

Abstract
Aim
Hearing loss is the most common sensory disorder that affects approximately one per 1000 live births. With this project, we aimed to identify gene variants that were common causes of hearing loss in Turkey to contribute to the planning of genetic screening programs for hearing loss, as well as to improve genetic counseling to affected families.
Material and Methods
Twenty-one families with at least two affected individuals and parental consanguinity who presented with non-syndromic severe-to-profound sensorineural hearing loss were included in this study. We first screened for mutations in GJB2 and mitochondrial DNA 12S RNA genes. Subsequently, we genotyped the TMIE c.250C>T and SNP markers flanking the SLC26A4, MYO7A, MYO15A, OTOF, CDH23, TMIE, TECTA, PCDH15, TMC1, TMPRSS3, TMHS genes in the remaining twelve families without mutations in GJB2.
Results
Screening for mutations in GJB2 gene showed c.[35delG];[35delG] mutation in four families, c.[35delG];[507C>A] mutation in two families, c.[35delG];[−23+1G>A] mutation in one family, and c.457G>A heterozygous mutation in one family. Genotyping SNP markers showed the c.[250C>T];[250C>T] mutation in TMIE in one family. A homozygous region with SNP genotypes was detected with the OTOF gene in one family, the TMPRSS3 gene in another family, and also a homozygous region was detected with TMHS, OTOF, and TMPRSS3 genes in another family.
Conclusions
Further research will be required to determine the genetic bases of hearing loss in families with non-syndromic hearing loss.
Keywords: Hearing loss, microarray, sequence, single nucleotide polymorphism
Introduction
Hearing loss is the most common sensory impairment that affects approximately one per 1000 live births. Genetic factors are responsible of approximately half of all cases and environmental factors are responsible for the other half (1). Environmental factors include prenatal maternal infections including rubella and cytomegalovirus, prematurity, use of ototoxic drugs, postnatal morbidities including meningitis, mastoiditis and chronic middle ear infection, and history of trauma (2).
Hearing loss originating from genetic factors is clinically classified in two groups including syndromic and non-syndromic hearing loss. Cases where no other organ system or laboratory findings accompany hearing loss are defined as non-syndromic hearing loss, and this group constitutes approximately 70–80% of hearing losses with a genetic origin (1). Syndromic hearing loss constitutes the remaining 20–30% (1). More than 400 syndromes that include hearing loss among the findings have been described until the present time. The most common syndromes that are accompanied by hearing loss include Pendred, Usher, Branchio-oto-renal (BOR), Waardenburg, and Alport syndromes (3). These syndomes constitute 15–20% of the population with hearing loss (3).
Autosomal recesive hearing loss constitutes 80% of non-syndromic hearing losses (4). Up to the present time, mutations in more than 60 genes have been shown to cause non-syndromic autosomal recessive hearing loss (5). The gene loci specified for non-syndromic hearing loss are designated DeaFNess (DFN), the autosomal dominant gene loci are designated DFNA, the autosomal recessive gene loci are designated DFNB, and the loci on the X chromosome are designated DFN. While one mutation in an autosomal gene may be recessive and another mutation may be dominant, the same gene may be responsible for both syndromic and non-syndromic hearing loss (6–8).
A significant portion of genetic hearing loss in many populations is explained with GJB2 gene mutations (9). The m.1555A>G mutation found in the 12S RNA gene of mitochondrial DNA is one of the non-syndromic hearing loss mutations, which is especially common in Spain and Far East countries. In a study conducted with Turkish patients, the prevalence of this mutation was reported as 1.8% (10).
Despite many studies, the etiology and molecular etiopathogenesis of hearing loss is still not clearly known. Involvement of proteins encoded by many different genes in the function of hearing is expected because the inner ear and hearing mechanism have a very complicated structure. When the intracellular functions of the genes identified were evaluated, it was observed that they may be adhesion molecules, enzymes, ion channels or carriers, and integral membrane proteins, and they may be involved in the cytoskeleton, extracellular matrix, nexuses, organizition of macromolecules, neurons or synapses, regulation of translation, and transcription and neurologic development (7). The data obtained in recent years suggest that at least 1% of human genes are necessary for hearing (8).
In our study, it was aimed to determine genetic changes that cause familial hearing loss in our community, and to contribute to genetic screening programs that would be established for hearing loss in the future and genetic counseling for the next generations. In addition, it will be possible to present the opportunity of preimplantation genetic diagnosis to patients with mutations that will be specified.
Material and Methods
The study was approved by the Erciyes University Ethics Committee (dated 04.01.2011, decision number: 2011/53) and supported by the Erciyes University Scientific Research Projects Unit with the Project number TSU-11-3483, and by the common Project of Miami University-Ankara University National Institute of Health (NIH) RO1 DC009645 (AU no: 2011ABH06739003). Informed consent was obtained from the families who accepted to participate in the study.
One hundred twenty-two individuals from 21 families who had two or more hearing-impaired children and consanguineous marriage, and whose pedigree suggested autosomal recessive inheritance were included in this study. Sixty-two of these individuals had hearing loss. On detailed clinical examination, patients with syndromes that accompanied hearing loss including Pendred, Usher, Branchio-oto-renal (BOR), Waardenburg, and Alport syndromes were excluded. The individuals who were affected had sensoryneural hearing loss that was congenital or had prelingual onset, and the severity ranged between mild to profound hearing loss. The diagnosis of sensoryneural hearing loss was made using standard audiometric tests.
