

Genome editing refers to a process of making precise and permanent changes in the genetic code of cells, tissues, and whole organisms. The first step in this process is to program an enzyme called a nuclease to bind to a precise DNA sequence, whereby the enzyme will cut the DNA. This double-stranded break will in turn induce the cell to make a repair at that site, which will change the DNA sequence. The repair mechanism can be utilized to either knock out or introduce selected genes. Given how rapidly gene editing systems are influencing diverse applications in biomedical research, biotechnology, and agriculture,1 clinical applications in neurology to repair genetic mutations causing disease, or to incorporate genes that might be of therapeutic benefit, are on the near horizon. The following cases illustrate how such an approach may be used to develop new ex vivo (case 1) and in vivo (case 2) therapies for neurologic disease, respectively, and highlight some of the translational challenges of using these molecular reagents safely and ethically in patients.
REPRESENTATIVE CASES
Case 1.
A 57-year-old woman presented to the neuromuscular clinic for an evaluation of lower limb weakness. She had a family history of amyotrophic lateral sclerosis (ALS) in her father. She described an insidious onset of right foot dorsiflexor weakness, in the absence of sensory symptoms, which had progressed rapidly over an 8-month period, and extended now to the contralateral leg, affecting her overall gait and stability. Her neurologic examination was notable for normal bulbar findings, mild weakness in the right intrinsic hand muscles with spared sensation, and bilateral, asymmetric generalized weakness of the lower limbs. She was weaker on the right than left and with distal predominance, and had associated right thigh and leg muscle fasciculation. The tendon reflexes were mildly hyperactive in the arms and markedly so at the knees with cross-adduction; ankle reflexes were absent but triggered contraction of the hamstrings. Plantar responses were absent. There was no sensory loss in the lower limb. She was unable to get up from her chair without pushing on it. On gait assessment, she had bilateral steppage, right more than left, with an inability to stand on her toes. EMG demonstrated a diffuse neurogenic process affecting motor neurons and their axons in the right cervical, thoracic, and lumbosacral segments, most consistent with progressive motor neuron disease. After inquiring about what clinical trials might be ongoing, she was screened and enrolled into a phase I trial evaluating the safety of autologous mesenchymal stem cell transplantation into the intrathecal space by lumbar puncture delivery.
Case 2.
A 4-year-old boy presented to the pediatric neurologist with his parents, who were concerned about difficulties he was having with running, climbing stairs, and frequent falls. There was a strong family history of sudden cardiac death. The boy could arise from a seated position only by pushing his arms against his thighs with his elbows locked and extending his hips. He had a normal height and weight at birth, but his growth curve fell below the normal centiles during his first year. His examination was notable for short stature, calf and lateral thigh hypertrophy, and scapular atrophy. Strength testing revealed mild weakness in the neck flexors, biceps, and triceps. He had more pronounced symmetric weakness in the muscles of the proximal lower extremity. He demonstrated the Gowers sign when getting up from the floor. Reflexes were reduced in the lower extremities. His sensory examination results were normal. His gait was waddling and his spine lordotic. A muscle biopsy of the left vastus lateralis showed prominence of necrotic fibers invaded by macrophages, groupings of regenerated fibers, wide-ranging fiber size variability, and hypercontracted fibers. There were dense endomysial and perimysial mononuclear cell infiltrates and areas of endomysial fibrosis. Immunohistochemical staining for the C- and N-terminal regions of dystrophin showed a background of marked sarcolemmal attenuation with rare fibers (<1%) staining positive. Genetic testing demonstrated a splice site mutation causing a frameshift mutation in the triple-helical-repeat domain in exon 54 of the dystrophin gene. Along with inquiring about the current clinical approaches to management, his parents asked about what strategies could potentially be available over their son's anticipated lifespan to correct the mutation.
GENERAL MECHANISMS OF GENOME EDITING
Nucleases.
There are 2 broad classes of nuclease enzymes for genome editing, each having a different mechanism of action (figure 1). Zinc finger nucleases and transcription activator-like effector nucleases (TALENs) are guided by DNA-binding protein arms. These systems work in pairs, with one arm binding a sequence upstream on the (+) strand and a second arm binding downstream on the (−) strand, and then the 2 arms come together in the middle to cut the DNA as a double-stranded break. The second, and more frequently utilized approach, uses clustered regularly interspaced short palindromic repeat (CRISPR)–associated (Cas) nucleases, which are DNA endonucleases guided by short segments of RNA, about 80 nucleotides long. The front end of the guide RNA has a 20-nucleotide sequence that is complementary to the target DNA, and the tail end serves to tether Cas nuclease to the target DNA for cutting.2
Figure 1. Genome editing systems.

