

Abstract
The early oligomerization of amyloid β-protein (Aβ) is a crucial step in the etiology of Alzheimer’s disease (AD), in which soluble and highly neurotoxic oligomers are produced and accumulated inside neurons. In search of therapeutic solutions for AD treatment and prevention, potent inhibitors that remodel Aβ assembly and prevent neurotoxic oligomer formation offer a promising approach. In particular, several polyphenolic compounds have shown anti-aggregation properties and good efficacy on inhibiting oligomeric amyloid formation. 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose is a large polyphenol that has been shown to be effective at inhibiting aggregation of full-length Aβ1-40 and Aβ1-42, but has the opposite effect on the C-terminal fragment Aβ25-35. Here, we use a combination of ion mobility coupled to mass spectrometry (IMS-MS), transmission electron microscopy (TEM) and molecular dynamics (MD) simulations to elucidate the inhibitory effect of PGG on aggregation of full-length Aβ1-40 and Aβ1-42. We show that PGG interacts strongly with these two peptides, especially in their N-terminal metal binding regions, and suppresses the formation of Aβ1-40 tetramer and Aβ1-42 dodecamer. By exploring multiple facets of polyphenol-amyloid interactions, we provide a molecular basis for the opposing effects of PGG on full-length Aβ and its C-terminal fragments.
Keywords: Alzheimer’s disease; amyloid β-protein; polyphenol; 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose; ion-mobility mass spectrometry; computational modeling
Graphical abstract
INTRODUCTION
Extracellular deposits of amyloid β-protein (Aβ) in the brain are a pathological hallmark of Alzheimer’s disease (AD), the most common type of dementia, which is characterized by neuronal cell loss leading to cognitive impairment.1–8 Aβ proteins are derived from sequential endoproteolytic cleavage of the amyloid precursor protein (APP) by β- and γ-secretase9–12 to generate alloforms of 37–43 amino acid. Aβ1-40 and Aβ1-42 (Scheme 1) are the dominant forms and the main targets in the amyloid cascade hypothesis. Aβ1-40 is the main constituent of all Aβ species present in the body (~90%), while Aβ1-42 (9%) is the more toxic form and has a higher aggregation propensity.13, 14 Compelling evidence has shown that the early, soluble amyloid oligomers are the primary pathologic agents in AD. Some of these amyloid oligomers are highly neurotoxic and accumulate inside neurons, causing a decline in synaptic functions.1, 15–21
Scheme 1.

(A) Primary sequence of Aβ1-40 and Aβ1-42 and three dimensional structure of Aβ1-42 (B) chemical structure of 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose (PGG).
Soluble oligomers are produced and accumulated during the conversion of soluble, non-toxic Aβ monomers to benign fibrils.22 They exist in equilibrium with other species, and may have relative short lifetimes; as a result, they are difficult to isolate and characterize. Of note, previous studies by IMS-MS show that Aβ1-40 and Aβ1-42 follow different pathways en route to fibrils: Aβ1-40 initially forms dimers and tetramers, whereas Aβ1-42 forms hexamers and dodecamers.9, 13 Among these oligomeric species, appearance of the dodecamer (56 kDa) has been linked to cognitive decline in both human AD brains and transgenic mice.23, 24 However, the etiology of AD is presently not well understood and no effective treatment is available. Current approved drugs for AD show only meager efficacy at mitigating disease progression.
An attractive therapeutic approach for AD treatment is to remodel the Aβ assembly pathway in a way that attenuates the neurotoxicity of the transient, early-stage soluble Aβ oligomers.9, 13, 25–28 In this context, many polyphenolic compounds have shown promise as potential therapeutic agents for AD treatment and prevention.29–32 This class of compounds is plentiful in nature; many can be extracted from plants and herbs.32, 33 More specifically, 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose (PGG) (Scheme 1), a large tannin-type polyphenol found in the traditional medicinal herb Paeonia suffruticosa, has been shown to be potent inhibitor for both Aβ1-40 and Aβ1-42 aggregation.34 Paradoxically, this same polyphenol has been recently shown to be an aggregation agonist for Aβ25-35, a cytotoxic fragment of Aβ, by promoting the formation of extended Aβ25-35 conformations.35 This discrepancy may be due to a mismatch between the size and shape of PGG and its amyloid targets; however, supporting evidence for this model remains inadequate.
More recently, the variation between different polyphenols in their inhibitory effects on Aβ oligomerization have been correlated to differences in polyphenol size and shape. For large polyphenolic compounds, hydrophobic interactions with Aβ were stronger than hydrophilic ones. This difference was attributed to incompatibility between the geometry of large polyphenols and intramolecular hydrogen bonds.32 In contrast, for small polyphenolic compounds both polar and nonpolar interactions were equally involved in the Aβ binding interfaces.32 Therefore, it is reasonable to propose that large polyphenols (e.g., PGG) are more selective for their targets, and that the inhibitory mechanisms of large polyphenols cannot be extrapolated from previous studies on small polyphenols.
Herein we utilize ion mobility spectrometry-mass spectrometry (IMS-MS),36, 37 transmission electron microscopy (TEM) and molecular dynamics (MD) simulations to characterize the Aβ self-assembly pathways and to determine the structural changes within amyloid systems promoted by PGG. We investigate the effects and binding motifs of this ligand with full-length Aβ1-40 and Aβ1-42, focusing on early homo- and hetero-oligomer (Aβ:PGG) formation and structure. Additionally, we evaluate the interactions of PGG with Aβ1-11 and Aβ11-22, which taken together with published data on Aβ25-35,35 provide insight into the molecular mechanism by which large polyphenols inhibit amyloid aggregation.
