

Abstract
Bacteria within Curli biofilms are protected from environmental pressures (e.g., disinfectants, antibiotics), and this is responsible for intractable infections. Understanding aggregation of the major protein component of Curli, CsgA, may uncover disease-associated amyloidogenesis mechanisms. Here, we report the application of pulsed hydrogen–deuterium exchange and mass spectrometry (HDX-MS) to study CsgA aggregation, thereby obtaining region-specific information. By following time-dependent peptide signal depletion, presumably a result of insoluble fibril formation, we acquired sigmoidal profiles that are specific for regions (region-specific) of the protein. These signal-depletion profiles not only provide an alternative aggregation measurement, but also give insight on soluble species in the aggregation. The HDX data present as bimodal isotopic distributions, one representing a highly disordered species whereas the other a well-structured one. Although the extents of deuterium uptake of the two species remain the same with time, the relative abundance of the lower mass, less-exchanged species increases in a region-specific manner. The same region-specific aggregation properties also pertain to different aggregation conditions. Although CsgA is an intrinsically disordered protein, within the fibril it is thought to consist of five imperfect β-strand repeating units (labeled R1–R5). We found that the exterior repeating units R1 and R5 have higher aggregation propensities than do the interior units R2, R3, and R4. We also employed TEM to obtain complementary information of the well-structured species. The results provide insight on aggregation and a new approach for further application of HDX-MS to unravel aggregation mechanisms of amyloid proteins.
Keywords: Curli, CsgA, protein aggregation, hydrogen deuterium exchange (HDX), Pulsed HDX, Mass Spectrometry, transmission electron microscopy, EX1-like
Graphical abstract

Introduction
Although amyloid proteins are commonly associated with Alzheimer’s1, Parkinson’s2, and other neuro-degenerative diseases, some play a critical role in normal cellular physiology.3, 4 Of particular interest are curli, an extracellular amyloid fiber produced by Salmonella spp., Escherichia coli, and other enteric bacteria.5, 6 These fibers are involved in the adhesion of bacteria and biofilm formation, protecting bacteria from the environmental stress of disinfectants and antibiotics.3, 5, 6. Biofilms are the cause of persistence and recurrence of many infections.7, 8 Understanding the curli amyloid production process provides a unique perspective on the molecular mechanism underlying amyloidogenesis and its related cellular toxicity and function.
The major component of the curli fiber is CsgA, initially synthesized as an intrinsically disordered protein with a molecular weight of 13 kDa. The 22 N-terminal residues serve as a signal peptide for CsgA secretion through the outer membrane. The remaining residues 23–131 correspond to an amyloid core region in the curli fiber and consist of five imperfect, repeating strand-loop-strand units. CsgA consists primarily of serine, glutamine, glycine, and asparagine residues.9 Its aggregation in vitro has been studied using various conventional techniques including Thioflavin T (ThT) fluorescence and circular dichroism (CD) spectroscopy.10-12 Cherny et al.13 identified prion-like hexa-repeats within CsgA and showed that chemically synthesized hexapeptides of CsgA with aromatic residues are able to self-associate into well-ordered fibrillary species. Wang et al.14 showed, by using either synthesized peptides corresponding to each repeating unit or proteins with each repeating unit deleted, that repeats R1, R3 and R5 of the five imperfect repeating units in the amyloid core of CsgA are the regions responsible for polymerization. The latter group also showed that mutations of four conserved residues in R1 and R5 slow CsgA aggregation in vitro and completely disable curli assembly in vivo.15
MS-based approaches have become a highly sensitive and effective tool for investigating biomolecules in their native or near-native state.16-18 HDX-MS, for example, allows experimentalists to probe the protein conformation at moderate structural resolution, providing critical insights into higher-order structures of protein complexes.16-18 By measuring deuterium exchange differences, one can characterize changes in protein dynamics and conformation induced by protein/ligand binding,19, 20 post-translational modification21 and aggregation22-25. This is applicable because protein backbone amide hydrogen undergo HDX based on solvent accessibility and hydrogen bonding. Regions that show greater amide hydrogen exchange rates represent flexible or solvent accessible areas, whereas those that show a smaller exchange rate are likely involved in a relatively rigid conformation or buried environment. In the work presented here, we focused on using hydrogen–deuterium exchange coupled with mass spectrometry (HDX-MS) to study the aggregation behavior of CsgA.
To follow CsgA conformational changes during the highly regulated amyloid production process, we chose a pulsed HDX-MS platform24 in which differences in deuterium exchange relate to various populations of molecules that may exist in the aggregation process. This platform was first developed to examine conformational changes of soluble oligomers during amyloid peptides Aβ aggregation in a physiologically relevant environment.24 Recently, we applied both continuous and pulsed HDX-MS studies to another curli family member, CsgE, to determine the regions involved in self-association, and we discovered a conformational rearrangement during the oligomerization process.26 Extension of this platform to regulated CsgA aggregation provides an opportunity to understand region-specific aggregation for the near-native state. In addition, we complemented pulsed HDX-MS with transmission electron microscopy (TEM) to probe both soluble and insoluble CsgA proteins.