DNA isolation was performed using the classic phenol chlorophorm method. In all affected individuals, primarily the encoding and non-encoding exons of the GJB2 (NM_004004) gene were reproduced using polymerase chain reaction (PCR) with appropriate primers under appropriate conditions and examined using sequence analysis (CEQ8800, Beckman Coulter, ABD). The mitochondrial DNA 12S RNA gene was reproduced using appropriate primers under appropriate conditions for m.1555A>G mutation, was cut with BsmAI (5′-GTCTCN↓N-3′) restriction endonuclease enzyme (NEB, USA) and band differences were examined in agarose gel electrophoresis. Screening for the SLC26A4 (NM_000441.1), MYO7A (NM_000260.3), MYO15A (NM_016239.3), OTOF (NM_194248.2), CDH23 (NM_022124.5), TMIE (NM_147196.2), TECTA (NM_005422.2), PCDH15 (NM_033056.3), TMC1 (NM_138691.2), TMPRSS3 (NM_024022.2), LHFPL5 (NM_182548.3) genes was performed using the open array method (TaqManR OpenArrayR) in 12 families in whom m. 1555A>G mutations were not found in the mitochondrial DNA 12S RNA gene and no mutations were found in the GJB2 gene. These genes were selected among the genes that most commonly lead to hearing loss in the Turkish population. The single nucleotide polymorphisms (SNP) located in the open array platform were determined from the web site http://pga.gs.washington.edu/. The SNPs for each gene were selected primarily from inside the genes and from the 5′ and 3′ sites up to 15 000 bases considering the gene localization. In this study, SNPs that had a minor allele frequency of 0.2 and above and did not have linkage disequilibrium (LD values below 0.8 were included in the study) were evaluated. Subsequently, the distribution of SNP genotypes obtained from affected and unaffected family members in the family were pursued and gene regions that showed autosomal recessive inheritance were determined. The c.250C>T (p.R48W) mutation was added to the platform instead of polymorphic points for the TMIE gene.
Results
In our study, homozygous c.35delG mutations were determined in the GJB2 gene in subjects who had hearing loss in four families, and heterozygous c.457G>A (p.V153I) mutations were found in subjects who had hearing loss in one family. Combined heterozygosity was shown in subjects who had hearing loss in three families; two c[35delG];[507C>A], and one c.[35delG];[−23+1G>A] (Table 1). m.1555A>G mutations were not identified in the mitochondrial DNA 12S RNA gene in 12 families in whom no mutations were found in the GJB2 gene as a result of sequence analysis studies. In the open array studies performed in families, a c.250C>T mutation was observed in the TMIE gene in subjects who had hearing loss in family number 902 (Table 2a). Homozygosity was seen in the OTOF gene in family number 909 (Table 2b), in the TMPRSS3 gene in family number 910 (Table 2c), and in the OTOF, TMPRSS3, and TMHS genes in family number 917 (Table 2d, 2e, 2f). No genetic change was observed with the methods performed in the scope of this study in the remaining eight families.
Table 1.
GJB2 gene sequence analysis findings
| Family number | Individuals who underwent GJB2 analysis | Mutation found | Zygosity |
|---|---|---|---|
| 901 | 101–102 | c.35delG / c.508 C>A | Combined heterozygosity |
| 903 | 101–103 | c.35delG / c.508 C>A | Combined heterozygosity |
| 905 | 101–102 | c.457G>A / WT | Heterozygous |
| 913 | 102–103 | c.[35delG];[−23+1G>A] | Combined heterozygosity |
| 914 | 101–102 | c.35delG / c.35delG | Homozygous |
| 916 | 102–103 | c.35delG / c.35delG | Homozygous |
| 918 | 101–102 | c.35delG / c.35delG | Homozygous |
| 919 | 101–103 | c.35delG / c.35delG | Homozygous |
Table 2a.
Genotypes obtained from the open array data for the TMIE gene rs28942097 (c.250C>T, p.R84W)
| Family number | Subject number | rs28942097 | Phenotype |
|---|---|---|---|
| 902 | 102 | CT | Healthy sibling |
| 902 | 103 | TT | Sibling with hearing loss |
| 902 | 104 | CT | Healthy sibling |
| 902 | 105 | TT | Sibling with hearing loss |
| 902 | 202 | CT | Healthy mother |
| 902 | 302 | CT | Healthy father |
| 902 | 501 | TT | First-degree cousin with hearing loss |
| 902 | 502 | TT | First-degree cousin with hearing loss |
| 902 | 503 | TT | First-degree cousin with hearing loss |
| 904 | 101 | CC | Sibling with hearing loss |
| 904 | 102 | CC | Healthy sibling |
| 904 | 103 | CC | Sibling with hearing loss |
| 904 | 104 | CC | Sibling with hearing loss |
| 904 | 201 | CC | Healthy father |
| 904 | 301 | CC | Healthy mother |
| 905 | 101 | CC | Sibling with hearing loss |
| 905 | 102 | CC | Sibling with hearing loss |
| 905 | 201 | CC | Healthy father |
| 905 | 301 | CC | Healthy mother |
| 905 | 401 | CC | Sibling with hearing loss |
| 905 | 501 | CC | Sibling with hearing loss |
| 907 | 101 | CC | Healthy sibling |
| 907 | 102 | CC | Healthy sibling |
| 907 | 103 | CC | Sibling with hearing loss |
| 907 | 104 | CC | Sibling with hearing loss |
| 907 | 105 | CC | Sibling with hearing loss |
| 907 | 201 | CC | Healthy father |
| 907 | 301 | CC | Healthy mother |
| 908 | 101 | CC | Sibling with hearing loss |
| 908 | 102 | CC | Sibling with hearing loss |
| 908 | 103 | CC | Sibling with hearing loss |
| 908 | 104 | CC | Sibling with hearing loss |
| 908 | 201 | CC | Healthy father |
| 908 | 301 | CC | Healthy mother |
| 909 | 101 | CC | Healthy sibling |
| 909 | 103 | CC | Sibling with hearing loss |
| 909 | 301 | CC | Healthy father |
| 910 | 101 | CC | Sibling with hearing loss |
| 910 | 102 | CC | Sibling with hearing loss |
| 910 | 201 | CC | Healthy mother |
| 910 | 301 | CC | Healthy father |
| 911 | 101 | CC | Sibling with hearing loss |
| 911 | 102 | CC | Sibling with hearing loss |
| 911 | 201 | CC | Healthy father |
| 911 | 301 | CC | Healthy mother |
| 912 | 101 | CC | Sibling with hearing loss |
| 912 | 102 | CC | Sibling with hearing loss |
| 912 | 201 | CC | Father with hearing loss |
| 912 | 301 | CC | Healthy mother |
| 917 | 101 | CC | Sibling with hearing loss |
| 917 | 102 | CC | Sibling with hearing loss |
| 917 | 201 | CC | Healthy father |
| 917 | 301 | CC | Healthy mother |
| 920 | 101 | CC | Sibling with hearing loss |
| 920 | 102 | CC | Healthy sibling |
| 920 | 103 | CC | Sibling with hearing loss |
| 920 | 201 | CC | Healthy mother |
| 920 | 301 | CC | Healthy father |
Table 2b.
