 6A). We observed that while Fgf21 ChoRE promoter accessibility tended to increase in response to high glucose concentrations in Pparα hep+/+ hepatocytes, it failed to increase in Pparα hep−/− hepatocytes (Figure 6A). To get insight into the molecular mechanisms involved, we hypothesized that PPARα could affect ChoRE accessibility through epigenetic processes and tested whether a pan-histone deacetylase (pan-HDAC) inhibitor (Imai et al., 2016) could increase Fgf21 promoter accessibility. Treatment of hepatocytes with a HDAC inhibitor further enhanced the difference observed in Fgf21 accessibility between Pparαhep+/+ and Pparαhep−/− hepatocytes (Figure 6A), which correlated with a potentiated effect of glucose on Fgf21 expression (Figure 6B). While no significant change in Fgf21 promoter accessibility was observed in Pparαhep −/− hepatocytes treated with the HDAC inhibitor (Figure 6A), a modest but significant effect was observed for Fgf21 mRNA levels measured under these conditions. However, treatment failed to fully rescue Fgf21 gene expression to control levels (Figure 6B). These data suggest that Fgf21 promoter accessibility at the ChoRE is altered in the absence of PPARα by a mechanism that may partly rely on histone acetylation. To determine whether the potentiating effect of the HDAC inhibitor on glucose-induced Fgf21 expression was due to enhanced ChREBP activity, experiments were performed in Chrebp+/+ and Chrebp−/− hepatocytes (Figure 6C). Similar to what was observed in Pparαhep+/+ hepatocytes (Figure 6B), the effect of glucose (25 mM) on Fgf21 expression was increased when Chrebp+/+ hepatocytes were treated with the HDAC inhibitor (Figure 6C). Chrebp, Chrebpβ, and Lpk followed a similar trend, with a significant effect observed for Chrebp (Figure 6C). Importantly, the potentiated effect of the HDAC inhibitor on Fgf21 expression was lost in Chrebp−/− hepatocytes (Figure 6C), indicating a dependence on ChREBP activity. Altogether, our results suggest that PPARα is essential to allow ChREBP to gain access to the promoter of Fgf21. Moreover, our data show that the use of HDAC inhibitors enhances the ChREBP-dependent effect of glucose on Fgf21 but cannot fully rescue the effect of PPARα deficiency.
Figure 6.

Fgf21 Promoter Accessibility Is Reduced in the Absence of PPARα
(A and B) Primary hepatocytes derived from adult Pparαhep+/+ or Pparαhep−/− mice were treated with 10 μM of the HADC inhibitor LBH589 or DMSO as a control for 24 hr in the presence of 5 or 25 mM glucose. (A) FAIRE-qPCR was performed as described in Experimental Procedures. (B) Fgf21 gene expression determined by qPCR. Data are presented as means ± SEM from 4 independent cultures performed in duplicate. Significance is based on two-way ANOVA followed by Bonferroni post hoc test (∗p % 0.05; ∗∗p % 0.01; ∗∗∗p % 0.001).
(C) Primary hepatocyte derived from adult Chrebp+/+ or Chrebp−/− mice were treated with 10 μM of the HADC inhibitor LBH589 or with DMSO as control for 24 hr in the presence of medium containing 5 or 25 mM glucose. Relative gene expression of Chrebp, Chrebpβ, Lpk, and Fgf21 was determined by qPCR.
Data are presented as means ± SEM from 4 independent cultures performed in duplicate. Significance is based on two-way ANOVA followed by Bonferroni post hoc test (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001).
Sucrose Intake Is Increased in Mice Lacking PPARα in the Liver
Finally, we determined whether the decrease in circulating FGF21 observed in glucose-challenged Pparα hep−/− mice (Figure 5B) paralleled with an increase in sucrose preference, since it was recently demonstrated that FGF21’s action on the paraventricular nucleus of the hypothalamus blocks sugar-seeking behavior and sugar intake in mice (Talukdar et al., 2016, von Holstein-Rathlou et al., 2016). First, we observed that there was no change in body weight or food or water intake in Pparα hep−/− compared to Pparα hep+/+ mice fed with standard chow (Figures 7A–7C). Sucrose preference was then evaluated by giving Pparα hep+/+ and Pparα hep+/+ littermates matched for age and body weight free choice between a bottle containing a 10% sucrose solution and water (Figure 7D). Consumption, which was measured daily for 3 days, demonstrated that Pparα hep−/− mice consumed 30% more sucrose solution than Pparα hep+/+ mice, while the volume of water drunk remained similar between genotypes (Figure 7E).