Materials and Methods
Materials
Wild-type CsgA containing a 6-His-tag at the C-terminus were expressed and purified as previously reported.11 CsgA stock was stored in solution containing 200 mM imidazole, 50 mM potassium phosphate buffer (pH 7.2), and 6 M guanidine hydrochloride (GdnHCl) at −20 °C to prevent aggregation. Prior to the HDX experiment, the protein solution was exchanged into phosphate buffer saline (PBS, pH 7.4) using a Sephadex G25 desalting column (HiTrap GE Healthcare, Pittsburgh, PA).
D2O was purchased from Cambridge Isotope Laboratories Inc. (Andover, MA). Angiotensin I peptide and all other chemicals were purchased from MilliporeSigma (St. Louis, MO).
Pulsed HDX-MS
Following removal of GdnHCl, 10 μM CsgA was incubated at 22 °C and monitored as a function of time. The first time point was approximately 1 min after protein was passed through the desalting column to remove the GdnHCl. Three different incubation conditions were tested: continuous stirring, quiescent (no stirring), and one following a freeze/thaw cycle of the buffer-exchanged CsgA. Samples (20 μL) obtained at different incubation times, were centrifuged at 13,000 rpm (Eppendorf Microcentrifuges 5418, Hamburg, Germany), at 4 °C for 10 min. All incubation times did not include the 10 min centrifugation at 4 °C. The top 10 μL of the supernatant was discarded to concentrate the large, soluble oligomers that will preferentially populate the lower solution after centrifugation.
Pulsed deuterium labeling was achieved by diluting the lower 10 μL solution into 40 μL of D2O buffer for 30 s at 4 °C. Ice-cold, reducing quench buffer containing 3 M urea with 1% trifluoroacetic acid (TFA) was added to each reaction solution at a ratio of 1:1 (v:v) to quench the HDX reaction. Angiotensin I peptide (60 pmol) was spiked into the quenched solution as an internal standard. Online pepsin digestion and HPLC separation were performed as previously described.26 Briefly, protein samples were digested while flowing through a custom-packed online pepsin column. The resulting peptides were trapped and desalted on a C8 trap column (Agilent Inc., Santa Clara, CA). Separation of the peptide mixtures was carried out on a 1.9 μm reversed-phase C18 column (Hypersil Gold Thermo Fisher Scientific, Waltham, MA) with a 5.5 min linear gradient. All experiments were performed in triplicate.
MS analysis of the eluted peptides was conducted on a MaXis quadrupole time-of-light instrument (Bruker Daltonics Inc., Billerica, MA) by using positive-ion electrospray ionization (ESI) mode. The ESI spray voltage was set to 4 kV. The temperature and nebulizer gas were 180 °C and 0.8 bar, respectively. The drying gas flow rate was 6 L/min.
HDX-MS Data Analysis
Identification (mapping) of peptic peptides was achieved in a preliminary LC/MS/MS experiment by submitting peptic peptides to collision-induced dissociation (CID) fragmentation in a data-dependent mode on a Thermo LTQ XL Orbitrap (San Jose, CA). Product-ion spectra were then submitted to MassMatrix (version 2.4.2)27 for identification, followed by manual inspection. Among all identified peptides, eight with relatively strong signals that represent the complete sequence were chosen for following the CsgA aggregation (Figure S1).
A custom-developed program implemented in MathCAD (v14, Parametric Technology Corp., MA) was used to fit the HDX isotopic envelopes of each CsgA peptide with two binomial distributions. The program outputted centroid masses and peak areas of both species. Peak area of Angiotensin I was obtained from a peak fit using a binomial distribution. Relative abundances of MS-detectable CsgA peptides with regard to the internal standard were calculated by normalizing peak areas of CsgA peptides to those of Angiotensin I ions of all charge states. The deuterium uptake percentage was calculated by using Eq. 1:
| (1) |
where mHDX and mcontrol are the centroid mass of the deuterated and non-deuterated peptides, respectively, 0.80 corresponds to the final D2O content of the buffer system, and (N − 2) represents the number of the amide hydrogens that are exchangeable, assuming first two residues retain no deuterium by the time of measurement.
The fractional species B (at lower m/z region) was calculated according to where IA and IB are the abundances of species A and species B, respectively. Curve fitting with four parameter sigmoidal curves, calculating the first-derivatives of the sigmoidal curves, and all data plottings were performed with Prism 6 (GraphPad, La Jolla, CA).