Open array data of the family number 909 who showed segregation in the OTOF gene
| Family number-Subject number/Phenotype | 909-101/Healthy sibling | 909-103/Hearing loss | 09-301/Healthy father |
|---|---|---|---|
| rs11674089 | AG | GG | AG |
| rs2280516 | GG | GG | GG |
| rs2272069 | No amplification observed | GG | CG |
| rs869440 | No amplification observed | AA | AA |
| rs939817 | CT | CC | CT |
| rs6746918 | AA | GG | AA |
| rs1879760 | AA | AA | AA |
| rs4665874 | CC | CC | CC |
| rs6547103 | GG | GG | GG |
| rs13029128 | GT | GG | GT |
| rs1011108 | CT | CC | CT |
| rs939815 | CC | CC | CC |
Table 2c.
Open array data of family number 910 showing segregation in the TMPRSS3 gene
| Family number-Subject number/Phenotype | 910-101/Hearing loss | 910-102/Hearing loss | 910-201/Healthy mother | 910-301/Healthy father |
|---|---|---|---|---|
| rs1079380 | GG | GG | GG | GG |
| rs424694 | TT | TT | CT | TT |
| rs2839490 | TT | TT | CT | TT |
| rs462149 | GG | GG | GG | GG |
| rs225310 | GG | GG | GT | GG |
| rs1078272 | TT | TT | TT | TT |
| rs1109352 | GG | GG | GG | GG |
| rs225430 | AA | AA | AA | AC |
| rs10887973 | TT | TT | TT | GT |
| rs9981624 | GG | GG | CG | GG |
| rs9980448 | CC | CC | CT | CT |
| rs13052676 | CC | CC | CC | CT |
Table 2d.
Open array data of family number 917 showing segregation in the OTOF gene
| Family number-Subject number/Phenotype | 917-101/Hearing loss | 917-102/Hearing loss | 917-201/Healthy mother | 917-301/Healthy father |
|---|---|---|---|---|
| rs11674089 | GG | GG | AG | GG |
| rs2280516 | GG | GG | GG | GG |
| rs2272069 | GG | GG | GG | CG |
| rs869440 | AA | AA | AA | AG |
| rs939817 | CC | CC | CC | CT |
| rs6746918 | GG | GG | AG | GG |
| rs1879760 | GG | GG | AG | GG |
| rs4665874 | AA | AA | AC | No amplification observed |
| rs6547103 | GG | GG | GG | CG |
| rs13029128 | GG | GG | GT | GG |
| rs1011108 | TT | TT | TT | CT |
| rs939815 | CC | CC | CC | CC |
Table 2e.
Open array data of family number 917 showing segregation in the TMPRSS3 gene
| Family number-Subject number/Phenotype | 917-101/Hearing loss | 917-102/Hearing loss | 917-201/Healthy mother | 917-301/Healthy father |
|---|---|---|---|---|
| rs1079380 | AG | AA | AG | AG |
| rs424694 | CC | CT | CT | CT |
| rs2839490 | CT | CT | CT | TT |
| rs462149 | AG | GG | GG | AG |
| rs225310 | TT | UND | GT | GT |
| rs1078272 | TT | TT | AT | TT |
| rs1109352 | GG | GG | GG | GG |
| rs225430 | AA | AA | AC | AA |
| rs10887973 | TT | TT | TT | TT |
| rs9981624 | GG | GG | GG | GG |
| rs9980448 | TT | TT | TT | TT |
| rs13052676 | CT | TT | TT | CT |
Table 2f.