RESULTS and DISCUSSION
Self-assembly of Aβ1-40 and Aβ1-42 by IMS-MS and TEM
The fundamental challenge in studies of transient, early-stage soluble Aβ oligomers is that the conformational transitions are not easily accessible by traditional bulk measurements. In contrast, IMS-MS has been demonstrated to successfully capture transient structural changes accompanying Aβ self-assembly38–40, as well as to successfully evaluate the efficacy of small-molecule inhibitors of amyloid assembly processes.9, 13, 31, 35, 41–44
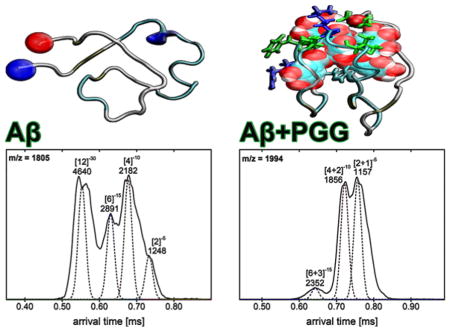
IMS-MS experiments with full-length Aβ1-40 and Aβ1-42 were performed in negative mode polarity because the natural charge states of those species are z = −3 (at pH 7). The nano-ESI-Q mass spectrum of pure Aβ1-40 (Figure 1A) reveals the presence of two major peaks at m/z 1081 and 1442, corresponding to Aβ monomers: [n]z = [1]−4 and [1]−3, respectively, where n is the oligomer number and z the charge. The two features in the ATDs at m/z 1731 are assigned to the Aβ dimer ([n]z = [2]−5) and tetramer ([n]z = [4]−10) (see Figure S2, Supporting Information). Similarly, the mass spectrum obtained for pure Aβ1-42 (Figure 1C) shows three peaks. The most abundant peaks are at m/z 1504 and m/z 1128 corresponding to the monomers [n]z = [1]−3 and [n]z = [1]−4, respectively. The ATDs obtained for the peak at m/z 1805 (shown in Figure S5, Supporting Information) show four different oligomeric species of Aβ1-42: dimer ([n]z = [2]−5), tetramer ([n]z = [4]−10), hexamer ([n]z = [6]−15) and dodecamer ([n]z = [12]−30), as previously reported.38, 40
Figure 1.

nano-ESI-Q mass spectra of (A) Aβ1-40, (B) Aβ1-40 incubated with PGG at 1:1 molar ratio, (C) Aβ1-42 and (D) Aβ1-42 incubated with PGG at 1:1 molar ratio. The Aβ concentrations were 10 μM in ammonium acetate buffer (10 mM, pH 7.4). The peaks are annotated as [n]z or [n + k]z where n is the number of Aβ molecules, k is the number of bound PGG molecules and z is the charge. Samples were immediately analyzed after the solution preparation.
To analyze the transient soluble Aβ oligomers, the experimental collisional cross sections (CCSs), obtained by measuring ATDs at different pressure/voltage (P/V) ratios (Equations S3 and S4), were compared to the ideal isotropic growth model, which approximates the cross sections (σ) of oligomers that grow equally distributed in all spatial dimensions (σn = σ1 × n2/3).45 From the CCSs obtained for Aβ1-40 and Aβ1-42 (Figure 2, panels A and D, and Supporting Information, Figures S2 and S5, Tables S1 and S3), positive deviations from the ideal isotropic model emerge at dimer indicating non-spherical growth consistent with previous results38, 40 that indicated the Aβ1-40 tetramer was near square planar and the Aβ1-42 hexamer was cyclic and planar and the dodecamer two stacked hexamers.
Figure 2.

Oligomer growth curves of (A) Aβ1-40 homo-oligomers, (B, C) Aβ1-40 homo- and hetero-oligomers, respectively, formed in the 1:1 mixture of Aβ1-40 and PGG, (D) Aβ1-42 homo-oligomers, (E, F) Aβ1-42 homo- and hetero-oligomers formed in the 1:1 mixture of Aβ1-42 and PGG. The cross section data are colored according to the intensities of the corresponding mass spectral peaks (darker or lighter colors indicate more or less intense features, respectively).
TEM images (Figure 3, panels A and D) obtained for aliquots taken from the same Aβ1-40 and Aβ1-42 samples used in IMS-MS experiments (10 μM in 10 mM ammonium acetate buffer, pH = 7.4) show more abundant fibrils for Aβ1-42 than for Aβ1-40. Fibrils of both peptides have a typical amyloid morphology (~10 nm in width). These results confirm that both systems eventually form fibrils, as previously observed46 indicating typical β-sheet structure.47
Figure 3.

Representative TEM images of (A) Aβ1-40 alone, (B, C) Aβ1-40 incubated with PGG at 1:1 and 1:10, (D) Aβ1-42 alone, (E, F) Aβ1-42 incubated with PGG at 1:1 and 1:10 molar ratios. The two scale bars shown in panel D apply to all panels. Samples were incubated for two days before the analyses.