Transmission Electron Microscopy (TEM)
CsgA solutions (2.5 μL, 4 μM) were transferred onto the surface of a glow-discharged carbon-coated 200-mesh copper grid (Electron Microscopy Sciences, Hatfield, PA). The grid surface was negatively stained with 1% uranyl acetate solution. Excess solution was removed by blotting with filter paper. The grid was left to air dry and then viewed using a JEOL 100CX Transmission Electron Microscope (Peabody, MA) with a 100 kV acceleration voltage.
Thioflavin T (ThT) Assay
Amyloid polymerization of CsgA was followed by the change in fluorescence of ThT as described in previous reports11. CsgA was 4 μM in 20 mM potassium phosphate buffer containing 150 mM NaCl and 25 μM ThT (pH 7.4), in a total volume of 1 mL in a glass tube and incubated at 25 °C with stirring. The ThT fluorescence change was recorded as a function of time on a spectrofluorometer (PTI, Photon Technology International, Edison, NJ) by using an excitation wavelength of 438 nm and emission 490 nm.
Results and Discussion
MS-detectable materials decrease as a result of aggregation
Insoluble materials (e.g., fibrils), are formed during the protein aggregation process. When HDX-MS is employed to follow the formation of intermediates or conformational changes during protein aggregation, DMSO can be used after deuterium labeling to dissolve insoluble materials before MS detection.22, 28 DMSO treatment, however, restricts HDX analysis to the global level as enzymes are not compatible with a high organic solvent content. To achieve higher resolution at the peptide level, one can either use a supersaturated urea solution to dissolve aggregates followed by significant dilution23, or focus on those soluble species that can be transferred directly into the mass spectrometer24, 29, 30. The latter may be the appropriate approach because soluble oligomers, rather than fibrils, are proposed to be toxic species in different neurodegenerative diseases,31, 32 making them potential targets for therapeutic drugs.2, 33 Thus, we focused on soluble species utilizing a MS-friendly quenching and digestion protocol in which some but not all fibrils are also investigated.24
To understand better the species sampled by HDX, we first investigated the depletion of peptide intensities as a function of time. The time course of soluble-species depletion reports the stages of CsgA aggregation and informs us when useful HDX observations should be made. Additionally, the depletion of soluble species as measured by MS should be considered as another measurement dimension for protein aggregation. We monitored the relative abundance of MS-detectable CsgA peptides with respect to an internal standard peptide (Figure 1). The large signal losses we see are likely caused by the formation of insoluble fibrils. We did not include data for peptide 94–105 because we were unable to follow it after peptide signals started to decrease significantly following the lag phase, owing to its low signal intensity and other interferences in its m/z region. All other peptides display sigmoidal profiles, which are similar to the ThT fluorescence profiles for CsgA aggregation.
Figure 1.

(A) Relative abundance of CsgA peptides with respect to the internal standard as a function of incubation time for peptide 1–25  , 26–37
, 26–37  , 38–48
, 38–48  , 49–69
, 49–69  , 70–83
, 70–83  , 84–93
, 84–93  and 106–137
and 106–137  . Data were fit with sigmoidal curves (dotted line). Error bars are standard deviation at each time point. (B) t1/2 value of each peptide extracted from sigmoidal curve fitting with errors indicating t1/2 ranges at 95% confidence level.
. Data were fit with sigmoidal curves (dotted line). Error bars are standard deviation at each time point. (B) t1/2 value of each peptide extracted from sigmoidal curve fitting with errors indicating t1/2 ranges at 95% confidence level.
All the depletion curves show an initial lag phase for approximately 60 min, followed by an exponential decrease for another 60 min; all peptides finally reach a plateau after 120 min. To characterize better each peptide, we fit our data with four-parameter sigmoidal curves (Figure 1A) and extracted t1/2 values for each peptide (Figure 1B). All peptides show nearly identical behavior with similar t1/2 values of approximately 72 min. We expected the behavior at the peptide level to be constant over the protein because each peptide is a representative of the whole protein. The sigmoidal curves derived from HDX-MS data break slightly earlier than that for ThT fluorescence (Figure S2). The aggregation of CsgA (4 μM) as followed by the ThT assay showed a sigmoidal curve with a lag phase of approximately 120 min, a growing phase of about 8 h to a plateau phase. An earlier paper reported that the ThT lag time of CsgA decreased 2-3 fold with a 20-fold increase in protein concentration, and the lag phase was approximately 90 min for 10 μM CsgA at neutral pH.10 The ThT lag time of CsgA aggregation varied from 1-3 h depending on conditions, as described elsewhere.10, 11, 14 Considering that the ThT assay follows formation of amyloids that are highly insoluble, our peptide-level quantitation results that report on soluble species depletion are consistent with those of the ThT assay, showing that both report on nucleation, elongation and fibrillation.