Figure 7.

Sucrose Intake Is Increased in Pparαhep−/− Mice
(A–C) Average body weight gain (A), food (B) and water (C) intake of age-matched Pparαhep+/+− and Pparαhep−/− mice (n = 12/group) over 6 weeks.
(D) Average body weight of age-matched male Pparαhep+/+ and Pparαhep−/− mice used for the sucrose preference test (n = 16/group).
(E) Pparαhep+/+ and Pparαhep−/− mice were given a 4-day adaptation period with two water bottles. A 10% sucrose solution was added to one of the water bottles for 3 days following adaptation and intake of sucrose solution and water was recorded daily for 3 days. Data are presented as means ± SEM from 5 to 6 individual mice. Significance is based on 2-way ANOVA followed by Bonferroni post hoc test (∗∗p ≤ 0.01).
Discussion
The regulation of FGF21 in the liver is complex due to the paradoxical regulation of this key hepatokine by fasting and glucose signals. In the current study, we uncovered a cross-talk between ChREBP and PPARα for the induction of hepatic FGF21 in response to a glucose challenge. The main finding of our study is that hepatic PPARα is necessary for the glucose-mediated induction of Fgf21 by ChREBP.
Key to β-oxidation and ketogenesis, FGF21 mobilizes energy in the liver for peripheral use, protecting against dyslipidemia and hepatosteatosis (Inagaki et al., 2007, Potthoff et al., 2009). First described as a fasting hormone regulated by PPARα, transcriptional regulation of Fgf21 in the context of excessive blood glucose has only recently been explored (Talukdar et al., 2016, von Holstein-Rathlou et al., 2016), despite early identification of the glucose-sensing region, ChoRE, on the Fgf21 promoter in both mouse (−74 to −52 bp) and human (−380 to −366 bp) (Iizuka et al., 2009). Our experiments confirm that FGF21 is significantly expressed and released by cultured mouse and human hepatocytes in response to glucose upon ChREBP binding to the ChoRE (Iizuka et al., 2009, Uebanso et al., 2011). Our study demonstrates that deletion of ChREBP blunts transcription and secretion of hepatic FGF21 in response to glucose. Importantly, hepatic rescue of ChREBP in global ChREBP knockout mice is sufficient to restore Fgf21 mRNA in the liver and protein in circulation, demonstrating the specificity of hepatic ChREBP activity on the induction of FGF21 in response to a glucose challenge. Interestingly, ChREBP rescue in the liver of Chrebp−/− mice significantly normalized blood glucose concentrations to control levels. This suggests that hepatic ChREBP activity and not peripheral ChREBP is essential for glucose-homeostasis maintenance. This result is consistent with a previous study in which hepatic ChREBP overexpression improved impaired glucose tolerance of high-fat-diet-challenged mice (Benhamed et al., 2012) and is in agreement with a recent study reporting that liver Chrebp knockout mice exhibit impaired insulin sensitivity and glucose intolerance (Jois et al., 2017). In this study, while hepatic Chrebp deletion protected against hepatic steatosis, it also resulted in gene expression changes in white and brown adipose tissues, suggesting inter-organ communication. The contribution of ChREBP to whole-body energy balance may therefore rely on its regulation of lipid species and/or hepatokine production that could contribute to inter-tissue coordination of energy homeostasis.