DNA nucleases may be guided to a specific genomic target sequence by proteins or RNA molecules. Zinc finger nucleases (ZFN) and transcription activator–like effector nucleases (TALENs) utilize endonuclease domains that are flanked by DNA-binding domains. Clustered regularly interspaced short palindromic repeat–associated (Cas) nucleases require single-guide (sg) RNA molecules, which link a short DNA recognition sequence of 20 nucleotides with a longer tail end to tether the genomic DNA and nuclease together into a single DNA-RNA-nuclease hybrid complex. In both systems, accurately targeted nucleases will cut the genomic DNA to create a blunt-ended, double-stranded break (DSB). Losing the structural integrity of its DNA will prompt the cell to repair that site using 1 of 2 mechanisms. Nonhomologous end joining (NHEJ) is the most frequently used mechanism, whereby the free DNA ends are rejoined together. This process usually requires the removal or insertion of nucleotides at the join, creating indel mutations that will shift the reading frame of the gene and lead to the expression of a nonsense or truncated protein. Homology-directed repair (HDR) uses a second DNA sequence as a template to bridge the gap by homologous recombination (HR). If a new DNA sequence is incorporated between flanking areas of homology, this strategy can be used to correct a mutated gene, or to introduce novel genes into the targeted genomic site. (Figure modified, with permission, from Heidenreich M, Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat Rev Neurosci 2016;17:36–443).
DNA repair mechanisms.
Once the DNA is cut, the most expedient way for cells to repair the double strand break is to stitch the 2 free ends back together. This process, called nonhomologous end joining (NHEJ), is usually not precise, and leads to base pair insertions or deletions (indels) at the join; this results in a shift in the reading frame of the gene and will typically knock out its function by creating a nonsense or truncated protein. The other repair pathway is mediated by homologous recombination between the ends of the cut sequence and an introduced second donor DNA template; this is known as a homology-directed repair (HDR) pathway. If the template includes a new sequence of DNA between the areas of homology, this pathway can be used to integrate that new sequence, with the aim of gene correction or replacement.3 Furthermore, 2 or more nucleases may be used at the same time to target DNA sites that are far apart from each other, and this approach is called multiplexing. The repair options are typically the same: either stitch the ends back together (but this time there is a larger chromosomal deletion), or again fill the gap using HDR to insert a new sequence.
RNA-GUIDED GENOME EDITING
Molecular basis.
RNA-guided genome editing is derived from an elegant system of adaptive immunity in bacteria.4 When bacteria are invaded by a virus, they are able to sample small fragments of the viral DNA, and insert these pieces into their own chromosome, essentially making a library, or a physical memory system of all prior infections. The structure of the library is such that the viral DNA pieces (termed protospacers) alternate with identical repeat-spacer element sequences; hence the name CRISPR.5 In order to destroy invading viruses, the CRISPR array library is first transcribed as full-length RNA sequences, which are then processed into shorter single pieces of RNA, each containing a copy of an individual viral sequence and a repeat-spacer sequence. These processed RNA sequences then serve as a guide, binding back to the original viral DNA (literally from whence it came), and cleaving the viral DNA in a complex with the Cas nuclease to inactivate the invader. Genome editing technologies are quickly evolving as more CRISPR types are discovered and harnessed to increase reagent efficiency and specificity.6 Known systems have been divided into 2 broad classes, which can further be divided into 6 types, and at least 19 subtypes.7 Class 2 systems are defined by having a single multifunctional protein effector.8 Requiring only one protein for RNA guidance and cleavage has made Class 2 systems particularly useful for genome editing; the canonical Cas9 (type II) versions from Streptococcus pyogenes (SpCas9) and Staphylococcus aureus (SaCas9) are Class 2 systems that are in widespread use. Other Class 2 systems, including type V (centromere and promoter factor 1),9,10 represent newer developments that utilize full-length, preprocessed CRISPR RNA arrays for simultaneous multiplexing. The type VI nucleases have recently opened a new frontier of targeting and editing RNA in cells (RNA-guided RNA editing).11,12
The CRISPR–Cas9 system has been adapted to eukaryotic cells,13 by humanizing the codon structure and by optimizing a linkage loop between the 2 functional ends of the RNA to create a single-guide RNA (sgRNA) molecule.2 All that is required of the researcher is to select the 20–base pair genomic target that is to be incorporated into the sgRNA. Once inside the cell, the sgRNA and Cas9 nuclease form targeting precomplexes, which then diffuse around the nucleus and interact with the genomic DNA (figure 2). The kinetics of this process are such that the complex will approach, bind, and release DNA quickly if there is not a complementary sgRNA match; if there is a partial match, the complex will take slightly longer to release; and if there is a full match, the sgRNA–nuclease complex will engage to cut the DNA.14 Full engagement of the complex requires the presence of a 2–6 nucleotide DNA sequence immediately next to the target, called the protospacer-adjacent motif (PAM) sequence.15,16 The full-length match next to the appropriate PAM sequence results in DNA unwinding into separate strands, each of which is cut by a separate enzyme domain. The cleaved DNA is then released from the RNA–nuclease complex to undergo repair.
