

Abstract
Background & objectives:
Glanzmann thrombasthenia (GT) is a rare, inherited autosomal recessive disorder characterized by qualitative or quantitative deficiency of integrin αIIbβ3 [glycoprotein IIb (GPIIb)/IIIa, CD41/CD61] diagnosed by absent or reduced platelet aggregation to physiological agonists, namely, collagen, adenosine-di-phosphate, epinephrine and arachidonic acid. The objective of this study was to quantitate platelet surface GPs, classify GT patients and relate the results with the severity of bleeding and platelet aggregation studies.
Methods:
Fifty one patients of GT diagnosed by platelet aggregation studies were evaluated for the expression of CD41, CD61, CD42a and CD42b on platelet surface by flow cytometry. The association between the clinical phenotype based on bleeding score and GT subtype on flow cytometric evaluation was assessed.
Results:
Twenty four (47%) patients of GT were classified as type I (as CD41/CD61 were virtually absent, <5%), six (11.8%) patients as type II (5-20% CD41/CD61) and 21 (41.2%) as type III or GT variants as they had near normal levels of CD41 and CD61. Type III GT patients had significantly lower numbers of severe bleeders (P=0.034), but the severity of bleeding did not vary significantly in type I and II GT patients. In all GT patients, mean CD41 expression was found to be lower than mean CD61 expression (P=0.002).
Interpretation & conclusions:
Type I GT was found most common in our patients and with lowered mean CD41 expression in comparison with CD61. Type III GT patients had significantly lower numbers of severe bleeders, but the severity of bleeding did not vary significantly in type I and II GT patients.
Key words: Flow cytometry, Glanzmann thrombasthenia, glycoproteins, platelet aggregation, platelet surface
Patients of inherited disorders of platelets are characterized by a prolonged clinical history of mucocutaneous bleeding. Glanzmann thrombasthenia (GT) is a rare, inherited autosomal recessive disorder characterized by qualitative or quantitative deficiency of integrin αIIbβ3 [glycoprotein IIa (GPIIb)/IIIa, CD41/CD61] diagnosed by absent or reduced platelet aggregation to physiological agonists, namely, collagen, adenosine diphosphate (ADP), epinephrine and arachidonic acid (AA)1. The prevalence of GT is estimated to be 1 in 1,000,000 population2, but it is more frequent in populations where consanguineous marriages are common, such as Indians, Iraqi Jews, Arabs and Iranians3. The two genes encoding GPIIb (ITGA2B) and GPIIIa (ITGB3) are closely associated at chromosome 17q214. Flow cytometric analysis with monoclonal antibodies specific for αIIb (GPIIb, CD41) and β3 (GPIIIa, CD61) provides a rapid and simple method for the diagnosis5,6,7. Based on their CD41/CD61 levels, GT patients have been classified into three types, type I with markedly low (<5%) levels, type II with reduced (5-20%) levels and type III (variant type) with approximately 80-100 per cent of control levels, due to dysfunctional GPIIb/IIIa receptors8,9,10.
To evaluate the role of flow cytometry in the evaluation of platelet GPs as a tool in the diagnosis of GT, we assessed unrelated patients of GT diagnosed by platelet aggregation studies. Since the analysis of platelet GPs have been done for carrier detection and molecular mutations in earlier studies from our centre6,9,10,11 and from centres in west and south India12,13,14, we quantitated the platelet GP levels and compared it with the bleeding severity in these patients. Though association between bleeding severity and CD41/CD61 expression has been done in a study9 from our centre, in the present study bleeding severity in GT patients was associated with the expression of platelet surface GPs CD41 and CD61.