Activated in response to free fatty acids liberated from adipocytes during fasting (Montagner et al., 2016), a role for hepatocyte PPARα in the response to glucose had not yet been identified. Our study reveals that the synergic induction of Fgf21 by glucose and the PPARα agonist Wy-14643 occurs in both mouse and human hepatocytes. We hypothesize that this cross-talk is specific to Fgf21, as we found no effect of glucose on other typical PPARα target genes. This Fgf21 specificity may be due to the proximity of ChREBP- and PPARα-binding sites on the Fgf21 proximal promoter, as both a PPRE and a ChoRE region coexist (−88 to −54 bp) (Girer et al., 2016). Microarray analysis comparing gene regulation during fasting and glucose challenge highlights a small subset of genes that are upregulated in both conditions. Of those genes Fgf21 is the most significantly induced. Therefore, FGF21 is a unique hepatic hormone showing dual regulation by fasting and carbohydrate signaling. Few publications have proposed a dialog between ChREBP and PPARs for the coordination of energy metabolism. Some examples of cross-talk and regulatory mechanisms have been described for lipid metabolism in brown adipose (Iizuka et al., 2013) and pancreatic β cell function (Boergesen et al., 2011). In the liver, it was also described that the PPARα co-factor PPAR γ coactivator-1β (PGC-1β) can act as a co-activator of ChREBP in response to glucose. PGC-1β is known to activate genes responsible for fatty acid oxidation and hepatic gluconeogenesis during fasting (Vega et al., 2000, Yoon et al., 2001), and it was also reported to upregulate de novo lipogenic genes during glucose stimulation in a ChREBP-dependent manner (Chambers et al., 2013). Altogether, these studies strongly suggest a direct and/or indirect interaction between ChREBP and the PPAR nuclear receptor family.
Here, we reveal a ChREBP-PPARα dialog in hepatocytes required for the induction of Fgf21 by glucose. Indeed, despite normal hepatic ChREBP activity toward glycolytic and lipogenic genes, we report that in the absence of PPARα, ChREBP binding to the Fgf21 ChoRE is significantly reduced, suggesting that hepatocyte PPARα is necessary for the ChREBP-mediated induction of Fgf21 in response to glucose. Our study also reports that Fgf21 promoter accessibility is reduced in the absence of PPARα. Deficient recruitment of ChREBP as well as Pol II was observed at the proximal Fgf21 promoter locus under elevated glucose conditions in the liver of mice lacking hepatocyte PPARα. The fact that epigenetic marks (i.e., H3K9ac) of active transcription were reduced at the Fgf21 promoter when PPARα was lacking in liver, under both fasting and glucose conditions, also supports the hypothesis of reduced promoter accessibility. Indeed, even under conditions of ChREBP overexpression, Fgf21 expression was not efficiently induced in the context of PPARα deficiency. Further experiments will be needed to determine whether PPARα acts as a “pioneer” transcription factor for Fgf21 transcription. Among several described functions, pioneer factors can trigger the opening and/or organization of the local chromatin, in turn allowing the binding of other transcription factors, histone-modification enzymes, chromatin modifiers, and/or nucleosome remodelers (Zaret and Carroll, 2011). Of note, an HDAC inhibitor potentiated a ChREBP-dependent effect of glucose on Fgf21 expression in primary hepatocytes. The HDAC inhibitor strategy in glucose-challenged PPARα-deficient hepatocytes only led to a modest restoration of FgF21 expression. While this approach was not sufficient to fully restore accessibility to the Fgf21 promoter, it does suggest that Fgf21 promoter accessibility at the ChoRE is altered in the absence of PPARα by a mechanism that may partly rely on histone acetylation. We employed FAIRE-qPCR analysis to determine Fgf21 promoter accessibility in response to glucose. A more sensitive approach, the assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) (Buenrostro et al., 2013) may have allowed us to determine whether Fgf21 promoter accessibility is indeed modified by an HDAC inhibitor strategy in Pparαhep−/− hepatocytes. While other molecular mechanisms are clearly involved, the implication of specific HDACs should be further investigated, since it was recently shown that PPARα prevents the recruitment of HDAC3 to the Fgf21 promoter in hepatocytes when concentrations of β-hydroxybutyrate, a key product of β-oxidation, are elevated (Rando et al., 2016).