Figure 2. Single-guide (sg) RNA–Clustered regularly interspaced short palindromic repeat–associated (Cas) 9 complex surveillance.

Cas9 proteins form ribonucleoprotein (RNP) complexes with sgRNA molecules in the cell nuclei prior to engagement with the target genomic DNA. The RNPs make frequent contact with accessible DNA, binding first wherever they encounter a matching protospacer-adjacent motif (PAM) and then making a rapid assessment of the first 5 or 6 genomic nucleotides (the “seed region”) for complementarity by initiating partial DNA unwinding. The length of time that the RNP will remain at the site, and thus the probability that the DNA will be cut, is proportional to the distance and content that is complementary to the guide RNA beyond the seed region and over the whole 20-nucleotide target stretch. Incorrect DNA matches will be released by the RNP, leaving the DNA intact and the RNP free to bind and assess other sites. The number of genomic areas that can be sampled simultaneously is dependent on the concentration of the RNPs within the nucleus. A full genomic match with the correct PAM sequence leads to full RNP engagement: the Cas9 employs a dual helicase and nuclease function, unwinding the target into separate strands, and cutting the DNA at a site that is typically 3 nucleotides away from the PAM sequence. Each strand is cut by separate nuclease domains, termed HNH for the DNA strand bound to the RNA guide, and RuvC for the freely separated strand. The RNP releases once the DNA is cut. (Panel right graphics are used with permission of Dr. Feng Zhang, McGovern Institute for Brain Research at the Massachusetts Institute of Technology; youtu.be/2pp17E4E-O8).
Challenges in RNA-guided DNA editing.
On-target effects are easy to measure, as it is known exactly where to look in the genome, based on the initial targeting design for the sgRNA. An efficient editing reagent will cut >50% of alleles, so the rate of indel formation and gene knockout is usually high. Efficient gene integration by HDR remains challenging—even with the best cutting reagents, it remains difficult to integrate a new sequence at rate higher than 5% of alleles. Cutting efficiencies in turn depend directly on how efficiently the reagents are delivered into the cells, and on how long they will be expressed vs how quickly the complexes will degrade. No genome editing technology is completely accurate, and at times the reagent will bind and cut DNA that was not the intended target. It is an enormous technical challenge to find the off-target changes and define their consequences, but this is probably the most important issue to consider in terms of patient safety.
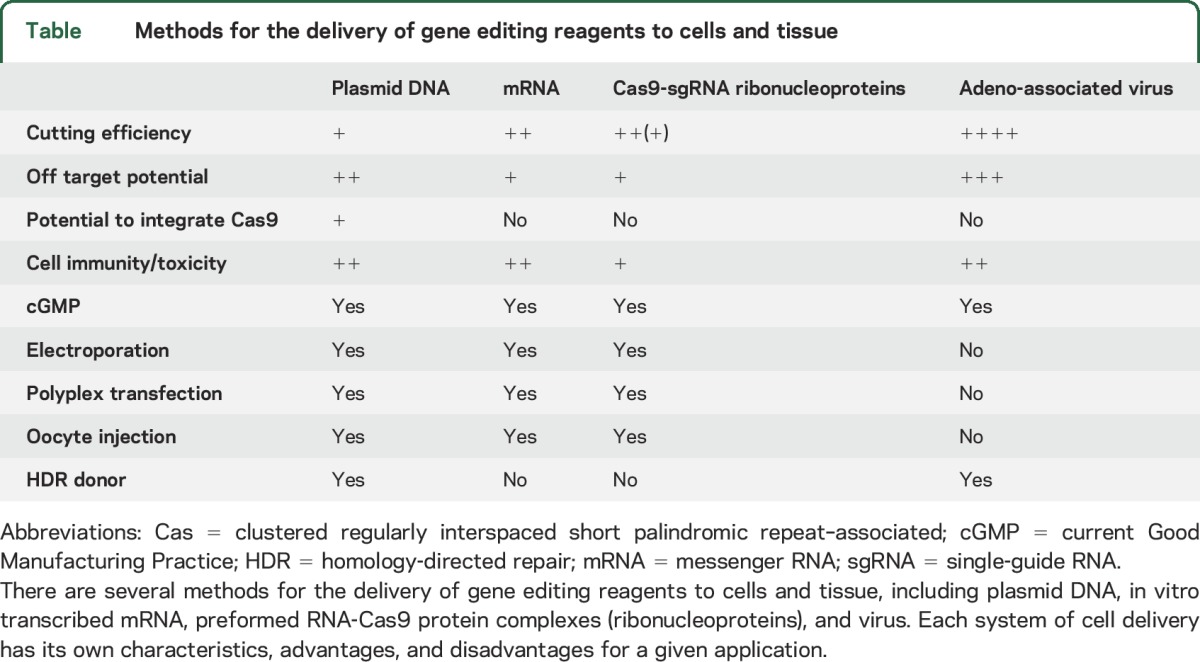
Currently, TALEN-based editing is more accurate than RNA-guided editing, as 2 independent binding arm events must coincide for DNA cleavage. Therefore, a major focus of RNA-guided genome editing is to fine-tune the engineering of Cas9 proteins to be more accurate.17 Off-target effects can be improved by using computer prediction models that help to design better guides18 and by adapting PAM specificity constraints.19 Furthermore, a number of next-generation sequencing modalities are being developed and applied to this problem to identify off-target effects when they occur.20,21 There are several ways to deliver the genome editing reagents to cells, such as using plasmid DNA, transcribed messenger RNA, and viruses (table). Ribonucleoproteins are preformed Cas9 guide complexes made in vitro using recombinant Cas9 protein and synthetic RNA.22 Ribonucleoproteins may have more favorable on- and off-target efficiencies than a plasmid-based delivery system due to their rapid targeting and degradation kinetics.