Material & Methods
In this cross-sectional study clinical records of 51 unrelated patients of GT consecutively enrolled at the Haematology Outpatient Department of All India Institute of Medical Sciences (AIIMS), New Delhi, India, between January 2009 to December 2012, with haemorrhage characteristic of qualitative or quantitative platelet disorders were reviewed. Diagnosis of GT was established based on history of mucocutaneous bleeding and absent or reduced platelet aggregation with agonists ADP, epinephrine, AA and/or ristocetin, with normal platelet count, prothrombin time (PT) and activated partial thromboplastin time (aPTT) and thrombin time11. Automated platelet count results were correlated with manual platelet counts for the same specimen. Fifty one healthy individuals (age range 19-50 yr, 42 males and 9 females) who had not taken medications or anti-platelet drugs for the preceding two weeks with normal platelet morphology and platelet counts were taken as controls. Controls were run in parallel with patient samples at the same time during flow cytometry evaluation. Clinical history of bleeding such as sites, severity and frequency of bleeding, trauma-related events, history of surgical procedures if any and history of menorrhagia in female patients above the pubertal age were recorded. History of packed red cells/platelet transfusions was recorded. In addition, family history such as consanguinity, bleeding complications in any of the parents/siblings, origin of the family were also noted. Patients with acquired bleeding, coagulation defects, platelet function defects other than GT and those on antiplatelet drugs were excluded from the study.
The study protocol was approved by the institutional ethics committee.
Grading of bleeding severity
The patient bleeding severity was graded by the analysis of clinical records of patients were evaluated using a questionnaire and checklist for bleeding patients in the outpatient clinic. Bleeds were classified in terms of severity according to the WHO bleeding assessment scale as used by others3,15. In this system, grade 1 (mild bleeders) was characterized by minor mucosal bleeds, petechiae, or retinal bleeding without vision impairment. Grade 2 (significant bleeders) patients gave history of gastrointestinal bleeds, haematuria or haemoptysis. Grade 3 (severe bleeders) included any bleeds requiring red cell or platelet transfusion support, and grade 4 bleeds included retinal and cerebral bleeds associated with morbidity or mortality.
Haematological workup
A routine haemogram was obtained from a five-part haematology cell analyzer (Sysmex XE2100, Kobe, Japan). Peripheral blood smears stained by Jenner-Giemsa were evaluated for platelet morphology and counts. Platelet aggregation studies were done using platelet-rich plasma in a platelet aggregometer (Bio/Data Corp., Horsham, USA) using final concentrations of ADP at 2 × 10−5 M AA at 500 μg/ml, epinephrine at 1.0 × 10−4 M and ristocetin at 2 mg/ml. Routine PT, aPTT and thrombin time were performed in all cases. Workup for von Willebrand disease (vWD) was done in selected cases.
Analysis for platelet receptor CD41, CD61, CD42a, CD42b by flow cytometry
Peripheral blood samples (2 ml) were collected in EDTA by fresh venepuncture for flow cytometry analysis from patients and healthy controls. Platelet count was adjusted to approximately 1.5 × 109/l in three tubes. Cells were incubated with monoclonal antibodies CD41 (FITC/Per CP-Cy-5.5) and CD61 in the first tube and CD42a (FITC) and CD42b (PE-A) in the second tube. Controls were run in parallel to test samples. All antibodies were obtained from BD Biosciences (San Jose, CA, USA). An unstained tube lacking the antibodies was processed similarly. After 20 min incubation, tubes were directly acquired on the flow cytometer (BD FACS Canto-6 colour), and all the analyses and interpretation were carried out post-acquisition by BD FACS Diva software (BD Biosciences, USA). Fluorescence measurement was expressed as the percentage of positive cells above a threshold set with the unstained tube acting as negative control. Compensation for fluorochrome spectral overlap was adjusted for analysis.
Statistical analysis
The association between the clinical phenotype based on bleeding score and GT subtype on flow cytometric evaluation was assessed using a Freeman-Halton extension of Fisher's exact test. Mean values of CD41 and CD61 were compared using a paired t test. All analyses were conducted using Stata v9.0 (StataCorp, Texas, USA) and SPSS v15.0 (SPSS Inc, Chicago, USA).
Results
Fifty one patients with GT (27 males and 24 females, median age four years, interquartile range 2-14 yr) were included in the study. Thirty three (64.7%) patients were diagnosed before the age of six years, 16 (31.4%) between 6 and 16 yr and only two (3.9%) subsequently. The family history of bleeding disorder was present in six (11.8%) patients. History of consanguinity could be elicited in 14 (27.5%) patients; however, the degree of consanguinity could not be ascertained.
