

ABSTRACT
Mitophagy is the process by which cells eliminate damaged or superfluous mitochondria by degrading them within lysosomes. We show that during development, selective removal of mitochondria by autophagy induces a metabolic shift toward glycolysis that is essential for differentiation of several cell types. These findings suggest potential applications of cell-fate manipulation strategies targeting mitophagy.
Keywords: Autophagy, metabolic reprogramming, hypoxia, BNiP3L
Metabolic reprogramming is a hallmark of cancer cells and, together with gene mutations, contributes to the initiation of tumorigenesis. In most cells, cell functions are sustained using energy derived from glycolysis coupled to oxidative phosphorylation. However, in cancer cells glucose transport is upregulated and glycolysis dramatically increased, even in the presence of oxygen, resulting in aerobic glycolysis (also known as the Warburg effect). This allows rapid production of ATP and the generation of glycolytic intermediates that are used to synthesize amino acids for new proteins, fatty acids for lipids, and other building blocks that sustain continuous growth.1 Tumor cells must also adjust their metabolism to different oxygen supply rates, which turn depend on the vascularization of the tumor and the distance of the cells from blood vessels. In conditions of hypoxia, oxidative phosphorylation is shut down and cells must rely in glycolysis as the sole means of generating ATP. In these conditions the transcription factor HIF1A/HIF-1 acts as a master regulator, orchestrating the cell's response to the lack of oxygen.2 In addition to hypoxic tumors, other areas such as stem cell niches and inflamed tissues also have low oxygen tension, and thus must generate ATP via anaerobic glycolysis. Metabolic pathways may be controlled by the same signals that control cell differentiation, which involves significant alterations in cell components and profound metabolic changes. While differentiated cells, such as muscle cells, adipocytes, and neurons, primarily rely on oxidative phosphorylation to generate ATP, embryonic stem cells and induced pluripotent stem cells rely more on glycolytic activity.3
Autophagy is a degradative pathway that mediates the degradation and recycling of cellular components inside lysosomes. These components can include whole organelles, such as mitochondria, in which case the process is known as mitophagy. Mitophagy is the only known pathway that can eliminate entire mitochondria, and defects in this process are implicated in diseases (e.g., neurodegenerative conditions) and in aging. Relatively few in vivo mitophagy studies have been performed due to a paucity of quantitative methods to assess mitophagy flux (the complete degradation of mitochondria inside lysosomes after their engulfment in autophagosomes). We recently developed a new method to quantify mitophagy flux in cells and tissues.4
The molecular mechanisms regulating mitophagy constitute an area of intense research interest. The most studied pathway is the PINK1/PRKN pathway, which is activated by stressors (e.g., after mitochondrial uncoupling in vitro), trigger ubiquitin-dependent autophagosome formation. Other mitophagy regulators such as NIX/BNIP3L and BNIP3 can also induce mitophagy by directly binding to the autophagosomal protein LC3, triggering engulfment of mitochondria by autophagosomes. While Pink1 and Prkn-deficient mice display a limited phenotype, Bnip3L/Nix-deficient mice develop anaemia due to alterations in red blood cell formation. During development reticulocytes eliminate all their mitochondria, a process that is altered in NIX-deficient animals.5 However, the role of Nix in vivo in other tissues remains unclear.
Because mitophagy is the only way to eliminate whole mitochondria, and since these organelles play a central role in controlling cellular metabolism, we postulated that mitophagy may contribute to changes in cell metabolism. We first demonstrated when mitotic arrest is induced in cancer cells using microtubule poisons, mitophagy is upregulated to induce a metabolic switch toward glycolysis, acting as a prosurvival mechanism.6,7 Taking these investigations one step further, we now demonstrate that this hypothesis also holds true in 2 physiologic settings: embryonic neural development and macrophage polarization.8
Our data demonstrate alterations in the nervous system and in tissue-resident macrophages in normal conditions in mitophagy-incompetent (Bnip3l/Nix- and Atg5-deficient) mice. Specifically, we demonstrate that during embryonic development, Nix-dependent mitophagy results in a metabolic shift toward glycolysis that controls neuronal differentiation (Fig. 1). This response is triggered by tissue hypoxia, which results in HIF-1 stabilization and increased mRNA expression of Nix and glycolytic enzymes, all of which are known HIF-1 target genes. Furthermore, our data suggest that mitophagy acts as the key regulator, since mitophagy-deficient retinas exhibit reduced expression of glycolytic enzymes, indicating that mitophagy is upstream of glycolysis. Interestingly, we show that neuronal differentiation is enhanced when glycolysis is forced by blocking pyruvate entry into the mitochondria and when autophagy is induced by mTOR inhibition with rapamycin. Conversely, in Atg5-deficient animals mitochondrial number is increased, while glycolysis and neuronal number are decreased. Together these data describe a physiologic pathway in which mitophagy degrades mitochondria and induces metabolic reprogramming and cell differentiation during embryonic neural development.
Figure 1.