Table.
Methods for the delivery of gene editing reagents to cells and tissue
APPLICATIONS OF GENOME EDITING IN NEUROLOGIC DISEASE
It is the ease of the approach and application of CRISPR–Cas9 technologies that has led to a rapid uptake in laboratories of all sizes and expertise and an acceleration in publications over the last 5 years.23 When the CRISPR–Cas9 system was first described in 2012, there were roughly 200 publications, which then increased to over 1,000 publications in in 2015 and over 1,600 in 2016.23 It is therefore easy to understand how a field that once focused on RNA processing and microbiology has now expanded rapidly into genetics, medicine, biotechnology, and agriculture.1
Gene editing for neurologic diseases will broadly proceed along 2 treatment paradigms: ex vivo and in vivo gene editing.
Ex vivo gene editing.
For ex vivo gene editing, RNA- or protein-guided nucleases are used to alter or add DNA in cells that are extracted from the patient and purified, gene-edited in cell culture conditions, selected and expanded in cell culture, and given back to the patient by cell infusion or tissue injection. In this paradigm, gene editing reagents are expressed and act only within the extracted cell populations. This approach provides the opportunity to control more fully the editing outcomes and establish critical measures of gene editing safety and efficiency. Cell selection is important in therapeutic strategies involving new gene integration or correction, where cells with successful gene insertion can be sorted from cells whose genomes are either uncut or cut without an insertion event.24 The difficulties of obtaining large, clean populations of genome-edited cells for ex vivo approaches will dictate the need to enrich for the edited cell population.
When using an ex vivo approach, furthermore, it is also possible to manipulate the cell cycle in culture conditions to improve the efficiencies by which genome cutting and HDR events take place to integrate the therapeutic genes of choice. The rates of HDR can be improved both by inhibiting the NHEJ pathways using small molecules,25–27 as well as enhancing the HDR pathway using drugs to either force cell cycle entry into, or maintain cells within, cell cycle phases where HDR selectively occurs, the G2 and S phases.28,29 Since the homology donor DNA has typically been delivered to the cells ex vivo as a plasmid source, repair efficiencies also depend critically on the efficiency of plasmid uptake, and its suitability as a template donor for eukaryotic cell repair mechanisms. Increasingly, high-efficiency donors now are being developed from viral sources, including coinfection of the cell with adeno-associated virus as the DNA donor template.30 Further challenges entail how to control whether an integration event will occur in one or both chromosomal alleles.31
CRISPR–Cas9 ex vivo gene-edited cell therapies are actively being used in hematology and oncology human clinical trials, and are paving the way forward for neuro-oncology applications, where the primary obstacle will be to improve the ability of modified T cells to kill large solid tumors in the CNS. Current trials involve IV infusions of CRISPR–Cas9-modified autologous T cells with knockdown of the gene encoding the programmed cell death protein 1 (PD-1); the aim is to increase the intensity of T-cell response by removing this inhibitory regulatory protein. These trials are being conducted in patients with small cell lung, bladder, prostate, and metastatic renal cancer.32–35 Chimeric antigen receptor (CAR) T cells have been used extensively in hematologic malignancies, particularly in chemotherapy-refractory B-cell acute lymphocytic leukemia, where the treatment has proven to be effective, as well as against neurologic tumors like glioma, neuroblastoma, and glioblastoma (reviewed in reference 36). CAR T cells will soon be used in conjunction with 3 CRISPR–Cas9 knockdown edits, including PD-1 and the endogenous T-cell receptor gene in clinical trials targeting myeloma, sarcoma, or melanoma.37 CAR T cells become active upon binding to a specific tumor antigen and signal for further T-cell recruitment, proliferation, and activation, thereby intensifying the immune response more precisely against that tumor type. Therefore, the first application of CRISPR–Cas9 genome therapies in neurology may well be for neuro-oncology applications, extending from these combined CAR T-cell genetic knockdown technologies.