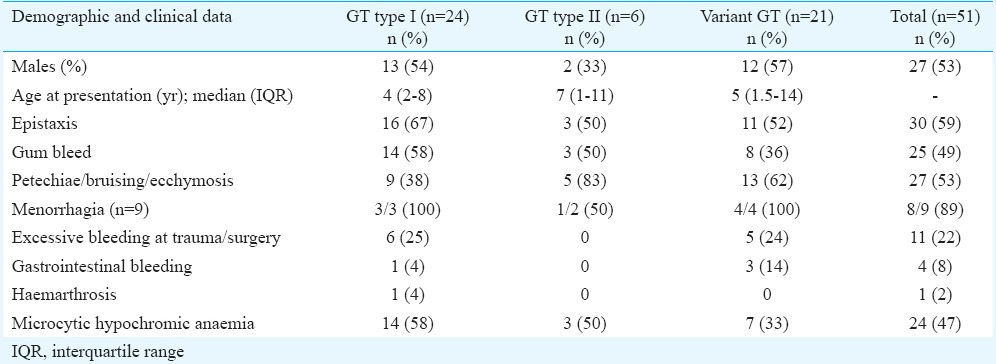
Table I shows that more than 50 per cent of type I GT patients had epistaxis or gum bleeding while petechiae, bruising and ecchymoses were more common in type III GT patients. Menorrhagia was the most common presentation in eight of nine (89%) females above pubertal age. Gastrointestinal bleeding and deep haematoma involving joint were rare and seen in four patients and one patient, respectively. None of the patients gave history of platelet transfusions. Packed red blood cells were given in five patients, of whom three belonged to GT I type and one each to GT II and GT III.
Table I.
Demographic and clinical profile of Glanzmann thrombasthenia (GT) patients
Haematological data
Twenty four (47%) patients had associated microcytic hypochromic anaemia with a median haemoglobin level of 9.3 g/dl (range 3.8-14.6 g/dl). The mean platelet count was 204±72 × 109/l. None of the GT patients had enlarged platelets on peripheral smear. The average total leucocyte count was 7.95±2.4 × 109/l. Absent aggregation was seen to ADP, epinephrine, and AA was seen in all patients. Incomplete ristocetin response comprising only primary wave aggregation was seen in 16 (31.4%) patients and normal response in 35 (68.6%) patients. vWD workup done in three patients where predominant mucocutaneous bleeding was present was normal. All three had only primary wave aggregation with ristocetin and belonged to the variant GT (type III) category on flow cytometry.
Bleeding score
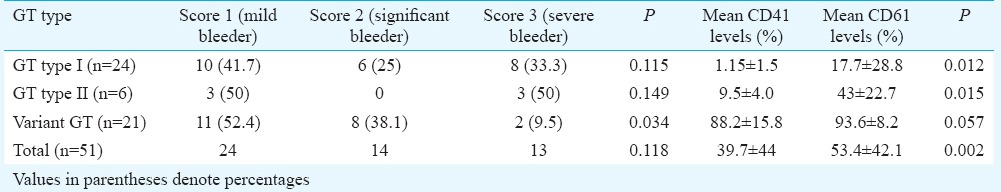
Association of GT type on flow cytometry with severity of bleeding is given in Table II. Symptoms of minor bleeding (score 1) were present in 24 (47%) patients, 14 (27.5%) had a clinically significant bleeding (score 2) and 13 (25.5%) had severe bleeding requiring transfusion (score 3). No patients had fatal or debilitating bleeding (score 4). Severe bleeders (WHO score 3) were found to be significantly lower in GT type III (2/21, 9.5%; P=0.034). No significant association between bleeding score and GT subtype was seen in type I and II GT patients (Table II).
Table II.