At the physiological level, we observed that Pparαhep−/− mice consumed more sucrose (+30%) than Pparαhep+/+ mice. Two studies have recently unraveled the mechanistic link between sucrose-derived FGF21 and nutrient preference (Talukdar et al., 2016, von Holstein-Rathlou et al., 2016). FGF21 production in response to carbohydrates was shown to markedly reduce sweet taste preference and suppress consumption of simple sugars by acting on specific regions of the brain (Talukdar et al., 2016, von Holstein-Rathlou et al., 2016). In these studies, hepatic FGF21 production in response to carbohydrate intake was attributed to ChREBP activity. The fact that PPARα is also involved in this liver-to-brain axis following simple sugar consumption opens new molecular path of regulation of macronutrient preference/intake and expands liver PPARα function from a fasting regulator to a modulator affecting the physiological sugar response.
In conclusion, we identify a transcriptional node in the control of hepatic FGF21 in response to glucose. The glucose sensor ChREBP requires PPARα for the induction of FGF21 in response to dietary sugar. This is a unique collaboration between transcription factors that are activated in response to distinct nutritional conditions (high glucose for ChREBP and fasting for PPARα). These data imply that drug targeting of PPARα may exert part of its beneficial effects on metabolic homeostasis by supporting the ChREBP-induced loop controlling sweet preference via FGF21.
Experimental Procedures
Generation of ChREBP Knockout Mice
Mice lacking exons 9–15 of the Chrebp gene were generated through homologous recombination. Correspondence regarding Chrebp−/− mice should be addressed to R.D. (renaud.dentin@inserm.fr) and C.P. (catherine.postic@inserm.fr). Experimental details are provided in Supplemental Experimental Procedures.
Animals
10- to 12-week-old adult male C57BL/6J, Chrebp+/+, Chrebp−/−, Pparαhep+/+, and Pparαhep−/− mice (Montagner et al., 2016) were used for all in vivo experiments. For hepatocyte cultures, male and female mice were used as described in the figure legends. Procedures were carried out according to the French guidelines for the care and use of experimental animals (animal authorization agreement number CEEA34.AFB/CP.082.12, Paris Descartes Ethical Committee). Mice were maintained in a 12-hr light/dark cycle with water and a standard diet (65% carbohydrate, 11% fat, and 24% protein) unless otherwise specified. Nutritional challenges details are described in Supplemental Experimental Procedures.
Injection of Adenovirus In Vivo
Adenovirus coding GFP and ChREBPCA (ChREBP isoform deleted of the LID domain) (Li et al., 2006) produced by Laboratoire de thérapie génique (Nantes, France; requests to R.D. at renaud.dentin@inserm.fr) were delivered through penis vein injection (3 × 109 [pfu]/mouse) to adult mice. Four days later, nutritional protocols were applied.
Primary Cultures of Human and Mouse Hepatocytes
Human hepatocytes were prepared from lobectomy segments resected from adult patients under the approval of the National Ethics Committee as described previously (Pichard et al., 2006, Marmier et al., 2015). Mouse hepatocytes were isolated as described previously (Dentin et al., 2004). Experimental details regarding culture conditions are provided in the figure legends and Supplemental Experimental Procedures.
ChIP Analysis
In vivo chromatin immunoprecipitation (ChIP) assays from mouse livers were performed by Active Motif. Briefly, genomic DNA regions of interest were isolated using antibodies against H3K9Ac (Active Motif), RNA Pol II (Active Motif), ChREBP (Novus), and PPARα (Santa Cruz Biotechnology). qPCR reactions were carried out in triplicate using SYBR Green Supermix (Bio-Rad) on a CFX Connect Real Time PCR system. Positive and negative control sites were tested for each factor as well as the sites of interest. The resulting signals were normalized for primer efficiency by carrying out qPCR for each primer pair using input DNA (pooled unprecipitated genomic DNA from each sample). Specific enrichment was expressed as percentage of input. Further details are provided in Supplemental Experimental Procedures.
In vitro ChIP assays from cultured hepatocytes were performed as described previously (Marmier et al., 2015). Briefly, genomic DNA regions of interest were isolated using antibodies against ChREBP (Novus) and immunoglobulin G (IgG) (Cell Signaling). DNA fragments were quantified by qPCR using primers described in Table S1. Results are expressed as fold enrichment. Further details are provided in Supplemental Experimental Procedures.
