

ABSTRACT
The endoplasmic reticulum (ER) stress sensor protein kinase RNA-like endoplasmic reticulum kinase (PERK) plays a major role during the unfolded protein response (UPR), mainly through eIF2α phosphorylation. We uncovered that PERK, by interacting with Filamin A, elicits F-actin remodeling required for ER-plasma membrane contact site formation after ER-Ca2+ depletion, through a UPR-independent mechanism.
KEYWORDS: Actin-cytoskeleton, calcium flux, contact sites, endoplasmic reticulum stress, Filamin A, PERK, STIM1, store operated calcium entry, unfolded protein response
Endoplasmic reticulum (ER) stress is caused by an imbalance in the ER protein folding machinery leading to a luminal increase in unfolded proteins, as the ER can no longer cope with protein folding demand. ER stress, which underlies pathologies like cancer, diabetes and neurodegeneration and is triggered by various intrinsic and extrinsic insults, is sensed by a dedicated signaling machinery, called the unfolded protein response (UPR). The UPR aims to restore ER protein-folding capacity, or when this initial attempt fails, drives cell death.1 UPR engages 3 main pathways activated by the Inositol Requiring Enzyme 1 (IRE1), activating transcription factor 6 (ATF6) and the protein kinase RNA-like endoplasmic reticulum kinase (PERK). The pro-survival role of the UPR entails PERK-mediated translational arrest through eukaryotic Initiation Factor 2 α (eIF2α) phosphorylation,2,3 and the activation of a transcriptional program regulated by 3 main transcription factors, namely ATF6, X-box binding protein 1 (XBP1) and ATF4.1
Recently, our understanding of these ER stress sensors in cellular homeostasis has greatly advanced, mainly through the characterization of the UPRosome.4 Most notably, IRE1α, which harbours kinase and endoribonuclease activities, was found to further fine-tune the UPR through its scaffolding and regulated Ire1-dependent decay (RIDD) functions.5 However, whether other ER stress sensors like PERK could have additional roles remains unclear. We previously unravelled that PERK has a tethering role at the ER-mitochondria juxtapositions,6 regulating cell death, inflammation and autophagy. Also PERK coordinates ER stress pathways leading to immunogenic cancer cell death, through mechanisms for which eIF2α phosphorylation could be dispensable, but which required Ca2+ signals and actin-cytoskeleton mediated exocytosis of danger signals.4 Moreover, PERK regulates intracellular Ca2+ fluxes and store operated calcium entry (SOCE),7,8 the main process used to replenish ER-Ca2+ levels upon ER-Ca2+ depletion, but the mechanism remained unknown.
Thus, we set out to find novel PERK interactors that could explain these findings. We used proximity-dependent biotin identification (BioID), and identified Filamin A (FLNA) as a PERK interactor. FLNA is a major orchestrator of the Filamentous-Actin (F-Actin) cytoskeleton, and forms crosslinked, orthogonal networks of F-Actin fibers. We mapped the binding of PERK to a small C-terminal stretch of 3 β-barrel domains on FLNA. In unstressed cells PERK deficiency caused a cortical relocation and increased polymerization of F-Actin, which reduced cell migration and focal adhesions, highlighting the link between PERK and FLNA. We then asked whether this PERK-mediated phenotype was relevant for Ca2+ signaling. We postulated that an increased presence of cortical F-Actin underneath the plasma membrane (PM) could have an effect on the formation of ER-PM contact sites triggered by ER-Ca2+ store depletion. Total internal reflection fluorescence (TIRF) microscopic experiments and electron microscopic analyses confirmed that the increase in cortical F-Actin caused by PERK ablation could prevent ER tubules from making contact with the PM.
In store-depleted cells, the ER membrane resident protein Stromal interaction molecule 1 (STIM1) homo-oligomerizes and translocates to the PM where it interacts with the Ca2+ channel Orai1 to induce SOCE.9 This interaction will cause Ca2+ to enter the cell and is one of the most studied biologic processes known to be regulated by ER-PM contacts.
PERK deficiency strongly blunted the re-localization of STIM1 to the PM. Re-expression of a kinase dead PERK significantly restored STIM1-mediated ER-PM contacts, indicating that PERK's regulation of ER-PM appositions is independent of its canonical role in the UPR.
To investigate if this PERK-mediated phenotype was restricted to STIM1-driven ER-PM contacts we studied another ER-PM tethering protein; Extended Synaptotagmin-1 (E-Syt1). E-Syt1 is an ER-transmembrane protein recently found to elicits ER-PM contacts after cytosolic Ca2+ elevations sensed by its Ca2+ sensitive C2 domains.10 In analogy with STIM1, PERK deficiency also abrogated E-Syt1 PM translocation upon ER-Ca2+ depletion and this phenotype was again rescued by the re-expression of kinase dead PERK. The formation of STIM1 and E-Syt1 mediated ER-PM contact sites required an intact PERK-FLNA axis and involved a tight spatial organization of ER-PM juxtapositions assisted by PM-localized F-Actin. Interestingly, this tight association was largely lost in PERK deficient cells. Furthermore, polymerizing or depolymerizing actin could inhibit and restore ER-PM contact site formation after ER-Ca2+ depletion, respectively.