In addition to T cells, other cell lines are being developed for potential ex vivo gene editing. These include autologous Schwann cell cultures derived from peripheral nerve biopsies for use in spinal cord injury38,39 and peripheral nerve injury40 (figure 3). Patient-derived induced pluripotent stem cells (iPSCs) can be subjected to ex vivo gene editing and differentiated into neuronal or glial lineages for transplantation. iPSC work in practice remains largely confined to in vitro disease modeling; concerns persist about the clonal homogeneity of differentiated cultures, phenotype persistence, and teratoma risks when considering human transplantation. iPSCs have been differentiated into sensory neurons41 for studying chemotherapy-induced peripheral neuropathy,42 oligodendrocyte precursor cells for remyelination in spinal cord injury repair,43,44 and Schwann cells for peripheral and spinal cord injury therapeutics.45 Autologous bone marrow or adipose tissue–derived mesenchymal stem cells (adMSCs) are emerging as a leading candidate cell platform for ex vivo therapies, given increasing evidence for the safety of unmodified cell transplantation in patients.46 Autologous mesenchymal stem cells are undergoing clinical trials at our institution in patients with ALS47 (case 1) and multiple system atrophy.48 A phase I trial involving 27 patients with ALS showed an excellent safety profile using a cell dose escalation design from 10 million to 100 million unmodified cells delivered intrathecally.47 A phase II, open-label study of safety and efficacy in 60 patients with ALS is currently under development using unmodified cells. The adMSCs obtained from fat biopsy are expanded up to billions of cells and then delivered to the patients via lumbar puncture or stored for future use. The early expansion period will provide an ideal opportunity to gene edit the cells using CRISPR–Cas9 or TALEN reagents and a neurotrophin gene donor for insertion into genomic safe harbor sites, where an insertion or deletion of DNA will not have a detrimental consequence to the cell. While neurotrophic factors are of therapeutic interest in ALS and other neurodegenerative diseases, the platform could be readily extended as a cell-based delivery system for other proteins, peptides, enzyme replacement therapy, and even small-fragment antibodies.
Figure 3. Ex vivo cell platforms.

Cells harvested from the patient may be modified in cell culture conditions using genome editing technology, and subsequently returned to the same patient as an autologous transplantation. This approach has first required the development of cell platforms using unmodified cells to establish important safety measures. Unmodified autologous Schwann cells and mesenchymal stem cells (MSCs) are in clinical trial use for neurologic disease. Autologous transplantation of cells that are differentiated from patient-induced pluripotent stem cells (iPSCs) is further away from clinical translation due to clonal variability, phenotype stability, and other safety concerns. ALS = amyotrophic lateral sclerosis; MSA = multiple system atrophy; NTF = neurotrophic factor.
In vivo genome editing.
For in vivo gene editing in neurologic disorders, the editing reagents will be delivered directly to cells in the CNS, peripheral nervous system, or muscle, where they will be expressed and make changes in the genomic DNA within the tissue. Potential gene editing applications include disrupting gain-of-function mutations and excision-repair approaches, provided that the target and a correction mechanism are clear. At least initially, these edits will probably be limited to defined, severe monogenic disorders. The advantages of in vivo therapy include correcting disease pathology at its source rather than treating secondary manifestations of the disease process.
There are several challenges of in vivo gene editing. These include achieving efficient reagent delivery to particular cell types and whole tissues; achieving gene correction in neurons and muscle where the target cells are quiescent, and where utilizing an HDR mechanism brings substantial complexity; and the limited number of efficient reagents that can act in a whole body milieu. One of the most attractive gene delivery vectors for the nervous system and muscle is adeno-associated virus (AAV), which does not integrate into the genome, and is not associated with any disease in humans. AAV produces excellent, high levels of long-term gene expression in quiescent neurons and muscle cells. AAV serotypes can be used that are more specific to one tissue type, and the expression of the gene editing reagents can be further put under the control of cell-specific promoters, as well as cell-state-specific promotors.49,50
There have been many clinical trials using AAV in the CNS in patients over the last 10 years, including the use of intrastriatal injections for Parkinson disease51,52 and basal forebrain injections for Alzheimer disease.53 These trials have generated good safety data about the overall viral approach, but do not yet help to determine the safety of delivering CRISPR–Cas9 reagents to the CNS. Similarly, there have been a number of clinical trials for AAV gene therapy in muscle, including limb-girdle muscular dystrophies, spinal muscle atrophy, and Becker and Duchenne muscular dystrophies (case 2).23,54 The correction of dystrophin coding mutations in Duchenne or Becker muscular dystrophies has emerged as a leading candidate for the first in vivo trials, not just in neurology, but in the field as a whole. The well-validated mouse mdx model of Duchenne muscular dystrophy is designed to have a premature stop codon in exon 23 of the dystrophin gene, and has a phenotype comparable to that in human disease. Three studies using a multiplexing CRISPR–Cas9 approach in this model to excise the mutated exon, with genome editing reagents delivered by either intramuscular or IV AAV serotype 9 injection, have shown de novo expression of dystrophin in a proportion of muscle fibers and improved muscle strength.55–57 The strategy for multiplex DNA excision may be suited for treatment of DNA repeat disorders, including Huntington disease, C9orf72-related disorders, Friedrich ataxia, and the spinocerebellar ataxias. Targeting and excising these repeats may be among the most important applications of gene editing in neurology.58 Multiplexing first within intronic and untranslated regions for the purposes of repeat expansion removal, exon skipping, and pathologic splice site modifications could establish important safety measures prior to targeting gene coding regions.