We used this quantitative approach on the mass spectra to avoid using the extracted ion chromatograms of the peptide signals during HDX (described next), which are complex owing to the broad isotopic envelopes characteristic of HDX. We also combined signals from all the charge states to eliminate charge variations at ESI. It is to note that peptides representing regions near the N-terminus generally provide higher ion signals than those close to the C-terminus. The differences in the initial relative abundances of each peptide likely result from differences either in ESI ionization efficiency or pepsin digestion but not from structure because each CsgA monomer lacks structure and exists as an intrinsically disordered protein (IDP).34 Furthermore, the protein is already denatured before digestion.
HDX reveals two species during CsgA aggregation
We next turned to HDX in an effort to follow region-specific aggregation, an opportunity easily achieved with MS. We first added 6 M GdnHCl to prevent the aggregation of CsgA and measured the extent of HDX as seen in the mass spectrum of peptide 106–137 in the presence of 6 M GdnHCl (Figure 2A). We complemented this measurement with those in the absence of GdnHCl (Figure 2B) incubated at selected time points. As all peptides show two species with a time-dependent abundance change during CsgA aggregation, we chose peptide 106–137 as a representative case for illustration and discussion. Although peptide signals decrease significantly with incubation times (Figure 1), the amount of soluble species remains detectable at the longest incubation time, a manifestation of the high dynamic range of most modern mass spectrometers. The abundance of peptide 106–137 relative to the internal standard during HDX (Figure 1), is also described in Figure 2 caption.
Figure 2.

HDX mass spectra of peptide 106–137 molecular ion (A) in the presence of GdnHCl and (B) in the absence of GdnHCl incubated for 0, 90, 120, 480, and 1440 min. The isotope cluster at higher m/z range is denoted as species A (blue) whereas the one at lower m/z range is denoted as species B (red) (C) Deuterium uptake of both species A (blue) and B (red) as a function of incubation time for all peptides. The relative abundance of peptide 106–137 to the internal standard is 21%, 4%, 1%, 0.9%, 0.5% at 0, 90, 120, 480, and 1440 min, respectively, as plotted in Figure 1.
In the presence of GdnHCl, a single, highly deuterium exchanged species exists that is the denatured monomer. The average deuterium uptake for all peptides of this denatured monomer is 70% (no back-exchange correction). Not one but two populations, denoted as species A and B, are present, however, and exhibit distinct deuterium uptake during CsgA aggregation. Species A, representing the highly exchanged population, has the same HDX behavior as that of the denatured CsgA monomer, indicating that these soluble species possess little or no defined structure. It is not surprising to observe a highly exchanged species A with this IDP protein. Weis and co-workers35 showed that IDP proteins undergo high HDX even on the time scale of seconds. In contrast to the disordered species A, of higher m/z, species B undergoes significantly lower deuterium uptake, suggesting a relatively more ordered structure.
To characterize species A and B more completely, we applied a two-binomial functional distributions to fit the HDX patterns of all the peptides. Deuterium uptake for both species A and B plotted as a function of incubation time are shown in Figure 2C. We have no data for peptide 94–105 after 240 min because its signal was no longer detectable. Our HDX results show that the deuterium uptake of both species A and B remain relatively constant during the aggregation process, but the levels are relatively constant with incubation time; specifically, the deuterium uptake of species A and B are ~ 60–80% and ~ 5–10%, respectively. Chapman and co-workers34 proposed that unfolded CsgA forms a transient oligomeric pool with an average molecular weight of approximately 66 kDa; this low-energy state contains dynamic and amorphous conformers once aggregation is triggered. Although species A shows the same HDX as the denatured monomer, we propose that species A is this dynamic, less structured oligomer. The subtle structural difference between the dynamic amorphous oligomer pool and completely unstructured monomer, however, is not revealed by HDX, possibly because the labeling time window is not sufficiently short for capturing small differences.
Species B, on the other hand, must be well-structured, not highly solvent-accessible because it undergoes a remarkably low extent of HDX. When carefully inspecting the mass spectra, we observed that species B is present immediately after GdnHCl is removed. This suggests that CsgA starts aggregating once GdnHCl is removed, consistent with previous reports.14, 34 As the incubation time increases, species B becomes relatively more abundant than species A (Figure 2B), indicating a change from an IDP to a more ordered structure accompanying aggregation. Species A remains detectable at the longest incubation time even though it has been seriously depleted (Figure 1), illustrating the high dynamic range of MS-based methods. However, MS detection has high bias towards the readily soluble materials. The actual abundance of species A relative to the total protein is low because majority of the protein has formed fibrils, most of which can’t be easily detected under our experimental setup. It is not possible, however, for us to distinguish whether species A at the end of aggregation process represents the original dynamic conformers that have not been converted to fibrils, or some products from the equilibrium between solid phase (i.e., fibril) and solution phase.