Association of Glanzmann thrombasthenia (GT) type on flow cytometry with severity of bleeding as per WHO score with comparison of mean CD41 and CD61 expression levels
Flow cytometry analysis
Twenty four (47%) patients were categorized as GT type I with absent or extremely reduced levels (<5%) of CD41 and CD61. Of these, 15 (62.5%) patients had reduced levels of both CD41 and CD61 (Fig. 1) while nine (37.5%) others had heterogeneous expression patterns with one patient showing reduced levels of CD61 only and eight patients showing reduced levels of CD41 only. Six (11.8%) patients were categorized as type II with median CD41 levels of 8.9 per cent (range 6.0-15.4) (Fig. 2) and median CD61 levels of 44.2 per cent (range 19.5-73.8). Twenty one (41.2%) patients exhibited a normal or near-normal amount of CD41 (median 96.1%; range 44.7-99.7) and CD61 (median: 96.2%; range 60.9-99.3) (Fig. 3).
Fig. 1.
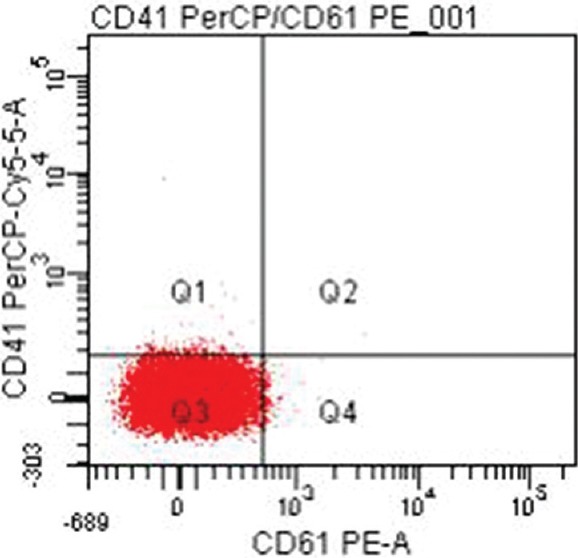
Type 1 Glanzmann thrombasthenia with reduced levels of both CD41 (PerCP-Cy5.5) and CD61 (PE-A) (<5%).
Fig. 2.
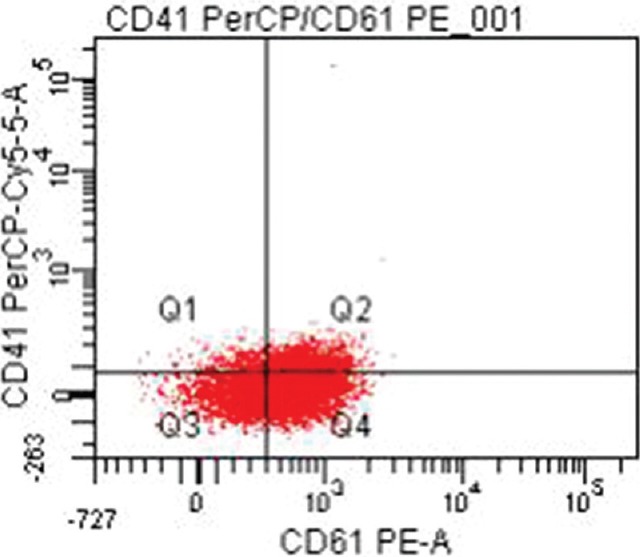
Type 2 Glanzmann thrombasthenia with reduced CD41 (PerCP-Cy5.5) and CD61 (PE-A) (5-50%).
Fig. 3.
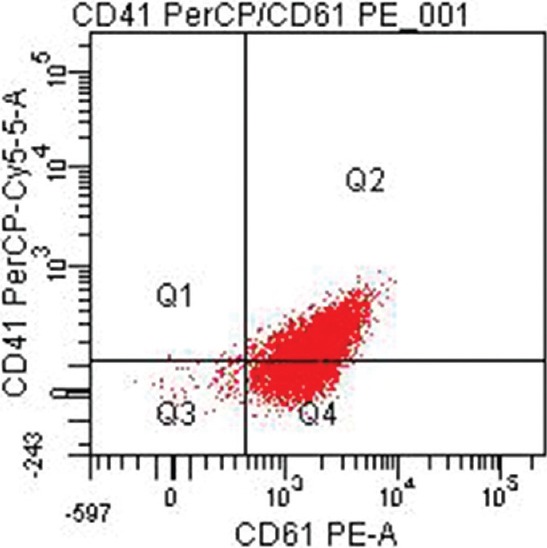
Type 3 Glanzmann thrombasthenia with near normal levels of CD41 (PerCP-Cy5.5) and CD61 (PE-A).