FAIRE-qPCR
FAIRE-qPCR was performed in Pparαhep+/+ and Pparαhep−/− hepatocytes using a protocol previously described (Simon et al., 2012). Briefly, cells were treated with 1% formaldehyde at room temperature for 5 min to form DNA-protein crosslinks, and the crosslinking was stopped by addition of glycine to a final concentration of 125 mM. FAIRE was analyzed by qPCR on genomic DNA using the ChoRE Fgf21 primers (Figure 5C) and calculated using relative enrichment for each amplicon using the comparative Ct method, such that a ratio is calculated for the signal from the FAIRE sample relative to the input control DNA signal. Results are expressed as fold enrichment. Experimental details are provided in Supplemental Experimental Procedures.
Gene Expression Analysis
Total cellular RNA was extracted using the SV total RNA isolation system (Promega). For qPCR analysis, total RNA samples (2 μg) were reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Primers for SYBR green assays are presented in Tables S2 and S3. Primers used to measure Chrebp expression were designed to detect the Chrebpα and Chrebpβ isoforms. A specific primer to detect only the β isoform was also used. Primers for Vnn1, Cyp4a10, and Cyp4a14 were previously described (Montagner et al., 2016). Amplifications were performed on an ABI Prism 7300 Real Time PCR System (Applied Biosystems). qPCR data were normalized by TATA-box binding protein (TBP) mRNA levels or 18S ribosomal (18S) for human samples and analyzed with LinRegPCR.22.
Transcriptomic profiles were obtained using Agilent SurePrint G3 Mouse V2 GE 8x60K (Design 074809) following the manufacturer’s instructions. Data were analyzed with R (http://www.r-project.org) using Bioconductor packages (http://www.bioconductor.org, v 2.12; Gentleman et al., 2004) as described in GEO: GSE26728. A model was fitted using the limma lmFit function (Wettenhall and Smyth, 2004). Correction for multiple testing was applied using a false discovery rate (Benjamini and Hochberg, 1995). Probes with adjusted p ≤ 0.05 were considered differentially expressed (n = 6). Hierarchical clustering was applied and the differentially expressed probes using 1 − Pearson correlation coefficient as distance and Ward’s criterion for agglomeration. The resulting dendrogram were illustrated as a heatmap. The enrichment of gene ontology (GO) biological processes was evaluated using a conditional hypergeometric test (GOstats package; Falcon and Gentleman, 2007).
Western Blotting Analysis
Proteins from hepatocytes and liver tissue were extracted from whole-cell lysates. Proteins were subjected to 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Rabbit polyclonal ChREBP (1:1,000, Novus Biologicals) and L-PK (1:1,500, a gift from Dr. Axel Kahn) antibodies were used. Protein β-actin (1:5,000) (Cell Signaling Technology) was used to normalize data.
Analytical Analysis
Blood glucose was measured from total blood using an Accu-Check glucometer (Roche). Liver triglycerides were measured with a colorimetric diagnostic kit (Triglycerides FS, Diasys). Serum insulin concentrations were determined using a rat insulin ELISA assay kit (Crystal Chem) with a mouse insulin standard. The mouse ELISA kit (Millipore) was used to measure FGF21 in cell culture medium (30 μL of medium was used) and mouse plasma (20 μL).
Statistical Analysis
Data represent at least three independent experiments, are reported as means ± SEM, and were analyzed with analysis of variance using Prism 5.0 (GraphPad) software. A Student’s t test was used when comparing two groups (followed by Mann-Whitney post hoc test) or two-way ANOVA when comparing three or more groups followed by a Bonferroni post hoc test. Statistical significance was defined as p < 0.05.