1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose attenuates fibril formation by remodeling the early Aβ1-40 and Aβ1-42 oligomerization and promoting hereto-oligomer formation
The mass spectrum of Aβ1-40 incubated with PGG at 1:1 molar ratio (Figure 1B) indicates that the ligand remodels Aβ assembly by modulating the early oligomer distribution and suppressing the formation of higher order oligomers. Of note, the peak at m/z 1731 corresponding to homo-Aβ dimer and tetramer is completely depleted. The mass spectral peaks at m/z 1081 and m/z 1442 are identified as Aβ1-40 monomers with [n]z = [1]−4 and [1]−3, respectively. Four new mass spectral peaks are observed and attributed to Aβ1-40:PGG hetero complexes, which are annotated as [n + k]z where k is the number of bound PGG molecules. These peaks are assigned as the quadruply charged monomer with one and two PGG molecules attached, the triply charged monomer with one PPG attached and the nominal quintuply charged dimer with one PPG attached. The ATD of the nominal dimer (see Figure S3 and Table S2, Supporting Information) indicates both an Aβ dimer and an Aβ tetramer complexed with one and two PGG molecules, respectively (Figure 4B). In terms of cross sections, the bound complexes are significantly more compact than the corresponding homo-Aβ1-40 dimer and tetramer (Figure 2 and Figure 4, panels A and B). The relative intensities of those two features are reversed compared to the pure peptide, indicating that the dimer is favored over the tetramer with PGG attached.
Figure 4.

Arrival time distributions (ATDs) of (A) Aβ1-40 peak (m/z = 1731, [n]z = [2]−5) for pure Aβ1-40, (B) Aβ1-40 and PGG hetero complex (m/z = 1920, [n + k]z = [2+1]−5) in the 1:1 mixture, (C) Aβ1-42 peak (m/z = 1805, [n]z = [2]−5) for pure Aβ1-42, (D) Aβ1-42 and PGG hetero complex (m/z = 1994, [n + k]z = [2+1]−5) in the 1:1 mixture.
At 1:10 Aβ1-40:PGG molar ratio (Figure S1, Supporting Information), the population of Aβ1-40:PGG hetero-oligomers increases, indicating that PGG competitively binds to Aβ1-40 and alters the Aβ1-40 assembly pathway. The CCSs obtained for Aβ1-40 homo- and hetero-oligomers (Figure 2, panels B and C, and Supporting information, Figure S3 and Table S2) correlate with isotropic growth and suggest the formation of globular aggregates in place of fibrils. These results are in a good agreement with TEM data (Figure 3, panels B and C) which show that the presence of PGG reduces the abundance of fibrillar aggregates compared to Aβ1-40 alone (Figure 3A). Moreover, globular aggregates become more abundant when PGG is in excess (e.g., at 1:10 Aβ1-40:PGG molar ratio).
Similar to Aβ1-40, the mass spectra of Aβ1-42 incubated with PGG at 1:1 and 1:10 Aβ1-42:PGG molar ratios (Figure 1D, and Supporting Information, Figure S4) show a decline in large Aβ homo-oligomer formation and an increase in Aβ1-42:PGG hetero complexes. The ATD of Aβ1-42:PGG nominal quintuply charged dimer peak at m/z 1994 (Figure 4D) shows dominant dimer and tetramer peaks and a weak hexamer peak in strong contrast to the corresponding ATD of pure Aβ1-42 where strong hexamer and dodecamer peaks are present (Figure 4C). Hence, PGG suppresses the formation of high-order Aβ1-42 homo- and hetero-oligomers. In the presence of PGG, the CCSs data of Aβ1-42 homo- and hetero-oligomers (Figure 2, panels E and F) are consistent with isotropic growth. TEM data (Figure 3, panels E and F) are consistent with the IMS-MS results and show an almost complete elimination of fibril formation and significant formation of globular aggregates.
We performed two additional experiments to examine the effect of PGG on preformed Aβ1-42 fibrils. In the first experiment Aβ1-42 was incubated for ~4 h on ice, followed by the addition of PGG at a 2:1 Aβ1-42:PGG molar ratio. The ATD corresponding to Aβ1-42 homo-oligomers at 1805 m/z was monitored. At t = 30 minutes after mixing only three features were observed corresponding to dimer, tetramer and hexamer but the dodecamer is completely gone (Figure 5, panels A and B). Further, at 6 h after PGG addition a significant decrease in the hexamer population is also observed (Figure 5C) indicating PPG remodels both dodecamer and hexamer formation. In a second experiment, PGG was added to a three-days incubated Aβ1-42 sample at molar ratios of 1:1 and 1:10 Aβ1-42:PGG. The samples were then incubated for two additional days. The TEM data (Figure 5, panels D-F) show that the addition of PGG to the aged Aβ1-42 sample reduces the population of mature amyloid fibrils. Even though fibrils remain in the final samples, they appear less clumped together than in the absence of PGG, and are replaced by globular aggregates in a concentration-dependent manner.
Figure 5.

PGG disassembles preformed Aβ1-42 aggregates. Arrival time distribution (ATD) of [n]z = [2]−5 Aβ1-42 peak (m/z = 1805) for (A) Aβ1-42 incubated alone for ~4 h, and (B, C) 30 min and 6 h, respectively, after the addition of PGG at a 2:1 Aβ1-42:PGG molar ratio. Representative TEM images of (D) Aβ1-42 incubated alone for three days at room temperature, and (E-F) mixed with PGG at 1:1 and 1:10 Aβ1-42:PGG molar ratios, respectively, and then incubated for an additional two days.