ETHICAL IMPLICATIONS OF GERMLINE EDITING
Germline editing refers to changing the DNA in human spermatocytes, oocytes, and their progenitor cells. Such editing could be done in a fertilized egg, early or later in embryonic development, or in somatic cells that have been induced to become germline cells. Germline editing produces irreversible genetic changes that will be inherited by subsequent generations and could potentially protect or rescue a child from severe inheritable disease. Prompted in part by a succession of studies using CRISPR–Cas9 in human zygotes obtained from fertility clinics,59,60 major bioethical reviews were convened in December 2015 and February 2017. In the International Summit on Human Gene Editing,61 which was jointly organized by the US Academy of Sciences, the US National Academy of Medicine, the UK Royal Society, and the Chinese Academy of Science, key issues posed by germline editing were identified. These included the risks of inaccurate editing (off-target effects), risks of incomplete editing (mosaicism), and difficulties in predicting the harm that new genetic changes may have when interacting with other genes and the environment. Overall, the obligations of the scientific community to consider the implications of editing on both the individual and future generations were emphasized, including the morality of purposefully altering wholescale human evolution, along with the potentially adverse ethnic, social, and economic implications. The recommendation from the first international meeting was that it would be irresponsible to proceed with germline editing until the relevant safety and efficacy issues are resolved, and that there was a better understanding and balancing of the risks, benefits, and alternatives. The second committee convened by the US National Academy of Sciences and the National Academy of Medicine concluded its scientific and ethical review with updated guidance statements that clinical trials involving germline editing might be permitted following more extensive research “for compelling reasons and under strict oversight” for couples where editing might represent truly a last reasonable option to bear a heathy child.62 The recent publication of a gene editing study in human embryos that demonstrated the correction of a MYBPC3 gene mutation, which causes hereditary hypertrophic cardiomyopathy, demonstrates that such research has accelerated, with surrounding academic controversy.63
PERSPECTIVE
Genome editing provides a novel therapeutic approach to neurologic disease with a high potential relevance. Further refinements are needed to address the multiple challenges of ex vivo and in vivo genome editing. The most critical factor affecting the repair outcomes is the efficiency of reagent delivery to cells ex vivo or to tissue in vivo. Research into improving genome editing reagent delivery will need to draw level with the current focus to enhance the molecular mechanisms of the editing reagents, in order for current Good Manufacturing Practice (cGMP) applications in patients to be fully realized. While improvements in cell delivery in turn may promote higher rates of genotypic change by NHEJ and HDR, minimizing the risks of off-target cutting, persistent reagent recutting, and unintended recombination events become more essential as a corollary. These points of genetic influence are the bottlenecks in the technology. There are some aspects that can be addressed or optimized in each laboratory, while there are other aspects that will advance only by means of the overall technology through large institutional laboratories, and through the availability of these advances to smaller laboratories. As such, the willingness, and the financial or proprietary incentives, to share research findings, facilitate replication studies, and supply reagents will become more confounded. The overall pace and intensity of genome editing research will continue to increase, providing more insights into and innovating new safety measures for targeting accuracy and genome-wide genotype change confirmation. All the while, the regulatory controls will continue to be outpaced, and the ethics will be challenged.