But how does PERK link ER-Ca2+ depletion to ER-PM contact site formation? We found that after ER-Ca2+ depletion PERK rapidly dimerizes and autophosphorylates independently of its ER stress sensing luminal domain. Furthermore, after ER-Ca2+ depletion, the interaction between PERK and FLNA increased.
Ca2+ induced PERK dimerization stabilized the PERK-FLNA interaction, leading to F-Actin remodelling and ER-PM contact site formation. Artificial triggering of PERK dimerization, with the commercial GSK inhibitor GSK2606414,11 increased PERK-FLNA interaction and led to increased PM recruitment of STIM1, indicating that PERK dimerization itself –but not PERK activity- is a major event leading up to FLNA assisted ER-PM contact site formation (Fig. 1).
Figure 1.
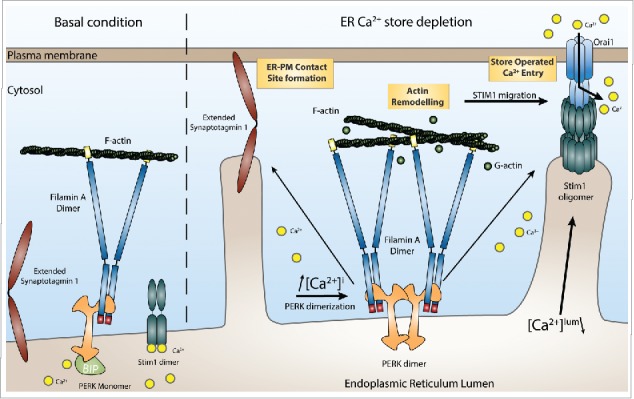
Functional consequences of the interaction between PERK and FLNA on ER-PM contact site formation. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) interacts with Filamin A (FLNA) in resting conditions. After endoplasmic reticulum (ER)-Ca2+ store depletion and subsequent cytosolic Ca2+ elevation, the cytosolic domain of PERK dimerizes. This dimerization event strengthens the interaction of PERK with FLNA. This leads to actin rearrangement and altered actin polymerization dynamics, allowing efficient Stromal interaction molecule 1 (STIM1) and Extended Synaptotagmin-1 (E-Syt1) translocation to the plasma membrane (PM) and the formation of ER-PM contacts.
Research into the UPR, and specifically on how to harness the UPR in the context of disease (cancer, cancer therapy, neurodegeneration) have focused only on the previously known role of PERK. This study shows that PERK has broader roles beyond the traditional UPR and regulates the functional connection between the ER and other crucial cellular compartments, like the PM. Since the dynamic regulation of contact sites between the ER and the PM affects critical cellular processes including but not limited to, PM signaling and lipid composition, endocytosis and exocytosis, this study may open up new and exciting avenues expanding on the role of PERK in ER stress.12
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8(7):519-29; PMID:17565364; https://doi.org/ 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 2.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397(6716):271-4; PMID:9930704; https://doi.org/ 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 3.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003; 23(20):7198-209; PMID:14517290; https://doi.org/ 10.1128/MCB.23.20.7198-7209.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, et al.. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31(5):1062-79; PMID:22252128; https://doi.org/ 10.1038/emboj.2011.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 2013; 23(11):547-55; PMID:23880584; https://doi.org/ 10.1016/j.tcb.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, et al.. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012; 19(11):1880-91; PMID:22705852; https://doi.org/ 10.1038/cdd.2012.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R, McGrath BC, Kopp RF, Roe MW, Tang X, Chen G, Cavener DR. Insulin secretion and Ca2+ dynamics in beta-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J Biol Chem 2013; 288(47):33824-36; PMID:24114838; https://doi.org/ 10.1074/jbc.M113.503664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Vliet AR, Agostinis P. When under pressure, get closer: PERKing up membrane contact sites during ER stress. Biochem Soc Trans 2016; 44(2):499-504; PMID:27068961; https://doi.org/ 10.1042/BST20150272 [DOI] [PubMed] [Google Scholar]
- 9.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005; 437(7060):902-5; PMID:16208375; https://doi.org/ 10.1038/nature04147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P. PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 2013; 153(7):1494-509; PMID:23791178; https://doi.org/ 10.1016/j.cell.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A 2001; 98(26):15294-9; PMID:11742096; https://doi.org/ 10.1073/pnas.261560998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, Rocha S, Houcine A, Derua R, Verfaillie T, et al.. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol Cell 2017; 65(5):885-99 e6; PMID:28238652; https://doi.org/ 10.1016/j.molcel.2017.01.020 [DOI] [PubMed] [Google Scholar]