1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose attenuates full-length Aβ amyloid formation primarily via interactions with the N-terminal metal binding and central hydrophobic core regions
In order to gain insight into the inhibitory mechanism of PGG on amyloid aggregation, we performed explicit solvent molecular dynamics (MD) simulations. We combined the molecular mechanics energies with generalized Born and surface area continuum solvation (MM-GBSA) calculations to investigate the binding motifs in a complex of a single PGG bound to an Aβ1-42 monomer and determine possible structures of the heterodimers. In the first two trajectories, the initial structures of Aβ1-42 are taken as extended conformation in order for the protein to be able to simultaneously fold and bind to PGG during the course of the simulation. The theoretical cross sections of the resulting modeled structures (σ = 840 and 884 Å2) are in good agreement with experimental values of [1+1]−4 (σ = 848 and 879 Å2, see Table S4), suggesting that the simulations provide reasonable models of heterodimers. The most stable complexes identified from the two trajectories are shown in Figure 6, panels A and B with binding energies of −41 and −44 kcal/mol, respectively.
Figure 6.

(A–D) Representative structures of Complexes 1–4 obtained from MD simulations. PGG is shown in space-filling representations. Aβ is shown in ribbon representation, and important residues interacting with PGG are shown in red: acidic, blue: basic, green: non-polar and cyan for histidine. The MM-GBSA binding energies and theoretical cross sections are also reported. (E) MM-GBSA per residue binding energy decomposition. Residues at the binding sites are annotated. The N-terminus and C-terminus are represented as blue and red beads, respectively.
Per residue binding energy decomposition (Figure 6E) shows that in Complex 1, PGG interacts with the N-terminal segment (Aβ1-11: side chains of Phe4, Asp7 and backbone atoms of Arg5), the first central hydrophobic core (Aβ12-22: Lys16, Val17 and Val18), the second hydrophobic segment (Aβ25-35: Ala30 and Ile32) and the C-terminal end (Aβ36-42: Val41 and Ala42). The C-terminal end of Aβ36-42 folds into a β-turn/hairpin.48 In Complex 2, the interactions of PGG with residues in the N-terminal Aβ1-11 are largely preserved, but Asp7 is replaced by His6. Although the interactions with the central hydrophobic core are weakened, the C-terminal end (Aβ36-42) adopts an extended conformation, preserving the strong PGG binding. In fact, MM-GBSA calculations reveal that the binding energy of Complex 2 is slightly stronger than that of Complex 1 (−44 vs. −41 kcal/mol, respectively). We performed two additional simulations starting with the most populated structures from Sgourakis et al.49 These simulations result in complexes where the ligand is strongly bound the N-terminal region of Aβ1-42 (Figure 6, panels C and D) with weaker binding energies (−14 and −13 kcal/mol). These simulations further highlight the importance of the N-terminal region of Aβ1-42 as a binding site of PGG.
The interactions between PGG and Aβ1-42 are not limited to hydrophobic/polar interactions, as acidic and basic residues also make substantial contributions. The contribution from residue side chains is equally as important as that from backbone atoms. Therefore, while polyphenols may complex non-specifically with a wide range of amyloid peptides,29, 30, 50 the binding of PGG to Aβ1-42 (and Aβ1-40) is primarily driven by interactions with the N-terminal metal binding and first central hydrophobic core regions. For that, it is first required that the full length Aβ adopts a folded conformation. However, the direct comparison between the binding energies derived from MD simulations obtained for complexes of truncated Aβ1-11 and Aβ11-22 peptides and PGG may not be the best parameter to evaluate the binding motifs of the system, since the Aβ fragments may not adopt the same conformations as they would adopt in full length Aβ. In order to verify the importance of these PGG-binding motifs experimentally, we directly assayed the effects of PGG on the assembly of the fragments Aβ1-11 and Aβ11-22.
Aβ1-11
The natural charge states of Aβ1-11 can be −2 to −3 in water, depending upon the protonation states of His6, which residue is more likely to be positive than neutral; hence the IMS-MS data were collected in negative mode polarity. The mass spectrum of Aβ1-11 (Figure 7A) shows three major peaks. The first mass spectral peak, at m/z 663, corresponds to a doubly charged Aβ1-11 monomer ([n]z = [1]−2), whereas the ATD and CCSs (Figure S8 and Table S5, Supporting Information) obtained for the latter peak, at m/z 1325, reveal the presence of four different species, Aβ monomer ([n]z = [1]−1), dimer ([n]z = [2]−2), trimer ([n]z = [3]−3) and tetramer ([n]z = [4]−4). The less intense peak at m/z 883 is assigned to a triply charged Aβ dimer ([n]z = [2]−3). The CCS data (Figure 8A) suggest that the Aβ1-11 oligomers in general adopt relatively globular conformations. The exception is the Aβ1-11 dimer that adopts two distinct conformations, one compact (σ = 414 Å2) and one extended (σ = 598 Å2). TEM imaging of the same sample used in IMS-MS analysis support these results, showing very few, amorphous aggregates (Figure 9A).
Figure 7.

nano-ESI-Q mass spectra of (A) Aβ1-11, (B) Aβ1-11 incubated with PGG at 1:1 molar ratio, (C) Aβ11-22, and (D) Aβ11-22 incubated with PGG at 1:1 molar ratio. Aβ concentrations were 100 μM in water. The peaks are annotated as [n]z or [n + k]z where n is the number of Aβ molecules, k is the number of bound PGG molecules and z is the charge. The asterisk denotes mass spectral peaks of impurities. Samples were incubated for one week before the analyses.
Figure 8.

Oligomer growth curve of (A) Aβ1-11 homo-oligomers, (B, C) Aβ1-11 homo- and hetero-oligomers, respectively, formed in the 1:1 mixture of Aβ1-11 and PGG, (D) Aβ11-22 homo-oligomers, (E, F) Aβ11-22 homo- and hetero-oligomers, respectively, formed in the 1:1 mixture of Aβ11-22 and PGG. The cross section data are colored according to the intensities of the respective peaks (darker and lighter colors indicate more and less intense features, respectively).