Open array data of family number 917 showing segregation in the TMHS gene
| Family number-Subject number/Phenotype | 917-101/Hearing loss | 917-102/Hearing loss | 917-201/Healthy mother | 917-301/Healthy father |
|---|---|---|---|---|
| rs1343796 | AG | No amplification observed | AG | AA |
| rs10807154 | GG | GG | GG | AG |
| rs2817012 | CC | CC | CG | CC |
| rs2817013 | AC | AC | AC | CC |
| rs6921084 | TT | TT | CT | CT |
| rs1049649 | AA | AA | AC | AA |
| rs2395637 | GG | GG | GG | GG |
| rs7752049 | AA | AA | AC | AA |
| rs2817057 | GG | GG | AA | GG |
| rs2817064 | AG | AG | AG | AG |
| rs9470094 | GG | GG | GG | GG |
| rs12211728 | AG | AA | No amplification observed | AG |
Discussion
Literature comparison of mutations observed in the GJB2 gene
It is known that mutations in the GJB2 gene have a significant role in non-syndromic hearing loss. Many different studies have been conducted in many different populations since mutations in the GJB2 gene were investigated for the first time in 1997 (9). This gene is responsible for almost half of non-syndromic hearing loss with autosomal recessive inheritance. One hundred fifty different mutations have been identified in the GJB2 gene to date and different mutations are noted in different populations. Most of the pathologic mutations are located in exon 2, which is the encoding region of the gene. Again, the c.35delG mutation observed in this exon is the most important cause of non-syndromic hearing loss in our country and throughout the world.
The most common mutations in the GJB2 gene in Caucasians and Asckenasi Jews is the c.35delG mutation, followed by the c.167delT mutation. In populations in East Asia, c.235delC mutations are also commonly observed in addition to these mutations (11).
In studies conducted in our country, the most common mutation found in the GJB2 gene among the other mutations is the c.−23+1G>A mutation (12). This is followed by c.360_362delGAT (p.delE120), c.71G>A (p.W24X), c.233delG, c.239A>G (p.Q80R), c.310_323del14, c.299–300delAT, c.167delT, c. 551G>C (p.R184P), c.269T>C (p.L90P), c. 517 C>T (p.P173S), c. 380G>A (p.R127H), c. 238C>A (p.Q80K) mutations (13, 14). The c.35delG mutation occurs as a result of deletion of a single guanine nucleotide in the sequence composed of six guanine nucleotides, and this leads to frameshift. Thus, a stop codon occurs (UGA) in the 13th position. The effect of the mutation at the protein level is occurence of a nonfunctional Cx26 protein composed of 12 amino acids instead of a normal protein composed of 226 amino acids (p.G12Vfs*2) (15).
When considered by country, the carrier frequency of this mutation is 3.4% in Italy, 3.5% in Greece, 2.75% in France, and 2.8% in Malta and Portugal, whereas it shows a marked decrease in countries in America and Asia (16). The carrier frequency in Turkey has been reported to range between 1.17% and 1.78% in different studies (17, 18). In other studies conducted in Turkey, the c.35delG mutation has been found in 5–53% of individuals with hearing impairment (7, 10). Although the frequency of this mutation is high in Central Anatolia and South Western Anatolia, it has been reported as 13.2% in sporadic cases and 22.1% in familial cases (10).
In parallel to variability of allele frequency in different populations, the frequency of non-syndromic hearing loss caused by homozygous c.35delG mutations shows great variance by races and populations. For example, homozygous c.35delG mutations have not been reported in studies conducted with individuals with non-syndromic hearing loss in China, Japan, Ghana, India, Korea, Pakistan, Taiwan, and Thailand (19, 20). On the other hand, the frequency of homozygous c.35delG mutations was found as 1.8% in a study conducted in Denmark, and 40% in a study conducted in Slovakia (19).
In Turkey, different studies have been conducted in this area. The results show variance by geographic distribution and the number of subjects included. In a study conducted by Barış et al. (21), homozygous c.35delG mutations were identified in 20.4% of the subjects, and heterozygous mutations were identified in 2.1%. In another study conducted by Tekin et al. (22), homozygous c.35delG mutations were reported in 15% of the subjects and heterozygous 35delG mutatione were observed in 7.81%.
Family studies have also been conducted in this area. In a study by Uyguner et al. (18) in which 60 families with autosomal recessive hearing loss were evaluated, homozygous c.35delG mutations were observed in 21.7% of families, and heterozygous c.35delG mutations were determined in 3.3% of families. In another study by Tekin et al. (12) in which familial cases were evaluated, homozygous c.35delG mutations was identified at a rate of 17.5% and heterozygous c.35delG was found with a rate of 1.9% (10). In a study by Kalay et al. (13) in which individuals with familial hearing loss were evaluated, homozygous c.35delG mutations were seen in 21.5% of subjects and heterozygous c.35delG was observed in 4.3% of subjects.
Hearing loss was present in 2 of 122 inividuals from 21 families included in our study (Figure 1a, b). Homozygous c.35delG mutations was identified in four families and heterozygous c.35delG mutations wers observed in three families. Studies have so far reported that GJB2 mutations generally cause severe (81–100 db) and profound (more than 100 db) prelingual sensoryneural hearing loss (23). In the group included in our study, the level of hearing loss was compatible with the literature and profound hearing loss was present in all subjects who were found to have mutations.
Figure 1. a, b.