Although this bimodal isotopic distribution is similar to that arising from an EX1 mechanism36, 37 that occurs when local protein refolding rates are slower than deuterium labeling rates, we do not invoke EX1 mechanism changes to explain the data. Similar phenomena were reported in other aggregation systems using pulsed HDX.26, 38 We hypothesize that pulsed HDX presents various populations of molecules that co-exist in solution; species A represents soluble, dynamic and amorphous conformers whereas species B the highly ordered. The origin and function of species B will be discussed below.
The five regions (R1–R5) of CsgA exhibit different aggregation propensities
Returning to a signal-depletion analysis, we investigated the B fractional species changing with time for each peptide (Figure 3) with the goal of obtaining region-specific information during conformational change from IDP to ordered fibrils. This species-dependent measurement is made possible by HDX but it has a more limited dynamic range than that for the data in Figure 1. The sigmoidal curve fitting of the data (Figure 3A) shows that, for all peptides, the fractional species B remains less than 3% before ~90 min and then increases afterwards. Peptides 26–37 and 106–137 display the most rapid increase in the fraction B species, followed by peptide 38–48. The last time points of peptides 84–93 and 106–137 are missing because their corresponding MS signals become very weak at long aggregation times. Although we are only able to follow peptide 94–105 for 240 min, the B fractional species appears to display a similar trend to that of the other peptides 1–25, 49–69, 70–83 and 84–93; that is, they all increase at a slower rate. We then compared the extracted t1/2 for each peptide (Figure 3B; error bars here indicate ranges at 95% confidence intervals). Peptides can be divided into two groups: peptides 26–37, 38–48, and 106–137 (group I) have significantly smaller t1/2 values compared to the peptides 1–25, 49–69, 70–83 and 84–93 (group II) at 95% confidence. Noticeably, peptides in group I have entered a stationary phase while peptides in group II have not completed aggregation (after 1000 min). If the slow aggregating regions, as represented by group II peptides could be measured to completion, their t1/2 values would be even longer, and the difference between the two groups of peptides, therefore, would be larger.
Figure 3.

(A) Fractional species B as a function of incubation time for peptide 1–25  , 26–37
, 26–37  , 38–48
, 38–48  , 49–69
, 49–69  , 70–83
, 70–83  , 84–93
, 84–93  , 94–105
, 94–105  and 106–137
and 106–137  . Data were fit with sigmoidal curves (dotted lines). (B) t1/2 value of each peptide extracted from sigmoidal curve fitting in A with errors indicating t1/2 ranges at 95% confidence level. Color were coded the same way as in A. (C) Increasing rates of fraction B species (i.e., the first derivative of fraction B species against incubation time) as a function of incubation time. Color coding is the same as in A.
. Data were fit with sigmoidal curves (dotted lines). (B) t1/2 value of each peptide extracted from sigmoidal curve fitting in A with errors indicating t1/2 ranges at 95% confidence level. Color were coded the same way as in A. (C) Increasing rates of fraction B species (i.e., the first derivative of fraction B species against incubation time) as a function of incubation time. Color coding is the same as in A.
To confirm the difference between two groups of peptides, we also analyzed the increasing rates of the fraction B species by calculating the first-derivatives of the fitting curves (Figure 3C). This method allows us to make a direct comparison between all peptides whether or not they have stationary phases. Peptides 26–37 and 106–137 have the largest maximum values, followed by peptide 38–48. The peptides of group II have similar and the small least increasing rates of the fraction B species. Both statistical methods show that regions represented by peptides from group I (26–37, 38–48, and 106–137) aggregate faster than the remaining regions of the protein. The observation that these peptides (26–37, 38–48, and 106–137) have significantly different abundance at starting points (Figure 1) ensures that their faster aggregation propensities are not artifacts caused by variable peptide signals.
To visualize the results, we also mapped the HDX results onto a predicted CsgA structure39 (Figure 4). This structure is a left-handed monomer subunit existing in the fibrils, and it has similar energy to one with a right-handed topology.39 Peptide 26–37 falls completely onto R1, 38–48 located at the end of R1 and the beginning of R2, and peptide 106–137 spans the end of R4 and the entire R5. Thus, our HDX results indicate that R1 and R5 become structured most rapidly among the five repeating units. This finding coincides with that of Chapman and co-workers14, indicating that R1, R3 and R5 are amyloidogenic, and R1 and R5 are the most significant contributors. The primary growth direction of CsgA fibrils is suggested to be perpendicular to the β-strands (dashed line), along with lateral fibril association induced by polar and acidic amino-acid side chains.39 This is consistent with the observation that R1 and R5 become ordered first, followed by the remaining regions.
Figure 4.