Author Contributions
A.I., A.M., F.B., C.P., and H.G. designed and performed experiments, analyzed the data, and wrote the manuscript. A.M., A.P., F.B., E.A., C.P., and H.G. revised the manuscript. E.A., A.P., E.F., M.R., C.L., V.F., S.M., and M.C. performed experiments and analyzed the data. E.T., M. Do-Cruzeiro, F.L., R.D., and C.P. generated ChREBP knockout mice. M. Daujat-Chavanieu and S.G.-C. provided human hepatocytes. Y.L. performed microarray analysis and biostatics analysis. R.D., S.G., A.-F.B., J.G., and W.W. provided critical reagents and comments.
Acknowledgments
We would like to thank Dr. B. Staels and Dr. P. Lefebvre (Inserm U1011, Lille), Dr. N. Venteclef (Inserm UMR_S1138, Paris), and Dr. D. Langin (Inserm UMR 1048, Toulouse) for scientific discussion. We also thank Dr. A. Kayali and Dr. T. Issad (Institut Cochin, Inserm U1016) for critical reading of the manuscript. We thank L. Francese, S. Topçu (Institut Cochin, Inserm U1016), C. Naylies (GeT-TRiX, INRA ToxAlim), and G.Michel for providing excellent technical assistance. We thank the animal facility staff (EZOP, INRA ToxAlim) for their excellent work. H. Guillou’s lab (TIM, ToxAlim) is supported by grants from Région Occitanie. W.W. was supported by a start-up grant from Lee Kong Chian School of Medicine, Nanyang Technological University. C. Postic’s lab (U1016-Institut Cochin) is supported by grants from ChroME Network (Marie Curie Sklodowska Action H2020-MSCA-ITN-2015-675610), the Foundation for the Medical Research (FRM) (DEQ20150331744), and the European Foundation for the Study of Diabetes (EFSD) Novonordisk. H. Guillou’s and C. Postic’s labs are also supported by grants from the National Agency for Research (ANR) (ANR-12BSV1-0025-ObeLiP and ANR-15-CE14-0026-Hepatokind). R. Dentin’s lab (U1016-Institut Cochin) is supported by the European Research Council (ERC-2013-StG-336629) and the city of Paris (Projet Emergences-2011). This project is performed in the context of the DHU authors (autoimmune and hormonal diseases).
Published: October 10, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.09.065.
Contributor Information
Renaud Dentin, Email: renaud.dentin@inserm.fr.
Hervé Guillou, Email: herve.guillou@inra.fr.
Catherine Postic, Email: catherine.postic@inserm.fr.
Data and Software Availability
The accession number for the microarray data reported in this paper is GEO: GSE92502.
Supplemental Information
References
- Abdul-Wahed A., Guilmeau S., Postic C. Sweet sixteenth for ChREBP: established roles and future goals. Cell Metab. 2017;26:324–341. doi: 10.1016/j.cmet.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Benhamed F., Denechaud P.-D., Lemoine M., Robichon C., Moldes M., Bertrand-Michel J., Ratziu V., Serfaty L., Housset C., Capeau J. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Invest. 2012;122:2176–2194. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300. [Google Scholar]
- Berglund E.D., Li C.Y., Bina H.A., Lynes S.E., Michael M.D., Shanafelt A.B., Kharitonenkov A., Wasserman D.H. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology. 2009;150:4084–4093. doi: 10.1210/en.2009-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boergesen M., Poulsen Ll., Schmidt S.F., Frigerio F., Maechler P., Mandrup S. ChREBP mediates glucose repression of peroxisome proliferator-activated receptor alpha expression in pancreatic beta-cells. J. Biol. Chem. 2011;286:13214–13225. doi: 10.1074/jbc.M110.215467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for multimodal regulatory analysis and personal epigenomics. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers K.T., Chen Z., Lai L., Leone T.C., Towle H.C., Kralli A., Crawford P.A., Finck B.N. PGC-1β and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol. Metab. 2013;2:194–204. doi: 10.1016/j.molmet.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulides C., Dyson P., Sprecher D., Tsintzas K., Karpe F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J. Clin. Endocrinol. Metab. 2009;94:3594–3601. doi: 10.1210/jc.2009-0111. [DOI] [PubMed] [Google Scholar]