Figure 9.

Representative TEM images of (A) Aβ1-11 alone, (B, C) Aβ1-11 incubated with PGG at 1:1 and 1:10, (D) Aβ11-22 alone, (E, F) Aβ11-22 incubated with PGG at 1:1 and 1:10 molar ratios. The scale bar for all panels is given in panel D. Samples were incubated for one week before the analyses.
In contrast, the mass spectrum obtained in the presence of an equimolar ratio of PGG (Figure 7B) displays new mass spectral peaks at high m/z, which are attributed to multiple complexes formed between Aβ1-11 and PGG. Further analyses on these mass spectral peaks reveal high-order hetero-oligomers up to nonamer (e.g., [n + k]z = [9+5]−10 and [9+4]−9). The high-order homo-Aβ1-11 oligomers decrease in population due to the competitive binding of PGG, but are not entirely depleted. Further, no additional peaks are found in the mass spectrum collected for the Aβ1-11:PGG mixture at 1:10 molar ratio (Figure S7, Supporting Information), but the mass spectral peaks of Aβ1-11:PGG complexes become more intense. The IMS-MS data suggest that multiple PGG molecules competitively bind to Aβ1-11 oligomers and induce compact conformations, as evidenced by the CCSs of both Aβ1-11 homo- and hetero-oligomers (Figure 8, panels B and C, and Supporting Information, Figure S9 and Table S6), which correlate with an isotropic growth curve. TEM imaging (Figure 9, panels B and C) is again consistent with the IMS-MS observations, revealing a PGG concentration-dependent increase in the abundance of the globular aggregates.
Aβ11-22
The natural charge states of Aβ11-22 can be +1 to −1 in water, taking in account the protonation states of His13 and His14. Because it is more likely to be positive than neutral in water, the IMS-MS experiments with this species were performed in positive mode polarity. The mass spectrum of pure Aβ11-22 (Figure 7C) shows intense mass spectral peaks at m/z 495 ([n]z = [1]+3), 742 ([n]z = [1]+2) and 1484 ([n]z = [1]+1, [2]+2 and [3]+3). The minor peaks are at m/z 989 ([n]z = [2]+3), 1113 ([n]z = [3]+4) and 1187 ([n]z = [4]+5). No higher order oligomers are detected, which is intriguing given that this fragment contains the central hydrophobic core segment 16KLVFFAE22, which played a key role in the original model of Aβ aggregation.15, 51–53 The CCS data for Aβ11-22 homo-oligomers (Figure 8D) correlate with an isotropic growth curve, with the exception of dimer and minor deviations for trimer and tetramer. These observations are consistent with TEM imaging (Figure 9D), which shows that PGG increases the formation of amorphous aggregates in a concentration-dependent manner. The presence of PGG promotes Aβ11-22 hetero-oligomer formation with complexes observed in the 1:1 mixture up to the nonomer. At 1:10 molar ratio (Figure S10, Supporting Information), the peaks of Aβ hetero-oligomers become more intense. Taken together, the CCS data of both Aβ11-22 homo- and hetero-oligomers (Figure 8, panels E and F, and Supporting Information, Figure S12 and Table S8) and the TEM imaging (Figure 9, panels E and F) indicate that the presence of PGG promotes a concentration-dependent increase in the formation of globular aggregates.
We notice that the presence of PGG promotes the formation of large hetero-oligomers of Aβ1-11 or Aβ11-22. Of note, there are no homo-oligomers of the same size n observed in the mass spectra. A possible explanation is that small PGG hetero-oligomers can self-associate into larger ones, a phenomenon that was previously observed in the study of EGCG and Aβ25-35.31 Since each PGG can form complexes with several Aβ1-11 or Aβ11-22, we postulate that the PGG molecules can associate to form a large core, from which they can complex to a higher number of Aβ1-11 or Aβ11-22 chains. Moreover, our data on these two fragments demonstrate that PGG competitively complexes with Aβ1-11 and Aβ11-22 monomers and oligomers, in agreement with the binding motifs identified in the MD simulations. These binding motifs explain the opposing effects of PGG on Aβ1-40/Aβ1-42 and Aβ25-35. The short peptide Aβ25-35 is induced by PPG to form amyloid aggregates by adopting extended conformations31, 54 while PGG suppresses fibril formation in the full-length Aβ peptides through selective interactions with specific residues in the N-terminal metal binding and first central hydrophobic core regions, sequences that are absent in Aβ25-35. Binding involves both hydrophobic and hydrophilic interactions between PGG and residue side chains and backbones. While previous studies suggest that polyphenol binding to amyloid peptides is non-selective, we show for the first time that PGG interacts specifically and strongly with the N-terminal metal binding domain of Aβ to suppress amyloid formation.