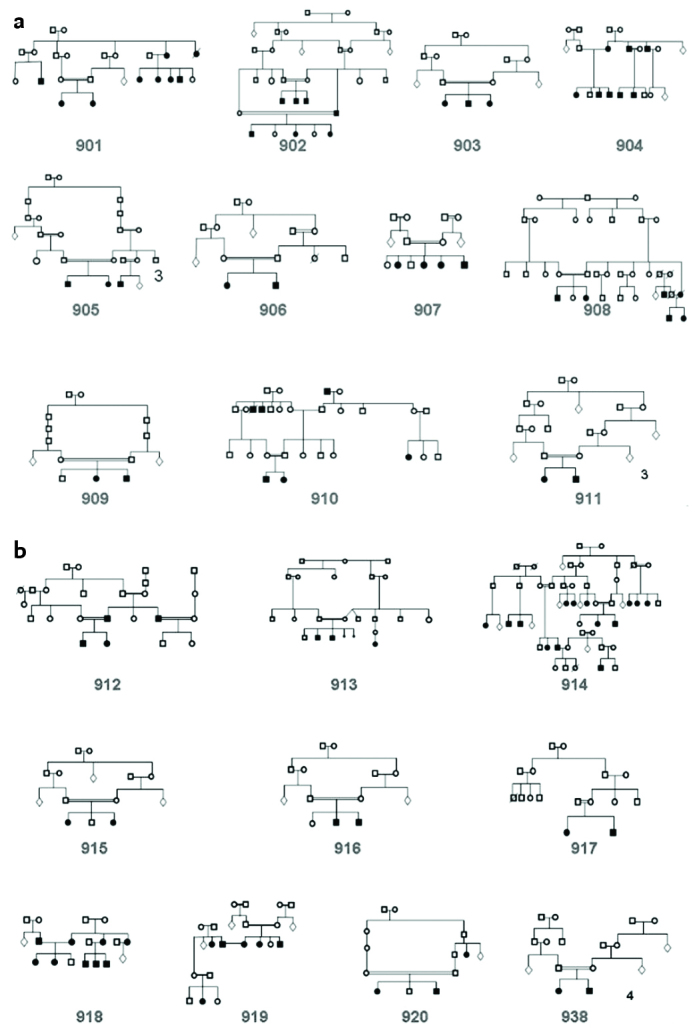
Pedigrees of the families included in the study
A combined heterozygous c.IVS1+1G>A mutation was found in a family who was found to have a heterozygous c.35delG mutation in the GJB2 gene. This explains the cause of hearing loss in the family. The c.−23+1G>A mutation was first reported by Denoyelle (24). The mutation is located in the first exon of the gene, which is non-encoding (rs80338940) and it was defined as −3170G>A in the years when it was initially demonstrated. It is predicted that it acts by impairing clipper function (24, 25). Although there is a limited number of studies related with this mutation in the literature, the frequency of this mutation has been reported in recent years in some populations, including Turkey. The c.IVS1+1G>A mutation has been reported more comonly in the Czech population (it has been found with a rate of 4% among all pathologic GJB2 mutations) (26). In a study conducted in Holland, the c.IVS1+1G>A mutation was observed as the third most common mutation following, c.35delG and del(GJB6-D13S1830) mutations (27). In all these studies, a heterozygous mutation was found in the second encoding exon of the GJB2 gene. Again, this mutation was investigated in affected individuals who carried heterozygous mutation in the GJB2 gene in a study conducted in China and reported in 1.89% of the subjects (28). In all these publications, it was recommended that this mutation should be investigated only in individuals carrying heterozygous mutations in exon 2. In addition, heterozygous c.IVS1+1G>A mutations were found in eight of 16 patients who had heterozygous mutations in exon 2 in a study by Tekin et al. (29). In another study, Padma et al. (30) observed this mutation in individuals who carried no pathologic homozygous or heterozygous mutations in exon 2 (0.3% of the study group) and it was emphasized that this mutation should be studied in all individuals with non-syndromic hearing loss.
In one family in our study group, p.V153I variability, which is also expressed as c.457G>A, was found heterozygously, again in the GJB2 gene. Controversial data related with this variability are present in the literature; some studies considered it a polymorphism, whereas other studies reported it as a pathologic mutation that could lead to hearing loss (31, 32). c.457G>A is reported to be a possible non-pathogenic SNP (rs111033186) in the National Center for Biotechnology Information (NCBI) website (33). The fact that it was also found in individuals who had no hearing loss in previous studies supports the view that is a polymorphism.
In our study, a combined heterozygous c.507C>A (p.C169X) mutation that has not been defined in the literature was found in two families that were found to have heterozygous c.35delG mutations in the GJB2 gene. “TGC” encodes the amino acid cysteine and a heterozygous mutation is present in the third base of the codon. This mutation, which leads to a stop codon, leads to the production of a 169 amino acid protein instead of a 226 amino acid protein encoded by the GJB2 gene.
In the present study, it was shown that the cause of hearing loss was GJB2 mutations in seven of 21 families with non-syndromic autosomal recessive hearing loss (Table 1). Tekin and Arıcı (12) reported that the rate of GJB2 homozygous mutations as 34.3% among patients with non-syndromic autosomal recessive hearing loss in Middle Anatolia.
Variabilities found in patients who were evaluated using the Open Array method
The TaqMan® OpenArray® technology used in our study is a platform with a wide area of application, with which a large number of genotyping procedures can be performed using a limited number of samples and consumables. It was preferred because of its high-quality data and resolution. The reason that this screening method was selected was that the possibility of mutations inherited from a common ancestor to be homozygous was high because the mothers and fathers were relatives in all families included in the study, and at least two children were affected. Thus, information about the location of mutation could be obtained by screening the homozygous regions in affected individuals. The analysis stage of the method is based on homozygous block screening in affected individuals or in individuals in regions where heterozygosity is present in the mother, father or healthy siblings and relatives. Interpretation has been made considering segregation in families. The gene may be ignored and missed even if there is mutation in that gene when the relevant homozygous blocks are small because the genome can be screened only in certain intervals. In addition, the method does not give a definite result, and confirmation with classic sequence analyses is required. However, it is a fast and inexpensive method that can be used to exclude known genes in candidate gene studies.