GLOSSARY
- AAV
adeno-associated virus
- adMSC
adipose tissue-derived mesenchymal stem cell
- ALS
amyotrophic lateral sclerosis
- CAR
chimeric antigen receptor
- Cas protein
CRISPR-associated protein
- CRISPR
clustered regularly interspaced short palindromic repeats
- HDR
homology-directed repair
- iPSC
induced pluripotent stem cell
- NHEJ
nonhomologous end joining
- PAM
protospacer adjacent motif
- PD-1
programmed cell death protein-1
- sgRNA
single-guide RNA
- TALEN
transcription activator-like effector nuclease
AUTHOR CONTRIBUTIONS
N.N. Madigan: conception and design of the project, acquisition and interpretation of data from the literature, drafting/revising the manuscript, critical revision for intellectual content, administrative and technical support. N.P. Staff and A.J. Windebank: drafting/revising the manuscript, critical revision for intellectual content, project supervision. E.E. Benarroch: conception and design of the project, drafting/revising the manuscript, critical revision for intellectual content, project supervision, administrative and technical support.
STUDY FUNDING
Research funding for the development and use of autologous adipose-derived MSCs in phase I and II trials in ALS at Mayo Clinic has been provided by The Bowen Family Foundation, The Judith and Jean Pape Adams Foundation, the Mayo Clinic Center for Regenerative Medicine, and Regenerative Medicine Minnesota.
DISCLOSURE
N.N. Madigan receives stipend support from the Mayo Clinic Graduate School of Medical Education. N.P. Staff receives grants from the National Cancer Institute and National Center for Advancing Translational Sciences, Regenerative Medicine Minnesota, and BrainStorm Cell Therapeutics. A.J. Windebank receives grants from the National Institute of Aging, National Center for Advancing Translational Sciences, Armed Forces Institute of Regenerative Medicine, Regenerative Medicine Minnesota, Morton Cure Paralysis Fund, Craig H. Neilsen Foundation, The Bowen Family Foundation, The Judith and Jean Pape Adams Foundation, Mayo Clinic Center for Regenerative Medicine, and research funding from BrainStorm Cell Therapeutics. E.E. Benarroch receives a stipend in his capacity as section editor of Clinical Implications of Neuroscience Research for Neurology®. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Doudna JA, Charpentier E. Genome editing: the new frontier of genome engineering with CRISPR-Cas9. Science 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- 2.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heidenreich M, Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat Rev Neurosci 2016;17:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature 2015;526:55–61. [DOI] [PubMed] [Google Scholar]
- 5.Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007;315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 6.Makarova KS, Wolf YI, Koonin EV. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res 2013;41:4360–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016;353:aad5147. [DOI] [PubMed] [Google Scholar]
- 8.Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 2015;13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shmakov S, Abudayyeh OO, Makarova KS, et al. Discovery and functional characterization of diverse Class 2 CRISPR-Cas systems. Mol Cell 2015;60:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zetsche B, Heidenreich M, Mohanraju P, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 2017;35:31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.East-Seletsky A, O'Connell MR, Knight SC, et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016;538:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.East-Seletsky A, O'Connell MR, Burstein D, Knott GJ, Doudna JA. RNA targeting by functionally orthogonal type VI-a CRISPR-Cas enzymes. Mol Cell 2017;66:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014;507:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014;513:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karvelis T, Gasiunas G, Young J, et al. Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol 2015;16:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016;529:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H, Xiao T, Chen CH, et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res 2015;25:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015;523:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan WX, Mirzazadeh R, Garnerone S, et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun 2017;8:15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crosetto N, Mitra A, Silva MJ, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods 2013;10:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeWitt MA, Corn JE, Carroll D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods 2017;121-122:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol 2016;34:933–941. [DOI] [PubMed] [Google Scholar]
- 24.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 2016;539:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 2015;33:538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu C, Liu Y, Ma T, et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015;16:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chu VT, Weber T, Wefers B, et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 2015;33:543–548. [DOI] [PubMed] [Google Scholar]
- 28.Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014;3:e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahrabi S, Sarkar S, Pfister SX, et al. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res 2016;44:5743–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaj T, Staahl BT, Rodrigues GM, et al. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res Epub 2017 Mar 2. [DOI] [PMC free article] [PubMed]