SUMMARY and CONCLUSIONS
The current study elucidates key aspects of the mechanism by which PGG inhibits amyloid formation, which can eventually be extended to other large polyphenolic compounds. The IMS-MS and MD data indicate that PGG interferes with the amyloid assembly of Aβ1-40 (Figure 1B and Figure 2, panels B and C) and Aβ1-42 (Figure 1D and Figure 2, panels E and F) by interacting with the N-terminal metal binding segment and the first central hydrophobic core. The result is a PGG concentration-dependent decrease in the abundance of both oligomeric and fibrillar Aβ1-40/Aβ1-42 aggregates. These results suggest the potential of PGG as a therapeutic agent may well alleviate the neurotoxicity of Aβ oligomers.34
METHODS
The materials and methods are described in detail on Section S1 of Supporting Information. Briefly, the full-length Aβ1-40 and Aβ1-42 samples (10 μM) were prepared in ammonium acetate buffer (10 mM, pH 7.4), whereas the Aβ fragments Aβ1-11 and Aβ11-22 were dissolved in water to the final concentration of 100 μM. The stock solution of PGG was prepared in water containing 20% (v/v) methanol and used in two different molar ratios (1:1 and 1:10) with respect to Aβ species. The IMS-MS experiments were performed in a home-built ion mobility spectrometer coupled to a mass spectrometer,36 where samples were loaded into a gold coated nano-ESI capillaries. For Aβ1-40, Aβ1-42 and Aβ1-11 a negative ionization mode was used, whereas a positive mode was employed to spray the Aβ11-22 segment. In the TEM analyses, aliquots of the same samples used in IMS-MS experiments were taken and adsorbed onto 300 mesh formvar/carbon copper grids (Electron Microscopy Sciences) and then imaged using a JEOL 123 microscope equipped with an ORCA camera and AMT Image Capture Software v. 5.24.
Explicit solvent MD simulations were performed using the Amber 12 package.55 The parameters of Aβ1-42 were taken from FF12SB and those of PGG were derived from RESP ESP charge Derive (http://upjv.q4md-forcefieldtools.org/RED/) as previously reported.56, 57 The initial structures were solvated in octahedral TIP3P water boxes. Detailed simulation protocols, MM-GBSA calculations and per residue binding energy decomposition are reported in Supporting Information.
Supplementary Material
HIGHLIGHTS.
PGG inhibits Aβ1-40/Aβ1-42 self-assembly.
PGG selectively interacts with N-terminal metal binding region of amyloid peptides.
Binding sites involve both hydrophilic and hydrophobic interactions.
Acknowledgments
We gratefully acknowledge support from the National Science Foundation (NSF) MCB- 1158577 (J.-E.S.), the National Institutes of Health Grant 1RO1AG047116- 01 (M.T.B.), the David and Lucile Packard Foundation (J.-E.S.), and a grant from Santa Barbara Cottage Hospital and the University of California, Santa Barbara (N.E.L). N.E.C.A. thanks Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for a post doctoral fellowship (204613/2014-0). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant OCI-1053575. We acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin for providing HPC resources through XSEDE Grant TG-MCA05S027 (J.- E.S.), and the NRI Microscopy Facility at UCSB. We acknowledge support from the Center for Scientific Computing at the CNSI and MRL via NSF MRSEC (DMR- 1121053) and NSF Grant CNS-0960316.
ABREVIATIONS USED
- AD
Alzheimer’s disease
- Aβ
amyloid β-protein
- Aβ1-11
amyloid β-protein (1-11)
- Aβ11-22
amyloid β-protein (11-22)
- Aβ12-22
amyloid β-protein (12-22)
- Aβ25-35
amyloid β-protein (25-35)
- Aβ36-42
amyloid β-protein (36-42)
- Aβ1-40
amyloid β-protein (1-40)
- Aβ1-42
amyloid β-protein (1-42)
- Ala
alanine
- APP
amyloid precursor protein
- Arg
arginine
- Asp
aspartic acid
- ATD
arrival time distribution
- CCS
collision cross sections
- His
histidine
- Ile
isoleucine
- IMS-MS
ion mobility spectrometry coupled to mass spectrometry
- Lys
lysine
- MD
molecular dynamics
- PGG
1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose
- Phe
phenylalanine
- TEM
transmission electron microscopy
- Val
valine
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. Detailed description of materials, sample preparation, methods, including ion mobility spectrometry-mass spectrometry (IMS-MS), transmission electron microscopy (TEM) and molecular dynamic (MD) simulations. Additional mass spectra, arrival time distributions (ATDs), collision cross sections (CCSs) and TEM images of Aβ1-42 incubated with and without 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose (PGG). This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 2.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Zaghi J, Goldenson B, Inayathullah M, Lossinsky AS, Masoumi A, Avagyan H, Mahanian M, Bernas M, Weinand M, Rosenthal MJ, Espinosa-Jeffrey A, de Vellis J, Teplow DB, Fiala M. Alzheimer disease macrophages shuttle amyloid-beta from neurons to vessels, contributing to amyloid angiopathy. Acta Neuropathologica. 2009;117:111–124. doi: 10.1007/s00401-008-0481-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-beta in Alzheimer disease: The null versus the alternate hypotheses. Journal of Pharmacology and Experimental Therapeutics. 2007;321:823–829. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 6.Jakob-Roetne R, Jacobsen H. Alzheimer’s disease: From pathology to therapeutic approaches. Angewandte Chemie-International Edition. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 7.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Skwarek-Maruszewska A, Horre K, Vandewyer E, Wolfs L, Snellinx A, Saito T, Radaelli E, Corthout N, Colombelli J, Lo AC, Van Aerschot L, Callaerts-Vegh Z, Trabzuni D, Bossers K, Verhaagen J, Ryten M, Munck S, D’Hooge R, Swaab DF, Hardy J, Saido TC, De Strooper B, Thathiah A. Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer’s disease mouse models. Science Translational Medicine. 2015;7:309ra164. doi: 10.1126/scitranslmed.aab3492. [DOI] [PubMed] [Google Scholar]