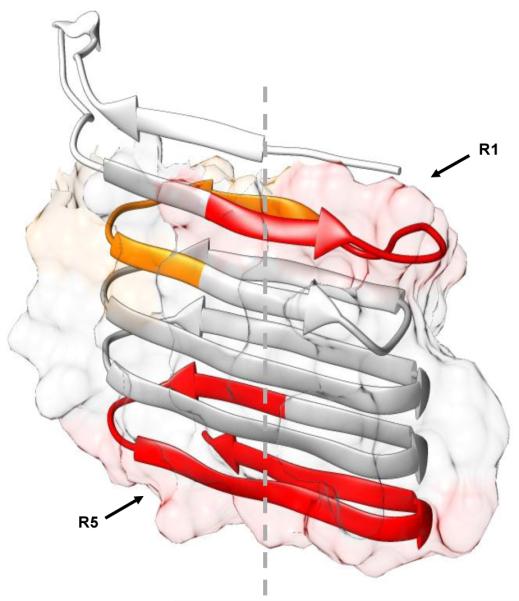
Relative increasing rates of fraction B species mapped onto a computed CsgA monomer structure. Colors were coded the same way as in Figure 3: 26–37 in red, 38–48 in orange, 106–137 in dark red, and the remaining peptides in gray. Surface areas of amyloid core (23–131) that contains five imperfect repeats, calculated in UCSF Chimera,40 are shown as the background to backbone structure. Dashed line indicates the proposed primary fibril growth direction.
TEM suggests origins of species B from large aggregates to fibrils
As discussed earlier, species A is likely to be a highly dynamic, amorphous oligomer that undergoes HDX within the pulse-labeling window as that of the denatured monomer. CsgA needs to escape from this low-energy pool to generate nucleation seeds for amyloid formation.34 Therefore, species A is of little interest, and we suggest that species B is more important. Both their abundances, relative to that of the internal standard, and the fraction B species of all peptides exhibit sigmoidal profiles as a function of incubation time. If species B is part of the nucleation seeds, one would expect the lag phase monitored by either method to be the same. The fractional species B, however, starts to increase significantly after 90 min when soluble materials are already largely depleted. In other words, region-specific aggregation propensities are observed when large amount of insoluble fibrils are already formed. Additionally, species B is formed immediately after denaturant removal. Although the fraction B species increases with incubation time, the absolute quantity of species B remains at a constant, low level. Thus, we suspect that species B represents different large components in the CsgA aggregation than does species A.
To test our hypothesis and capture a more complete picture of CsgA aggregation, we employed TEM as a complementary analysis (Figure 5). MS detects soluble materials whereas negative stain and TEM provide morphology of protein molecules from large oligomers to aggregates, especially the relatively large, insoluble species that are unlikely to be detected by MS. The protein aggregates observed in the early phase of CsgA aggregation (Figure 5A and 5B) are morphologically different than the amyloid fibrils formed at the plateau phase (Figure 5C). There are a few large, amorphous aggregates and clumps of different sizes observed at the lag phase (Figure 5A, 30 min). Beside the amorphous aggregates, needle-like short fibrils (Figure 5B, right panel) start appearing at the early growth phase (Figure 5B, 1 h). In a previous study, the authors described the formation of needle-like protofibrils at the early process of CsgA aggregation.10 The appearance of short fibrils in the sample incubated for 1 h indicates formation of potential nucleation speices;10 that time correlates with the time course determined by large depletion of the peptide signals. Based on both HDX-MS and TEM experiments, we propose that species B represents a soluble part of the large amorphous aggregates at early time points, possibly as protofibrils or fibrils along the aggregation pathway. It is unclear whether species B signals growth-competent protofibrils (i.e., on-pathway oligomers). Nevertheless, our HDX results show that R1 and R5 fold faster than the remaining regions, suggesting they have higher amyloidogenic properties.
Figure 5.

TEM images of CsgA incubated with continuous stirring at 30 min, 1 h, and 5 days. Two figures at each incubation time point were provided with a different scale.
CsgA aggregation depends on conditions
We also followed CsgA aggregation at two other sample handling conditions by monitoring changes in the fraction B species. We did not detect significant changes in that fraction for all the peptides when incubating CsgA without stirring for as long as 144 h (data not shown). It is not surprising that the fractional species B under quiescent conditions increases more slowly for each region than under agitation where seed fibrils are fragmented under mechanical stress. Interestingly, one flash freeze-and-thaw cycle of freshly purified CsgA (denoted as freeze/thaw) followed by incubation under the quiescent condition resulted in the appearance of a bimodal distribution much more rapidly than for freshly prepared CsgA under the same quiescent incubation. The fraction B species for all peptides shows a significant increase after 120 min (Figure S3). This finding suggests that one freeze/thaw cycle of CsgA monomers promotes seed formation, thereby shortening the lag phase. In a manner similar to that of freshly prepared CsgA studied under continuous stirring, fractional species B for peptides 26–37, 38–48, and 106–137 show more rapid increases than do the remaining peptides. This result further supports the finding that R1 and R5 have higher aggregation propensities than the interior regions.