In our study, a homozygous p.R84W mutation was identified in the TMIE gene with TaqMan® OpenArray® genotyping in one family. The TMIE gene encodes a protein named transmembrane inner ear protein. The gene is composed of 4 exons in the 3p21 chromosomal region (34). The inner ear pathology observed in studies performed with mice in which the TMIE gene was affected showed that the gene was essential for maturation of sensory hair cells in the cochlea and development of steriocilia in the maturation process following a normal delivery (35). To date, seven mutations have been found in this gene encoding a 156 amino acid protein in relation with non-syndromic hearing loss including two in the extracellular region (4-BP INS, 125CGCC, p.E31G), two in the intronic region (6-BP DEL/1-BP INS in intron 1, c.212-2A>C in intron 2), and three in the cytoplasmic region (p.R81C, p.R84W, p.R92W) (36). When studies related with the TMIE gene were examined, mutations in the TMIE gene were reported at a rate of 6.6% in a study conducted in Turkey with 49 families who were not related to one another and had non-syndromic hearing loss (37). In a large-scale study performed in Pakistan, mutations in the TMIE gene were identifed at a rate of 1.7%.
If the c.250C>T (p.R84W) mutation is to be evaluated alone, it was found in a family from North India for the first time (38). In a subsequent study performed with a Turkish population in which 51 families were evaluated primarily, this mutation was determined in four families and afterwards 254 families were evaluated and the c.250C>T mutation was found in four more families. When the regional distribution was evaluated, the highest percentage was observed in Southeastern Anatolia (10.3%). It was thought that this mutation, which was observed in exon 3, occured more commonly as a result of the founder effect in Anatolia and it was recommended that the mutation should be added to non-syndromic hearing loss screening programs in Middle Eastern populations (34). The fact that this mutation was found in a limited study group consisting of 21 families supports this recommendation.
In the group in which we performed screening, segregation was found in the OTOF gene in two families. The OTOF gene, which encodes the otoferlin protein, is expressed highly in the internal hair cells in the organ of Corti, utriculus, and sacculus and in the brain. It was determined that mutations in this gene led to non-syndromic recessive auditory neuropathy, which is a special type of hearing loss. Up to this point, more than 40 pathologic allelic variants have been reported (39).
In a large-scale study that included 557 families in Pakistan, OTOF mutations were found in 13 families and reported to occur with a frequency of 2.3% (40). In a study by Chiu et al. (41), the OTOF gene was reported to be one of the most common causes of this type of hearing loss in Asian populations. In Spain, the prevalence of OTOF mutations was reported as 5% in all cases of non-syndromic autosomal recessive prelingual hearing loss (42). In a study by Duman et al. (37), the most common genes observed in the Turkish population in non-syndromic autosomal recessive hearing loss following the GJB2 gene were reported and the OTOF gene was reported to be the fourth relevant gene with mutations with a frequency of 5%, following MYO15A (9.9%), TMIE (6.6%), and TMC1 (6.6%).
In our study, segregation was found in the TMPRSS3 gene in two families. The TMPRSS3 gene encodes transmembrane serin protease 3 and is expressed in the Dieters cells of the organ of Corti and in the stria vascularis (43). It is thought that the protein is involved in sodium reuptake from the endolymph in amiloride-sensitive sodium channels (44). In a study by Guipponi et al. (45), it was emphesized that mutations in the TMPRSS3 gene were important in terms of enlightening the mechanism of hearing, though these mutations are not observed very commonly in populations with hearing loss. In a study by Wattenhofer et al. (46), a total of 448 families from Spain, Italy, Greece, and Australia were examined and mutations were found in the gene in question with a frequency of 0.45%. Again, the same study reported the frequency to be approximately 0.38% in Caucasians. In a study conducted in Pakistan in which 159 families were evaluated, mutations were found in the relevant gene in four families and the frequency in the Pakistani population was reported as approximately 2.5%.
In a study by Duman et al. (37) in the Turkish population, families with consanguineous marriage and with at least three children with hearing loss were screened and mutations in the TMPRSS3 gene were found with a frequency of 1.7%. Although segregation was found in two families in our study group, the TMPRSS3 gene is not one of the genes that commonly leads to hearing loss in light of the data obtained from the literature. It has been recommended that it should be studied in cases where variability is absent in genes in which mutations are commonly found, or screened with classic methods by adding array platforms in families who are found to have seggregation.
In our study, segreggation was found in the TMHS gene in one family. The TMHS gene encodes the tetraspan membrane protein in the hair cell sterocillia. It is thought that it is involved in morphogenesis of hair cells, and mutations in this gene may lead to vestibular dysfunction, corti injury, and anomalies in hair coils of the inner ear (47). The effect of mutations in this gene on hearing loss was defined in mice for the first time and later by Shabbir et al. (48) in subsequent studies in two families including one from Pakistan and one from India. Subsequently, two different mutations (c.649delG, p.Glu216Argfs*26 and c.494C>T, p.Thr165Met) were found in the TMHS gene in a study conducted with two large Turkish families by Kalay et al. (13). In the same study, a c.649delG carrier state was found in control screenings. It is notable that only four mutations have been found in the TMHS gene in the literature, and two of these were observed in the Turkish population. There is a limited number of studies related with this gene. Therefore, it was added to the open array platform and included in our study.
Hearing loss was present in 62 of 122 individuals from 21 families included in our study. Homozygous c.35delG mutations were found in four families, heterozygous c.35delG mutations were found in three families and a c.457G>A polymorphism was found in one family in our study group. In one family, which was found to have a heterozygous 35delG mutation in the GJB2 gene, a heterozygous c.−23+1G>A mutation was also present. Heterozygous p.C169* (c.507C>A) mutations, which have not been described previously, were found in the other two families that were found to have heterozygous 35delG mutation in our study. Twelve families that were not found to have mutations in the GJB2 gene were examined in terms of 11 genes, which are known to lead to hearing loss, using the open array method. The mutation in question was investigated by adding one mutation (c.250C>T) to the platform instead of SNP, only for the TMIE gene. The c.250C>T mutation was found in the TMIE gene in one family. Informative homozygosity was found for the OTOF gene in one family, for the TMPRSS3 gene in one family, and for all three genes including the TMHS, OTOF, and TMPRSS3 genes in one family. We plan to perform sequence analysis in further studies in families that were determined to have informative homozygosity in our study. No genetic variability was found in eight of 21 families evaluated in this study and it we plan to investigate the genetic variabilities leading to hearing loss in further studies using more advanced and comprehensive techniques in these families and in the family that was found to have a c.457G>A polymorphism.