Mean values of CD41 were significantly lower than those of CD61 in type I (P=0.012) and type II GT patients (P=0.015) and all patients taken together (P=0.002) (Table II). Median fluorescent intensity of CD41 and CD61 was also compared and found to be significantly different. Mean values of CD41 were lower than CD61 in the variant GT subgroup also, however, this was not significant.
In the 51 age- and sex-matched unrelated controls, mean values of CD41 and CD61 were 97.1 and 98.2 per cent, respectively. There were no differences in the mean expression of CD41 and CD61 in these cases. In all GT patients, levels of CD42a and CD42b were found to be within normal limits (CD42a: 96.1±3.8%; CD42b: 96.4±3.2%).
Discussion
This study was undertaken to evaluate Indian patients with GT, quantitate their platelet surface GPs, CD41 and CD61 and compare their GT subtype on flow cytometry with their clinical bleeding score. Based on the flow cytometric quantification of CD41 and CD61 in GT patients, 47 per cent were classified as GT type I, 11.8 per cent as GT type II and 41.2 per cent showed normal expression of CD41 and CD61. The data conformed to the previous studies from the same centre that type I GT was most common in Indian patients9,16. Similar results were reported from Iran3.
In this study, 16 (31.4%) patients showed only primary wave aggregation to ristocetin. The reason for this may be the use of low-dose ristocetin in our screening. The response to high-dose ristocetin was normal in these patients. Although ristocetin-induced aggregation is normal in GT patients, the maximal increase in light transmission reached is often reduced due to defective, secondary, platelet-to-platelet contact-induced aggregation4. At lower concentrations of ristocetin, secondary aggregation response has been reported to be absent in GT and afibrinogenemia17,18.
In our study, epistaxis and petechial bleeding were the most common symptoms while menorrhagia was the most common presentation in pubertal female patients. These were similar to other reports in literature19,20.
History of consanguinity could be elicited in 27.5 per cent patients, of whom only six gave family history of bleeding disorder; all of them had a similar history in a sibling and not a parent. Expression is autosomal recessive, consisting either of true homozygosity of a single genetic abnormality or double heterozygous expression of separate abnormalities8,10. Although screening of parents could not be carried out in all cases, it was evident from the data that heterozygous GT might be higher in the Indian population.
Patients in our study had significantly lower mean values of CD41 as compared to CD 61. Spurious false increase of CD61 expression due to platelets adhering to fragments of WBCs was excluded by leaving debris and WBCs outside on gating. Heterogeneous expression of CD41 and CD61 on flow cytometry has been reported earlier21. Western blot analysis performed on the platelet lysates in a study from Iran showed increased expression of GPIIIa as compared to GPIIb in type I patients3. We also found higher mean expression of CD61, corresponding to GPIIIa. In the Indian population, mutations in the ITGA2B gene have been reported to be predominant12. This may be responsible for lower CD41 expression in type I and II patients in this study. However, this needs to be confirmed by larger studies involving molecular mutation analysis.
No significant association between bleeding score and GT subtype was seen in type I and II GT patients. Earlier studies have reported no correlation between the severity of bleeding of the patients and the GT subtype8,9,21. The lower incidence of severe bleeders in variant GT may be related more to the higher level of CD 41 and 61 expressions. Conditions other than GT may have similar results of platelet aggregation studies with normal GP levels on flow cytometry. Some of these patients may represent acquired GT caused by GPIIb/GPIIIa specific autoantibodies. Employing the PAC-1 antibody or other conformation-dependent monoclonal antibodies that recognize activated GPIIb/IIIa complex would permit the identification of elusive forms of GT characterized by functional impairment7. However, antibody detection studies were not carried out in type III GT patients in this study. Type III GT may also include patients with inherited bleeding disorders with gene defects other than ITGA2B and ITGB322. These may include defects in intracellular signalling pathways as shown by the reports of mutation in the in the RAS guanyl-releasing protein-2 gene coding for calcium and DAG-regulated guanine exchange factor-122. Exome or whole-genome sequencing would be of interest for type III GT patients. Although none of our patients gave history of being transfused platelets, many GT patients who have received platelet infusions eventually may produce anti-GPIIb/IIIa antibodies23. This points towards the stringency in performing flow cytometry at diagnosis and not after platelet transfusion.