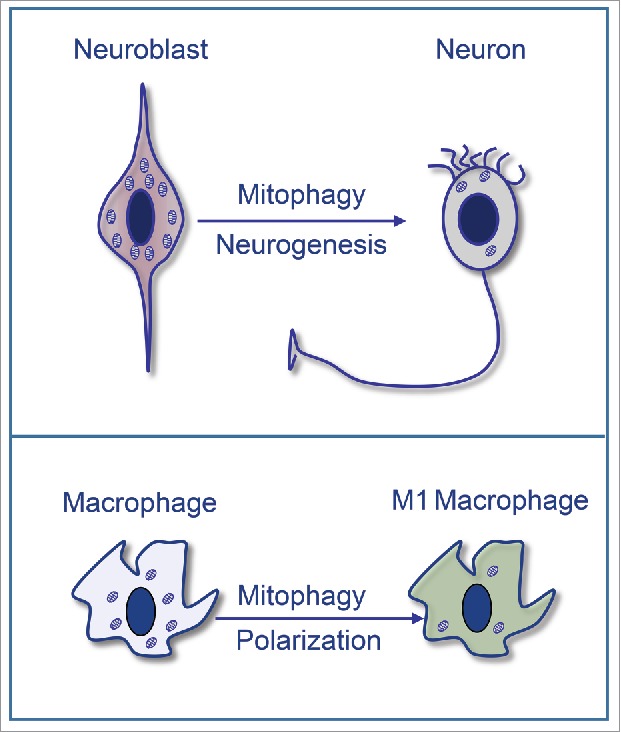
Programmed mitophagy regulates neurogenesis and macrophage polarization: Mitophagy triggers a metabolic shift toward glycolysis that regulates the differentiation of retinal ganglion cells in the embryonic mouse retina. In addition, mitophagy regulates glycolysis to sustain the proinflammatory response during M1 macrophage polarization.
Another metabolic reprogramming scenario is the polarization of macrophages to the proinflammatory M1 phenotype. These macrophages also use energy derived from glycolysis to enable a rapid immune response.9 We show that peritoneal macrophages also require mitophagy to modulate glycolysis and generate these proinflammatory responses. In Nix-deficient macrophages, as well as normal macrophages subjected to autophagy or mitophagy inhibition, glycolysis is attenuated and the expression of inflammatory regulators such as Tnf, Il1b, and Nos2 is reduced (Fig. 1). The requirement of mitophagy for metabolic reprogramming coupled with cell differentiation therefore appears to be a universal phenomenon, as suggested by its occurrence in 2 very different cell types (macrophages and neurons).
The ubiquity of mitochondrial dysfunction in neurodegenerative and aging-associated diseases suggests that new therapies targeting mitophagy and metabolic reprogramming could hold significant potential. Moreover, our data support the view that metabolic interventions targeting mitophagy could also be used to attenuate inflammatory responses in diseases such as diabetes and atherosclerosis, in which aberrant and chronic inflammation exacerbates the disease condition. Finally, our data suggest potential applications of cell-fate manipulation strategies that target mitophagy. This metabolic role of autophagy and mitophagy could be targeted to potentiate the generation of retinal ganglion cells from stem cells, with possible applications for the treatment of diseases such as glaucoma, which is characterized by the loss of ganglion cells and their axons. Indeed, one of the first symptoms of some mitochondrial diseases is the loss of vision associated with alterations in ganglion cells, which are highly dependent on proper mitochondrial function. The molecular mechanisms associated with the death of retinal ganglion cells, remain largely unknown. However, strategies that improve mitochondrial quality through the modulation of mitophagy could be used to develop new therapies for this disease. In conclusion, our data point to mitophagy as a central modulator of metabolism and cell fate, and a key determinant of neuron differentiation and macrophage polarization.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Research in the P.B. laboratory is supported by grants BFU2015–65623 and BFU2015–71869-REDT from Spain's Ministerio de Economía y Competitividad, COST Action Transautophagy (CA15138), and ITN MSCA-ITN-ETN 765912.
References
- 1.Chen X, Qian Y, Wu S. The warburg effect: Evolving interpretations of an established concept. Free Radic Biol Med 2015; 79:253-63; PMID:25277420; https://doi.org/ 10.1016/j.freeradbiomed.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012; 148:399-408; PMID:22304911; https://doi.org/ 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development 2013; 140:2535-47; PMID:23715547; https://doi.org/ 10.1242/dev.091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mauro-Lizcano M, Esteban-Martinez L, Seco E, Serrano-Puebla A, Garcia-Ledo L, Figueiredo-Pereira C, Vieira HL, Boya P. New method to assess mitophagy flux by flow cytometry. Autophagy 2015; 11:833-43; PMID:25945953; https://doi.org/ 10.1080/15548627.2015.1034403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ney PA. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta 2015; 1853:2775-83; PMID:25753537; https://doi.org/ 10.1016/j.bbamcr.2015.02.022 [DOI] [PubMed] [Google Scholar]
- 6.Domenech E, Maestre C, Esteban-Martinez L, Partida D, Pascual R, Fernandez-Miranda G, Seco E, Campos-Olivas R, Pérez M, Megias D, et al.. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol 2015; 17:1304-16; PMID:26322680; https://doi.org/ 10.1038/ncb3231 [DOI] [PubMed] [Google Scholar]
- 7.Esteban-Martinez L, Domenech E, Boya P, Salazar-Roa M, Malumbres M. Mitophagy in mitosis: More than a myth. Autophagy 2015; 11:2379-80; PMID:26565111; https://doi.org/ 10.1080/15548627.2015.1108509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esteban-Martinez L, Sierra-Filardi E, McGreal RS, Salazar-Roa M, Marino G, Seco E, Durand S, Enot D, Graña O, Malumbres M, et al.. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. Embo J 2017; 36:1688-760; PMID:28465321; https://doi.org/ 10.15252/embj.201695916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van den Bossche J, O'Neill LA, Menon D. Macrophage Immunometabolism: Where are we (Going)? Trends Immunol 2017; 38:395-406; PMID:28396078; https://doi.org/ 10.1016/j.it.2017.03.001 [DOI] [PubMed] [Google Scholar]