There are still unknown aspects of the etiology and development of hearing loss despite the high number of studies. It is to be expected that many different genes are involved because the inner ear and the mechanism of hearing have an extremely complex structure. As supported by our study, it is recommended that the GJB2 gene should be screened primarily using sequence analysis in candidate gene studies directed at hearing loss, and the known genes should subsequently be excluded using panels that have been used in recent years. Preparation of panels should be made according to populations, and the genes and mutations that are observed more commonly in different populations should have priority. The fact that the c.250C>T mutation in the TMIE gene, which is observed rarely, was found in our study group of just 21 families supports this recommendation.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this study from the ethics committee of Erciyes University (04.01.2011, 2011/53).
Informed Consent: Signed informed consent was obtained from each participant.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - A.S., D.D., A.S., G.B., F.C., M.A.S., M.E., M.T., M.D.; Design - A.S., D.D., A.S., G.B., F.C., M.A.S., M.E., M.T., M.D.; Supervision - A.S., D.D., A.S., G.B., F.C., M.A.S., M.E., M.T., M.D.; Resources - M.T., M.D.; Materials - A.S., M.T., M.D.; Data Collection and/or Processing - A.S., D.D., A.S., G.B., F.C., M.A.S., M.E., M.T., M.D.; Analysis and/or Interpretation - A.S., M.T., M.D.; Literature Search - A.S., D.D., M.T., M.D.; Writing Manuscript - A.S., D.D., M.T., M.D.; Critical Review - A.S., D.D., M.T., M.D.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: This work was supported by the both Erciyes University, Scientific Research Projects Office’s project (the project number TSU-11-3483) and a joint project of Miami University (National Institutes of Health Grant number R01DC009645) and Ankara University (the project number 2011ABH06739003).
References
- 1.Kalatzis V, Petit C. The fundamental and medical impacts of recent progress in research on hereditary hearing loss. Hum Mol Genet. 1998;7:1589–97. doi: 10.1093/hmg/7.10.1589. https://doi.org/10.1093/hmg/7.10.1589. [DOI] [PubMed] [Google Scholar]
- 2.Willems PJ. Genetic causes of hearing loss. N Engl J Med. 2000;342:1101–9. doi: 10.1056/NEJM200004133421506. https://doi.org/10.1056/NEJM200004133421506. [DOI] [PubMed] [Google Scholar]
- 3.Gorlin RJ, Toriello HV, Cohen MM, editors. Hereditary hearing loss and its syndromes. Oxford: Oxford University Press; 1995. pp. 337–9. [Google Scholar]
- 4.Morton CC1, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354:2151–64. doi: 10.1056/NEJMra050700. https://doi.org/10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 5.The Hereditary Hearing loss Homepage [Internet] Available from: http://hereditaryhearingloss.org. [Edited 13 Dec 2011]
- 6.The Hereditary Hearing loss Homepage [Internet] Available from: http://hereditaryhearingloss.org/main.aspx?c=.HHH&n=86162. [Edited 13 Dec 2011]
- 7.Tekin M, Arnos K, Pandya A. Advances in hereditary deafness. Lancet. 2001;358:1082–90. doi: 10.1016/S0140-6736(01)06186-4. https://doi.org/10.1016/S0140-6736(01)06186-4. [DOI] [PubMed] [Google Scholar]
- 8.Calapoğlu NŞ. Sendromik olmayan işitme kaybının genetiği. S.D.Ü Tıp Fak Derg. 2006;13:37–46. [Google Scholar]
- 9.Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–3. doi: 10.1038/387080a0. https://doi.org/10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 10.Tekin M, Duman T, Bogoclu G, et al. Spectrum of GJB2 mutations in Turkey comprises both Caucasian and Oriental variants: roles of parental consanguinity and assortative mating. Hum Mutat. 2003;21:552–3. doi: 10.1002/humu.9137. https://doi.org/10.1002/humu.9137. [DOI] [PubMed] [Google Scholar]
- 11.Choi SY, Lee KY, Kim HJ, et al. Functional evaluation of GJB2 variants in nonsyndromic hearing loss. Mol Med. 2011;17:550–6. doi: 10.2119/molmed.2010.00183. https://doi.org/10.2119/molmed.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tekin M, Arici ZS. Genetic epidemiological studies of congenital/prelingual deafness in Turkey: population structure and mating type are major determinants of mutation identification. Am J Med Genet A. 2007;143A:1583–91. doi: 10.1002/ajmg.a.31702. https://doi.org/10.1002/ajmg.a.31702. [DOI] [PubMed] [Google Scholar]
- 13.Kalay E, Caylan R, Kremer H, Brouwer APM, Karagüzel A. GJB2 mutations in Turkish patients with ARNSHL: prevelance and two novel mutations. Hear Res. 2005;203:88–93. doi: 10.1016/j.heares.2004.11.022. https://doi.org/10.1016/j.heares.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Denoyelle F, Weil D, Maw MA, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997;6:2173–7. doi: 10.1093/hmg/6.12.2173. https://doi.org/10.1093/hmg/6.12.2173. [DOI] [PubMed] [Google Scholar]