The limitation of the present study was that patients could not be evaluated for molecular mutations.
In conclusion, our results showed that GT type III had significantly lower numbers of severe bleeders (WHO score 3). Type I GT was found most common in our patients akin to previous data, with lowered mean CD41 expression in comparison with CD61. Since flow cytometry is now being increasingly used to confirm the presence or absence of platelet GPs, it must be performed early in disease diagnosis, for accurate subtyping of GT and before platelet transfusion to avoid diagnostic dilemmas.
Acknowledgment
Authors acknowledge Dr V. Sreenivas for helping in statistical analysis.
Footnotes
Conflicts of Interest: None.
References
- 1.Nurden AT, Pillois X, Wilcox DA. Glanzmann thrombasthenia: State of the art and future directions. Semin Thromb Hemost. 2013;39:642–55. doi: 10.1055/s-0033-1353393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diz-Küçükkaya R. Inherited platelet disorders including Glanzmann thrombasthenia and Bernard-Soulier syndrome. Hematology Am Soc Hematol Educ Program. 2013;2013:268–75. doi: 10.1182/asheducation-2013.1.268. [DOI] [PubMed] [Google Scholar]
- 3.Farsinejad A, Farajollahi MM, Kazemi A, Saemi N, Faranoush M. Different biochemical expression pattern of platelet surface glycoproteins suggests molecular diversity of Glanzmann's thrombasthenia in Iran. Blood Coagul Fibrinolysis. 2013;24:613–8. doi: 10.1097/MBC.0b013e328360a558. [DOI] [PubMed] [Google Scholar]
- 4.Franchini M, Favaloro EJ, Lippi G. Glanzmann thrombasthenia: An update. Clin Chim Acta. 2010;411:1–6. doi: 10.1016/j.cca.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Nair S, Ghosh K, Kulkarni B, Shetty S, Mohanty D. Glanzmann's thrombasthenia: Updated. Platelets. 2002;13:387–93. doi: 10.1080/0953710021000024394. [DOI] [PubMed] [Google Scholar]
- 6.Kannan M, Saxena R. Glanzmann's thrombasthenia: An overview. Clin Appl Thromb Hemost. 2009;15:152–65. doi: 10.1177/1076029608326165. [DOI] [PubMed] [Google Scholar]
- 7.Miller JL. Glycoprotein analysis for the diagnostic evaluation of platelet disorders. Semin Thromb Hemost. 2009;35:224–32. doi: 10.1055/s-0029-1220330. [DOI] [PubMed] [Google Scholar]
- 8.Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: A review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118:5996–6005. doi: 10.1182/blood-2011-07-365635. [DOI] [PubMed] [Google Scholar]
- 9.Kannan M, Ahmad F, Yadav BK, Anand M, Jain P, Kumar R, et al. Glanzmann's thrombasthenia in North Indians: Sub classification and carrier detection by flow cytometry. Platelets. 2009;20:12–5. doi: 10.1080/09537100802434853. [DOI] [PubMed] [Google Scholar]
- 10.Kannan M, Ahmad F, Yadav BK, Kumar P, Jain P, Kumar R, et al. Carrier detection in Glanzmann thrombasthenia: Comparison of flow cytometry and Western blot with respect to DNA mutation. Am J Clin Pathol. 2008;130:93–8. doi: 10.1309/HYE4AP9961CEP0C0. [DOI] [PubMed] [Google Scholar]
- 11.Kannan M, Ahmad F, Yadav BK, Kumar R, Choudhry VP, Saxena R. Molecular defects in ITGA2B and ITGB3 genes in patients with Glanzmann thrombasthenia. J Thromb Haemost. 2009;7:1878–85. doi: 10.1111/j.1538-7836.2009.03579.x. [DOI] [PubMed] [Google Scholar]