Conclusion
Unlike protein aggregation and amyloid formation in neurodegenerative diseases,1, 2, 4 CsgA aggregation into a functional amyloid occurs by a well-regulated mechanism5. To compensate for the usual limitation that HDX detects principally soluble species without regard to their relative abundance, we followed the abundance of each peptide relative to an internal standard. These profiles are sigmoidal with a lag phase consistent with that determined by ThT fluorescence. HDX-MS reveals two species during the aggregation with distinctly different extents of labeling. One species (denoted A) exchanges rapidly similar to that of a denatured monomer, whereas the other species (denoted B) is structured and well-protected. Although HDX extents of both species are nearly constant with respect to incubation time, the fraction of the ordered species B increases significantly after the lag phase. We also found that among fractional species B, certain regions of the protein (26–37, 38–48, and 106–137) develop structure more quickly than others, in agreement with the observation that CsgA nucleation specificity is encoded mainly by repeating units R1 and R5.14 Thus, we see that HDX and MS can make significant contributions to understanding amyloid formation.
TEM, which probes large or insoluble materials that are less likely to be observed by mass spectrometry, shows large amorphous aggregates during the lag phase whereas mass spectrometry detects mainly dynamic conformers and a small fraction of high-order structure. The protofibrils and then the mature fibrils can be seen by TEM during the time that fractional species of ordered-structure B starts to increase noticeably. Based on these data, we hypothesize that the species B uncovered by HDX-MS represents heterogeneous large components in the CsgA aggregation. Studies under different aggregation conditions further confirm that region-specific aggregation information can be extracted from the fractional species B.
Supplementary Material
Highlights.
Amyloid formation of CsgA protein in curli followed by pulsed HDX and MS
Amyloid formation of CsgA also characterized by MS quantification of soluble protein
One highly structured and one disordered species coexist during aggregation of CsgA
Regions of CsgA that determine aggregation identifiable by HDX and MS
Protein species involved in various stages of aggregation seen by TEM
Acknowledgements
This work was supported by NIH National Institutes of General Medical Sciences: 2P41GM103422 to MLG and 1R01AI099099 to CF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Giacobini E, Gold G. Alzheimer disease therapy--moving from amyloid-beta to tau. Nat Rev Neurol. 2013;9:677–686. doi: 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- [2].Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- [4].Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- [5].Evans ML, Chapman MR. Curli biogenesis: order out of disorder. Biochim Biophys Acta. 2014;1843:1551–1558. doi: 10.1016/j.bbamcr.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- [8].Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- [9].Wang X, Chapman MR. Curli provide the template for understanding controlled amyloid propagation. Prion. 2008;2:57–60. doi: 10.4161/pri.2.2.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dueholm MS, Nielsen SB, Hein KL, Nissen P, Chapman M, Christiansen G, Nielsen PH, Otzen DE. Fibrillation of the major curli subunit CsgA under a wide range of conditions implies a robust design of aggregation. Biochemistry-Us. 2011;50:8281–8290. doi: 10.1021/bi200967c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shu Q, Crick SL, Pinkner JS, Ford B, Hultgren SJ, Frieden C. The E. coli CsgB nucleator of curli assembles to beta-sheet oligomers that alter the CsgA fibrillization mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6502–6507. doi: 10.1073/pnas.1204161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nenninger AA, Robinson LS, Hammer ND, Epstein EA, Badtke MP, Hultgren SJ, Chapman MR. CsgE is a curli secretion specificity factor that prevents amyloid fibre aggregation. Mol Microbiol. 2011;81:486–499. doi: 10.1111/j.1365-2958.2011.07706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cherny I, Rockah L, Levy-Nissenbaum O, Gophna U, Ron EZ, Gazit E. The formation of Escherichia coli curli amyloid fibrils is mediated by prion-like peptide repeats. J Mol Biol. 2005;352:245–252. doi: 10.1016/j.jmb.2005.07.028. [DOI] [PubMed] [Google Scholar]