- 9.Zheng X, Wu C, Liu D, Li H, Bitan G, Shea JE, Bowers MT. Mechanism of C-Terminal Fragments of Amyloid β-Protein as Aβ Inhibitors: Do C-Terminal Interactions Play a Key Role in Their Inhibitory Activity? The Journal of Physical Chemistry B. 2016;120:1615–1623. doi: 10.1021/acs.jpcb.5b08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 11.Haass C, Selkoe DJ. Alzheimer’s disease - A technical KO of amyloid-beta peptide. Nature. 1998;391:339–340. doi: 10.1038/34800. [DOI] [PubMed] [Google Scholar]
- 12.O’Brien RJ, Wong PC. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annual Review of Neuroscience. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng X, Liu D, Klaerner FG, Schrader T, Bitan G, Bowers MT. Amyloid beta-protein assembly: The effect of molecular tweezers CLR01 and CLR03. Journal of Physical Chemistry B. 2015;119:4831–4841. doi: 10.1021/acs.jpcb.5b00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roychaudhuri R, Zheng X, Lomakin A, Maiti P, Condron MM, Benedek GB, Bitan G, Bowers MT, Teplow DB. Role of species-specific primary structure differences in Aβ42 assembly and neurotoxicity. Acs Chemical Neuroscience. 2015;6:1941–1955. doi: 10.1021/acschemneuro.5b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 16.Sciacca MFM, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophysical Journal. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Somavarapu AK, Kepp KP. Direct correlation of cell toxicity to conformational ensembles of genetic Aβ Variants. Acs Chemical Neuroscience. 2015;6:1990–1996. doi: 10.1021/acschemneuro.5b00238. [DOI] [PubMed] [Google Scholar]
- 18.Teplow DB. On the subject of rigor in the study of amyloid beta-protein assembly. Alzheimers Research & Therapy. 2013;5:39. doi: 10.1186/alzrt203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, Baumketner A, Shea JE, Urbanc B, Cruz L, Borreguero J, Stanley HE. Elucidating amyloid beta-protein folding and assembly: A multidisciplinary approach. Accounts of Chemical Research. 2006;39:635–645. doi: 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- 20.Walsh DM, Selkoe DJ. A beta Oligomers - a decade of discovery. Journal of Neurochemistry. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 21.Hayden EY, Teplow DB. Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Research & Therapy. 2013;5:60. doi: 10.1186/alzrt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nature Reviews Molecular Cell Biology. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 23.Gong YS, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 25.Mason JM, Kokkoni N, Stott K, Doig AJ. Design strategies for anti-amyloid agents. Current Opinion in Structural Biology. 2003;13:526–532. doi: 10.1016/s0959-440x(03)00100-3. [DOI] [PubMed] [Google Scholar]
- 26.Bartolini M, Andrisano V. Strategies for the inhibition of protein aggregation in human diseases. Chembiochem. 2010;11:1018–1035. doi: 10.1002/cbic.200900666. [DOI] [PubMed] [Google Scholar]
- 27.Liu T, Bitan G. Modulating self-assembly of amyloidogenic proteins as a therapeutic approach for neurodegenerative diseases: Strategies and mechanisms. Chemmedchem. 2012;7:359–374. doi: 10.1002/cmdc.201100585. [DOI] [PubMed] [Google Scholar]
- 28.Feng BY, Toyama BH, Wille H, Colby DW, Collins SR, May BCH, Prusiner SB, Weissman J, Shoichet BK. Small-molecule aggregates inhibit amyloid polymerization. Nature Chemical Biology. 2008;4:197–199. doi: 10.1038/nchembio.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chemical Biology & Drug Design. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 30.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nature Structural & Molecular Biology. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 31.Bleiholder C, Do TD, Wu C, Economou NJ, Bernstein SS, Buratto SK, Shea JE, Bowers MT. Ion mobility spectrometry reveals the mechanism of amyloid formation of A beta(25–35) and its modulation by inhibitors at the molecular level: Epigallocatechin gallate and scyllo-inositol. Journal of the American Chemical Society. 2013;135:16926–16937. doi: 10.1021/ja406197f. [DOI] [PubMed] [Google Scholar]
- 32.Hayden EY, Yamin G, Beroukhim S, Chen B, Kibalchenko M, Jiang L, Ho L, Wang J, Pasinetti GM, Teplow DB. Inhibiting amyloid -protein assembly: Size-activity relationships among grape seed-derived polyphenols. Journal of Neurochemistry. 2015;135:416–430. doi: 10.1111/jnc.13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bravo L. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutrition Reviews. 1998;56:317–333. doi: 10.1111/j.1753-4887.1998.tb01670.x. [DOI] [PubMed] [Google Scholar]
- 34.Fujiwara H, Tabuchi M, Yamaguchi T, Iwasaki K, Furukawa K, Sekiguchi K, Ikarashi Y, Kudo Y, Higuchi M, Saido TC, Maeda S, Takashima A, Hara M, Yaegashi N, Kase Y, Arai H. A traditional medicinal herb Paeonia suffruticosa and its active constituent 1,2,3,4,6-penta-O-galloyl-beta-d-glucopyranose have potent anti-aggregation effects on Alzheimer’s amyloid beta proteins in vitro and in vivo. Journal of Neurochemistry. 2009;109:1648–1657. doi: 10.1111/j.1471-4159.2009.06069.x. [DOI] [PubMed] [Google Scholar]