- 12.Nelson EJR, Nair SC, Peretz H, Coller BS, Seligsohn U, Chandy M, et al. Diversity of Glanzmann thrombasthenia in Southern India: 10 novel mutations identified among 15 unrelated patients. J Thromb Haemost. 2006;4:1730–7. doi: 10.1111/j.1538-7836.2006.02066.x. [DOI] [PubMed] [Google Scholar]
- 13.Peretz H, Rosenberg N, Landau M, Usher S, Nelson EJR, Mor-Cohen R, et al. Molecular diversity of Glanzmann thrombasthenia in Southern India: New insights into mRNA splicing and structure-function correlations of alphaIIbbeta3 integrin (ITGA2B, ITGB3) Hum Mutat. 2006;27:359–69. doi: 10.1002/humu.20304. [DOI] [PubMed] [Google Scholar]
- 14.Nair S, Ghosh K, Shetty S, Mohanty D. Mutations in GPIIIa molecule as a cause for Glanzmann thrombasthenia in Indian patients. J Thromb Haemost. 2005;3:482–8. doi: 10.1111/j.1538-7836.2005.01159.x. [DOI] [PubMed] [Google Scholar]
- 15.D'Andrea G, Margaglione M. Glansmann's Thrombasthemia Italian Team (GLATIT). Glanzmann's thrombasthenia: Modulation of clinical phenotype by alpha2C807T gene polymorphism. Haematologica. 2003;88:1378–82. [PubMed] [Google Scholar]
- 16.Kannan M, Ahmed RPH, Jain P, Kumar R, Choudhry VP, Saxena R. Type I Glanzmann thrombasthenia: Most common subtypes in North Indians. Am J Hematol. 2003;74:139–41. doi: 10.1002/ajh.10395. [DOI] [PubMed] [Google Scholar]
- 17.De Marco L, Girolami A, Zimmerman TS, Ruggeri ZM. Von Willebrand factor interaction with the glycoprotein IIb/IIa complex. Its role in platelet function as demonstrated in patients with congenital afibrinogenemia. J Clin Invest. 1986;77:1272–7. doi: 10.1172/JCI112430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Marco L, Girolami A, Russell S, Ruggeri ZM. Interaction of asialo von Willebrand factor with glycoprotein Ib induces fibrinogen binding to the glycoprotein IIb/IIIa complex and mediates platelet aggregation. J Clin Invest. 1985;75:1198–203. doi: 10.1172/JCI111816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Minno G, Coppola A, Di Minno MND, Poon MC. Glanzmann's thrombasthenia (defective platelet integrin alphaIIb-beta3): Proposals for management between evidence and open issues. Thromb Haemost. 2009;102:1157–64. doi: 10.1160/TH09-04-0225. [DOI] [PubMed] [Google Scholar]
- 20.Toogeh G, Sharifian R, Lak M, Safaee R, Artoni A, Peyvandi F. Presentation and pattern of symptoms in 382 patients with Glanzmann thrombasthenia in Iran. Am J Hematol. 2004;77:198–9. doi: 10.1002/ajh.20159. [DOI] [PubMed] [Google Scholar]
- 21.Farsinejad A, Abolghasemi H, Kazemi A, Aghaiipour M, Hadjati E, Faranoush M, et al. Classification of Iranian patients with Glanzmann's thrombasthenia using a flow cytometric method. Platelets. 2011;22:321–7. doi: 10.3109/09537104.2011.556275. [DOI] [PubMed] [Google Scholar]
- 22.Canault M, Ghalloussi D, Grosdidier C, Guinier M, Perret C, Chelghoum N, et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J Exp Med. 2014;211:1349–62. doi: 10.1084/jem.20130477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh K, Kulkarni B, Shetty S, Nair S. Antiplatelet antibodies in cases of Glanzmann's thrombasthenia with and without a history of multiple platelet transfusion. Indian J Hum Genet. 2009;15:23–7. doi: 10.4103/0971-6866.50866. [DOI] [PMC free article] [PubMed] [Google Scholar]