- Coskun T., Bina H.A., Schneider M.A., Dunbar J.D., Hu C.C., Chen Y., Moller D.E., Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- Dentin R., Pégorier J.P., Benhamed F., Foufelle F., Ferré P., Fauveau V., Magnuson M.A., Girard J., Postic C. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J. Biol. Chem. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- Falcon S., Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Fisher F.M., Estall J.L., Adams A.C., Antonellis P.J., Bina H.A., Flier J.S., Kharitonenkov A., Spiegelman B.M., Maratos-Flier E. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology. 2011;152:2996–3004. doi: 10.1210/en.2011-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gälman C., Lundåsen T., Kharitonenkov A., Bina H.A., Eriksson M., Hafström I., Dahlin M., Amark P., Angelin B., Rudling M. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 2008;8:169–174. doi: 10.1016/j.cmet.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girer N.G., Murray I.A., Omiecinski C.J., Perdew G.H. Hepatic aryl hydrocarbon receptor attenuates fibroblast growth factor 21 expression. J. Biol. Chem. 2016;291:15378–15387. doi: 10.1074/jbc.M116.715151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I., Hager G.L. Transcriptional and chromatin regulation during fasting: the genomic era. Trends Endocrinol. Metab. 2015;26:699–710. doi: 10.1016/j.tem.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M.A., Peroni O.D., Villoria J., Schön M.R., Abumrad N.A., Blüher M., Klein S., Kahn B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K., Takeda J., Horikawa Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009;583:2882–2886. doi: 10.1016/j.febslet.2009.07.053. [DOI] [PubMed] [Google Scholar]
- Iizuka K., Wu W., Horikawa Y., Saito M., Takeda J. Feedback looping between ChREBP and PPARα in the regulation of lipid metabolism in brown adipose tissues. Endocr. J. 2013;60:1145–1153. doi: 10.1507/endocrj.ej13-0079. [DOI] [PubMed] [Google Scholar]
- Imai Y., Ohta E., Takeda S., Sunamura S., Ishibashi M., Tamura H., Wang Y.H., Deguchi A., Tanaka J., Maru Y., Motoji T. Histone deacetylase inhibitor panobinostat induces calcineurin degradation in multiple myeloma. JCI Insight. 2016;1:e85061. doi: 10.1172/jci.insight.85061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Iroz A., Couty J.P., Postic C. Hepatokines: unlocking the multi-organ network in metabolic diseases. Diabetologia. 2015;58:1699–1703. doi: 10.1007/s00125-015-3634-4. [DOI] [PubMed] [Google Scholar]
- Jaeger D., Schoiswohl G., Hofer P., Schreiber R., Schweiger M., Eichmann T.O., Pollak N.M., Poecher N., Grabner G.F., Zierler K.A. Fasting-induced G0/G1 switch gene 2 and FGF21 expression in the liver are under regulation of adipose tissue derived fatty acids. J. Hepatol. 2015;63:437–445. doi: 10.1016/j.jhep.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jois T., Chen C., Howard V., Harvey R., Youngs K., Thalmann C., Saha P., Chan L., Cowley M.A., Sleeman M.W. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017 doi: 10.1016/j.molmet.2017.07.006. Published online July 18, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T., Takenoshita M., Kabashima T., Uyeda K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA. 2001;98:13710–13715. doi: 10.1073/pnas.231370798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S., Seydoux J., Peters J.M., Gonzalez F.J., Desvergne B., Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A., Adams A.C. Inventing new medicines: The FGF21 story. Mol. Metab. 2013;3:221–229. doi: 10.1016/j.molmet.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A., Wroblewski V.J., Koester A., Chen Y.F., Clutinger C.K., Tigno X.T., Hansen B.C., Shanafelt A.B., Etgen G.J. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774–781. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- Kroetz D.L., Yook P., Costet P., Bianchi P., Pineau T. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J. Biol. Chem. 1998;273:31581–31589. doi: 10.1074/jbc.273.47.31581. [DOI] [PubMed] [Google Scholar]
- Leone T.C., Weinheimer C.J., Kelly D.P. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.V., Chang B., Imamura M., Poungvarin N., Chan L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes. 2006;55:1179–1189. doi: 10.2337/db05-0822. [DOI] [PubMed] [Google Scholar]