- 35.de Almeida NEC, Do TD, Tro M, LaPointe NE, Feinstein SC, Shea J-E, Bowers MT. Opposing effects of cucurbit[7]uril and 1,2,3,4,6-penta-O-galloyl-β-d-glucopyranose on amyloid β25–35 assembly. ACS Chemical Neuroscience. 2016;7:218–226. doi: 10.1021/acschemneuro.5b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wyttenbach T, Kemper PR, Bowers MT. Design of a new electrospray ion mobility mass spectrometer. International Journal of Mass Spectrometry. 2001;212:13–23. [Google Scholar]
- 37.Wyttenbach T, Bowers M. Gas-phase conformations: The ion mobility/ion chromatography method. In: Schalley C, editor. In Modern Mass Spectrometry. Springer; Berlin, Heidelberg: 2003. pp. 207–232. [Google Scholar]
- 38.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nature Chemistry. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gessel MM, Bernstein S, Kemper M, Teplow DB, Bowers MT. Familial Alzheimer’s disease mutations differentially alter amyloid beta-protein oligomerization. ACS Chemical Neuroscience. 2012;3:909–918. doi: 10.1021/cn300050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein SL, Wyttenbach T, Baumketner A, Shea JE, Bitan G, Teplow DB, Bowers MT. Amyloid beta-protein: Monomer structure and early aggregation states of A beta 42 and its Pro(19) alloform. Journal of the American Chemical Society. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 41.Zheng X, Gessel MM, Wisniewski ML, Viswanathan K, Wright DL, Bahr BA, Bowers MT. Z-Phe-Ala-diazomethylketone (PADK) disrupts and remodels early oligomer states of the Alzheimer disease A beta 42 protein. Journal of Biological Chemistry. 2012;287:6084–6088. doi: 10.1074/jbc.C111.328575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young LM, Saunders JC, Mahood RA, Revill CH, Foster RJ, Tu LH, Raleigh DP, Radford SE, Ashcroft AE. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry-mass spectrometry. Nature Chemistry. 2015;7:73–81. doi: 10.1038/nchem.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S, Zheng X, Krishnamoorthy J, Savelieff MG, Park HM, Brender JR, Kim JH, Derrick JS, Kochi A, Lee HJ, Kim C, Ramamoorthy A, Bowers MT, Lim MH. Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer’s disease. Journal of the American Chemical Society. 2014;136:299–310. doi: 10.1021/ja409801p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soper MT, DeToma AS, Hyung SJ, Lim MH, Ruotolo BT. Amyloid-beta-neuropeptide interactions assessed by ion mobility-mass spectrometry. Physical Chemistry Chemical Physics. 2013;15:8952–8961. doi: 10.1039/c3cp50721a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bleiholder C, Dupuis NF, Wyttenbach T, Bowers MT. Ion mobility-mass spectrometry reveals a conformational conversion from random assembly to beta-sheet in amyloid fibril formation. Nature Chemistry. 2011;3:172–177. doi: 10.1038/nchem.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Economou NJ, Giammona MJ, Do TD, Zheng X, Teplow DB, Buratto SK, Bowers MT. Amyloid beta-Protein Assembly and Alzheimer’s Disease: Dodecamers of A beta 42, but Not of A beta 40, Seed Fibril Formation. Journal of the American Chemical Society. 2016;138:1772–1775. doi: 10.1021/jacs.5b11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis - Detection of a protofibrillar intermediate. Journal of Biological Chemistry. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 48.Roychaudhuri R, Yang M, Deshpande A, Cole GM, Frautschy S, Lomakin A, Benedek GB, Teplow DB. C-terminal turn stability determines assembly differences between A beta 40 and A beta 42. Journal of Molecular Biology. 2013;425:292–308. doi: 10.1016/j.jmb.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. The Alzheimer’s peptides A beta 40 and 42 adopt distinct conformations in water: A combined MD/NMR study. Journal of Molecular Biology. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soreghan B, Kosmoski J, Glabe C. Surfactant properties of Alzheimer’s A-beta peptides and the mechanism of amyloid aggregation. Journal of Biological Chemistry. 1994;269:28551–28554. [PubMed] [Google Scholar]
- 52.Tjernberg LO, Pramanik A, Bjorling S, Thyberg P, Thyberg J, Nordstedt C, Berndt KD, Terenius L, Rigler R. Amyloid beta-peptide polymerization studied using fluorescence correlation spectroscopy. Chemistry & Biology. 1999;6:53–62. doi: 10.1016/S1074-5521(99)80020-9. [DOI] [PubMed] [Google Scholar]
- 53.Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Amyloid fibril formation by A beta(16–22), a seven-residue fragment of the Alzheimer’s beta-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 54.Larini L, Shea JE. Role of beta-hairpin formation in aggregation: The self-assembly of the amyloid-beta(25–35) peptide. Biophysical Journal. 2012;103:576–586. doi: 10.1016/j.bpj.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Case DA, Darden T, Cheatham TE, Simmerling C, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts BP, Hayik S, Roitberg A, Seabra G, Swails J, Götz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. Amber 12. University of California; San Francisco: 2012. [Google Scholar]
- 56.Bleiholder C, Wyttenbach T, Bowers MT. A novel projection approximation algorithm for the fast and accurate computation of molecular collision cross sections (I). Method. International Journal of Mass Spectrometry. 2011;308:1–10. [Google Scholar]
- 57.Bleiholder C, Contreras S, Do TD, Bowers MT. A novel projection approximation algorithm for the fast and accurate computation of molecular collision cross sections (II). Model parameterization and definition of empirical shape factors for proteins. International Journal of Mass Spectrometry. 2013;345:89–96. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.