- 31.Paquet D, Kwart D, Chen A, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016;533:125–129. [DOI] [PubMed] [Google Scholar]
- 32.US National Library of Science. Knockout engineered T cells for metastatic non-small cell lung cancer. ClinicalTrials.gov [online]. 2016. Available at: clinicaltrials.gov/ct2/show/NCT02793856. Accessed September 7, 2017. [Google Scholar]
- 33.US National Library of Science. PD-1 knockout engineered T cells for muscle-invasive bladder cancer. ClinicalTrials.gov [online]. 2016. Available at: clinicaltrials.gov/ct2/show/NCT02863913. Accessed September 7, 2017. [Google Scholar]
- 34.US National Library of Science. PD-1 knockout engineered T cells for castration resistant prostate cancer. ClinicalTrials.gov [online]. 2016. Available at: clinicaltrials.gov/ct2/show/NCT02867345. Accessed September 7, 2017. [Google Scholar]
- 35.US National Library of Science. PD-1 knockout engineered T cells for metastatic renal cell carcinoma. ClinicalTrials.gov [online]. 2016. Available at: clinicaltrials.gov/ct2/show/NCT02867332. Accessed September 7, 2017. [Google Scholar]
- 36.Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol 2016;13:370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheridan C. CRISPR therapeutics push into human testing. Nat Biotechnol 2017;35:3–5. [DOI] [PubMed] [Google Scholar]
- 38.Bunge MB, Monje PV, Khan A, Wood PM. From transplanting Schwann cells in experimental rat spinal cord injury to their transplantation into human injured spinal cord in clinical trials. Prog Brain Res 2017;231:107–133. [DOI] [PubMed] [Google Scholar]
- 39.Anderson KD, Guest JD, Dietrich WD, et al. Safety of autologous human Schwann cell transplantation in subacute thoracic spinal cord injury. J Neurotrauma Epub 2017 Mar 21. [DOI] [PubMed]
- 40.Gersey ZC, Burks SS, Anderson KD, et al. First human experience with autologous Schwann cells to supplement sciatic nerve repair: report of 2 cases with long-term follow-up. Neurosurg Focus 2017;42:E2. [DOI] [PubMed] [Google Scholar]
- 41.Chambers SM, Qi Y, Mica Y, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol 2012;30:715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staff NP, Grisold A, Grisold W, Windebank AJ. Chemotherapy-induced peripheral neuropathy: a current review. Ann Neurol 2017;81:772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terzic D, Maxon JR, Krevitt L, et al. Directed differentiation of oligodendrocyte progenitor cells from mouse induced pluripotent stem cells. Cell Transpl 2016;25:411–424. [DOI] [PubMed] [Google Scholar]
- 44.Parr AM, Walsh PJ, Truong V, Dutton JR. cGMP-compliant expansion of human iPSC cultures as adherent monolayers. Methods Mol Biol 2016;1357:221–229. [DOI] [PubMed] [Google Scholar]
- 45.Ma MS, Boddeke E, Copray S. Pluripotent stem cells for Schwann cell engineering. Stem Cell Rev 2015;11:205–218. [DOI] [PubMed] [Google Scholar]
- 46.Dietz A, van Wijnen A, Butler GW, et al. A consistent, and predictable drug: the first 100 patients treated with autologous adipose derived mesenchymal stromal cells (MSCs) at the Mayo Clinic. Cytotherapy 2017;19:S155. [Google Scholar]
- 47.Staff NP, Madigan NN, Morris J, et al. Safety of intrathecal autologous adipose-derived mesenchymal stromal cells in patients with ALS. Neurology 2016;87:2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singer W, Low PA. Optimizing clinical trial design for multiple system atrophy: lessons from the rifampicin study. Clin Auton Res 2015;25:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Betley JN, Sternson SM. Adeno-associated viral vectors for mapping, monitoring, and manipulating neural circuits. Hum Gene Ther 2011;22:669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sjulson L, Cassataro D, DasGupta S, Miesenbock G. Cell-specific targeting of genetically encoded tools for neuroscience. Annu Rev Genet 2016;50:571–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Christine CW, Starr PA, Larson PS, et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009;73:1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marks WJ Jr, Ostrem JL, Verhagen L, et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol 2008;7:400–408. [DOI] [PubMed] [Google Scholar]
- 53.CERE-110 in subjects with mild to moderate Alzheimer's disease. ClinicalTrials.gov [online]. 2004. Available at: clinicaltrials.gov/ct2/show/NCT00087789. Accessed September 7, 2017. [Google Scholar]
- 54.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet 2011;12:341–355. [DOI] [PubMed] [Google Scholar]
- 55.Long C, Amoasii L, Mireault AA, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016;351:400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016;351:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tabebordbar M, Zhu K, Cheng JK, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016;351:407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long C, Amoasii L, Bassel-Duby R, Olson EN. Genome editing of monogenic neuromuscular diseases: a systematic review. JAMA Neurol 2016;73:1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang P, Xu Y, Zhang X, et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015;6:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang L, Zeng Y, Du H, et al. CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein. Mol Genet Genomics 2017;292:525–533. [DOI] [PubMed] [Google Scholar]
- 61.The National Academies of Sciences, Engineering and Medicine, Human Gene-Editing Initiative. International Summit on Human Gene Editing: A Global Discussion [online]. Available at: nationalacademies.org/gene-editing/Gene-Edit-Summit/. Accessed September 7, 2017. [Google Scholar]
- 62.Kaiser J. A yellow light for embryo editing. Science 2017;355:675. [DOI] [PubMed] [Google Scholar]
- 63.Ma K, Marti-Gutierrez N, Park SW, et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017;548:413–419. [DOI] [PubMed] [Google Scholar]