- Lundåsen T., Hunt M.C., Nilsson L.-M., Sanyal S., Angelin B., Alexson S.E.H., Rudling M. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007;360:437–440. doi: 10.1016/j.bbrc.2007.06.068. [DOI] [PubMed] [Google Scholar]
- Markan K.R., Naber M.C., Ameka M.K., Anderegg M.D., Mangelsdorf D.J., Kliewer S.A., Mohammadi M., Potthoff M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. 2014;63:4057–4063. doi: 10.2337/db14-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmier S., Dentin R., Daujat-Chavanieu M., Guillou H., Bertrand-Michel J., Gerbal-Chaloin S., Girard J., Lotersztajn S., Postic C. Novel role for carbohydrate responsive element binding protein in the control of ethanol metabolism and susceptibility to binge drinking. Hepatology. 2015;62:1086–1100. doi: 10.1002/hep.27778. [DOI] [PubMed] [Google Scholar]
- Montagner A., Polizzi A., Fouché E., Ducheix S., Lippi Y., Lasserre F., Barquissau V., Régnier M., Lukowicz C., Benhamed F. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65:1202–1214. doi: 10.1136/gutjnl-2015-310798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong K.L., Rye K.-A., O’Connell R., Jenkins A.J., Brown C., Xu A., Sullivan D.R., Barter P.J., Keech A.C., FIELD study investigators Long-term fenofibrate therapy increases fibroblast growth factor 21 and retinol-binding protein 4 in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 2012;97:4701–4708. doi: 10.1210/jc.2012-2267. [DOI] [PubMed] [Google Scholar]
- Pichard L., Raulet E., Fabre G., Ferrini J.B., Ourlin J.-C., Maurel P. Human hepatocyte culture. Methods Mol. Biol. 2006;320:283–293. doi: 10.1385/1-59259-998-2:283. [DOI] [PubMed] [Google Scholar]
- Potthoff M.J., Inagaki T., Satapati S., Ding X., He T., Goetz R., Mohammadi M., Finck B.N., Mangelsdorf D.J., Kliewer S.A., Burgess S.C. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. USA. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando G., Tan C.K., Khaled N., Montagner A., Leuenberger N., Bertrand-Michel J., Paramalingam E., Guillou H., Wahli W. Glucocorticoid receptor-PPARα axis in fetal mouse liver prepares neonates for milk lipid catabolism. eLife. 2016;5:e11853. doi: 10.7554/eLife.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez J., Palou A., Picó C. Response to carbohydrate and fat refeeding in the expression of genes involved in nutrient partitioning and metabolism: striking effects on fibroblast growth factor-21 induction. Endocrinology. 2009;150:5341–5350. doi: 10.1210/en.2009-0466. [DOI] [PubMed] [Google Scholar]
- Simon J.M., Giresi P.G., Davis I.J., Lieb J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012;7:256–267. doi: 10.1038/nprot.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S., Owen B.M., Song P., Hernandez G., Zhang Y., Zhou Y., Scott W.T., Paratala B., Turner T., Smith A. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016;23:344–349. doi: 10.1016/j.cmet.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebanso T., Taketani Y., Yamamoto H., Amo K., Ominami H., Arai H., Takei Y., Masuda M., Tanimura A., Harada N. Paradoxical regulation of human FGF21 by both fasting and feeding signals: is FGF21 a nutritional adaptation factor? PLoS ONE. 2011;6:e22976. doi: 10.1371/journal.pone.0022976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega R.B., Huss J.M., Kelly D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Holstein-Rathlou S., BonDurant L.D., Peltekian L., Naber M.C., Yin T.C., Claflin K.E., Urizar A.I., Madsen A.N., Ratner C., Holst B. FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the liver. Cell Metab. 2016;23:335–343. doi: 10.1016/j.cmet.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettenhall J.M., Smyth G.K. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004;20:3705–3706. doi: 10.1093/bioinformatics/bth449. [DOI] [PubMed] [Google Scholar]
- Yoon J.C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C.R., Granner D.K. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- Zaret K.S., Carroll J.S. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.