- [14].Wang X, Smith DR, Jones JW, Chapman MR. In vitro polymerization of a functional Escherichia coli amyloid protein. J Biol Chem. 2007;282:3713–3719. doi: 10.1074/jbc.M609228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang X, Chapman MR. Sequence determinants of bacterial amyloid formation. J Mol Biol. 2008;380:570–580. doi: 10.1016/j.jmb.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Konermann L, Pan JX, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chemical Society reviews. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- [17].Engen JR. Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Analytical chemistry. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Engen JR, Wales TE. Analytical Aspects of Hydrogen Exchange Mass Spectrometry. Annu Rev Anal Chem. 2015;8:127–148. doi: 10.1146/annurev-anchem-062011-143113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee JJ, Park YS, Lee KJ. Hydrogen-deuterium exchange mass spectrometry for determining protein structural changes in drug discovery. Arch Pharm Res. 2015;38:1737–1745. doi: 10.1007/s12272-015-0584-9. [DOI] [PubMed] [Google Scholar]
- [20].Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomic. 2011;8:43–59. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11:527–540. doi: 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Carulla N, Zhou M, Giralt E, Robinson CV, Dobson CM. Structure and intermolecular dynamics of aggregates populated during amyloid fibril formation studied by hydrogen/deuterium exchange. Acc Chem Res. 2010;43:1072–1079. doi: 10.1021/ar9002784. [DOI] [PubMed] [Google Scholar]
- [23].Paslawski W, Mysling S, Thomsen K, Jorgensen TJ, Otzen DE. Co-existence of two different alpha-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry. Angewandte Chemie. 2014;53:7560–7563. doi: 10.1002/anie.201400491. [DOI] [PubMed] [Google Scholar]
- [24].Zhang Y, Rempel DL, Zhang J, Sharma AK, Mirica LM, Gross ML. Pulsed hydrogen–deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proceedings of the National Academy of Sciences. 2013;110:14604–14609. doi: 10.1073/pnas.1309175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mysling S, Betzer C, Jensen PH, Jorgensen TJ. Characterizing the dynamics of alpha-synuclein oligomers using hydrogen/deuterium exchange monitored by mass spectrometry. Biochemistry-Us. 2013;52:9097–9103. doi: 10.1021/bi4009193. [DOI] [PubMed] [Google Scholar]
- [26].Wang H, Shu Q, Rempel DL, Frieden C, Gross ML. Continuous and Pulsed Hydrogen–Deuterium Exchange and Mass Spectrometry Characterize CsgE Oligomerization. Biochemistry-Us. 2015;54:6475–6481. doi: 10.1021/acs.biochem.5b00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xu H, Freitas MA. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 2009;9:1548–1555. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kheterpal I, Lashuel HA, Hartley DM, Walz T, Lansbury PT, Jr., Wetzel R. Abeta protofibrils possess a stable core structure resistant to hydrogen exchange. Biochemistry-Us. 2003;42:14092–14098. doi: 10.1021/bi0357816. [DOI] [PubMed] [Google Scholar]
- [29].Pan J, Han J, Borchers CH, Konermann L. Structure and dynamics of small soluble Abeta(1-40) oligomers studied by top-down hydrogen exchange mass spectrometry. Biochemistry-Us. 2012;51:3694–3703. doi: 10.1021/bi3002049. [DOI] [PubMed] [Google Scholar]
- [30].Pan J, Han J, Borchers CH, Konermann L. Conformer-specific hydrogen exchange analysis of Abeta(1-42) oligomers by top-down electron capture dissociation mass spectrometry. Analytical chemistry. 2011;83:5386–5393. doi: 10.1021/ac200906v. [DOI] [PubMed] [Google Scholar]
- [31].Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- [32].Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- [33].Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- [34].Evans ML, Chorell E, Taylor JD, Aden J, Gotheson A, Li F, Koch M, Sefer L, Matthews SJ, Wittung-Stafshede P, Almqvist F, Chapman MR. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol Cell. 2015;57:445–455. doi: 10.1016/j.molcel.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Keppel TR, Weis DD. Analysis of disordered proteins using a simple apparatus for millisecond quench-flow H/D exchange. Analytical chemistry. 2013;85:5161–5168. doi: 10.1021/ac4004979. [DOI] [PubMed] [Google Scholar]
- [36].Ferraro DM, Lazo N, Robertson AD. EX1 hydrogen exchange and protein folding. Biochemistry-Us. 2004;43:587–594. doi: 10.1021/bi035943y. [DOI] [PubMed] [Google Scholar]
- [37].Arrington CB, Robertson AD. Kinetics and thermodynamics of conformational equilibria in native proteins by hydrogen exchange. Methods Enzymol. 2000;323:104–124. doi: 10.1016/s0076-6879(00)23363-6. [DOI] [PubMed] [Google Scholar]
- [38].Serra-Vidal B, Pujadas L, Rossi D, Soriano E, Madurga S, Carulla N. Hydrogen/deuterium exchange-protected oligomers populated during Abeta fibril formation correlate with neuronal cell death. Acs Chem Biol. 2014;9:2678–2685. doi: 10.1021/cb500621x. [DOI] [PubMed] [Google Scholar]
- [39].Tian P, Boomsma W, Wang Y, Otzen DE, Jensen MH, Lindorff-Larsen K. Structure of a functional amyloid protein subunit computed using sequence variation. J Am Chem Soc. 2015;137:22–25. doi: 10.1021/ja5093634. [DOI] [PubMed] [Google Scholar]
- [40].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.