- 15.Cohn ES, Kelley PM, Fowler TW, et al. Clinicalstudies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1) Pediatrics. 1999;103:546–50. doi: 10.1542/peds.103.3.546. https://doi.org/10.1542/peds.103.3.546. [DOI] [PubMed] [Google Scholar]
- 16.Van Laer L, Cryns K, Smith RJ, Van Camp G. Nonsyndromic hearing loss. Ear Hear. 2003;24:275–88. doi: 10.1097/01.AUD.0000079805.04016.03. https://doi.org/10.1097/01.AUD.0000079805.04016.03. [DOI] [PubMed] [Google Scholar]
- 17.Tekin M, Akar N, Cin S, et al. Connexin 26 (GJB2) mutations in the Turkish population: implications for the origin and high frequency of the 35delG mutation in Caucasians. Hum Genet. 2001;108:385–9. doi: 10.1007/s004390100507. https://doi.org/10.1007/s004390100507. [DOI] [PubMed] [Google Scholar]
- 18.Uyguner O, Emiroglu M, Uzumcu A, et al. Frequencies of gap- and tight-junction mutations in Turkish families with autosomal-recessive non-syndromic hearing loss. Clin Genet. 2003;64:65–9. doi: 10.1034/j.1399-0004.2003.00101.x. https://doi.org/10.1034/j.1399-0004.2003.00101.x. [DOI] [PubMed] [Google Scholar]
- 19.Petersen MB, Willems PJ. Non-syndromic, autosomal-recessive deafness. Clin Genet. 2006;69:371–92. doi: 10.1111/j.1399-0004.2006.00613.x. https://doi.org/10.1111/j.1399-0004.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 20.Liu XZ, Xia XJ, Ke XM, et al. The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum Genet. 2002;111:394–7. doi: 10.1007/s00439-002-0811-6. https://doi.org/10.1007/s00439-002-0811-6. [DOI] [PubMed] [Google Scholar]
- 21.Baris I, Kilinc MO, Tolun A. Frequency of the 35delG mutation in the connexin 26 gene in Turkish hearing-impaired patients. Clin Genet. 2001;60:452–5. doi: 10.1034/j.1399-0004.2001.600608.x. https://doi.org/10.1034/j.1399-0004.2001.600608.x. [DOI] [PubMed] [Google Scholar]
- 22.Tekin M, Bogoclu G, Arican ST, et al. Evidence for single origins of 35delG and delE120 mutations in the GJB2 gene in Anatolia. Clin Genet. 2005;67:31–7. doi: 10.1111/j.1399-0004.2004.00334.x. https://doi.org/10.1111/j.1399-0004.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- 23.Sobe T, Vreugde S, Shahin H, et al. The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population. Hum Genet. 2000;106:50–7. doi: 10.1007/s004390051009. https://doi.org/10.1007/s004390051009. [DOI] [PubMed] [Google Scholar]
- 24.Denoyelle F, Marlin S, Weil D, et al. Clinical features of the prevalent form of the childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet. 1999;353:1298–303. doi: 10.1016/S0140-6736(98)11071-1. https://doi.org/10.1016/S0140-6736(98)11071-1. [DOI] [PubMed] [Google Scholar]
- 25.Shahin H, Walsh T, Sobe T, et al. Genetics of congenital deafness in the Palestinian population: multiple connexin26 alleles with shared origins in the Middle East. Hum Genet. 2002;110:284–9. doi: 10.1007/s00439-001-0674-2. https://doi.org/10.1007/s00439-001-0674-2. [DOI] [PubMed] [Google Scholar]
- 26.Seeman P, Sakmoryova I. High prevalence of the IVS1+1 G-A/GJB2 mutation among Czech hearing impaired patients in the coding region of GJB2. Clin Genet. 2006;69:410–3. doi: 10.1111/j.1399-0004.2006.00602.x. https://doi.org/10.1111/j.1399-0004.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- 27.Santos R, Aulchenko Y, Huygen P, et al. Hearing impairment in Dutch patients with connexin 26 (GJB2) and connexin 30 (GJB6) mutations. Int J Pediatr Otorhinolaryngol. 2005;69:165–74. doi: 10.1016/j.ijporl.2004.08.015. https://doi.org/10.1016/j.ijporl.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Yuan Y, Yu F, Wang G, et al. Prevalence of the GJB2 IVS1+1G >A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2. J Transl Med. 2010;8:127. doi: 10.1186/1479-5876-8-127. https://doi.org/10.1186/1479-5876-8-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sırmacı A, Akcayoz-Duman D, Tekin M. The c.IVS1+1G>A mutation in the GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J Genet. 2006;85:213–6. doi: 10.1007/BF02935334. https://doi.org/10.1007/BF02935334. [DOI] [PubMed] [Google Scholar]
- 30.Padma G, Ramchander PV, Nandur UV, Padma T. GJB2 and GJB6 gene mutations found in Indian probands with congenital hearing impairment. J Genet. 2009;88:267–72. doi: 10.1007/s12041-009-0039-5. https://doi.org/10.1007/s12041-009-0039-5. [DOI] [PubMed] [Google Scholar]
- 31.Bayazit YA, Cable BB, Cataloluk O, et al. GJB2 gene mutations causing familial hereditary deafness in Turkey. Int J Pediatr Otorhinolaryngol. 2003;67:1331–5. doi: 10.1016/j.ijporl.2003.08.003. https://doi.org/10.1016/j.ijporl.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Meşe G, Londin E, Mui R, Brink PR, White TW. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum Genet. 2004;115:191–9. doi: 10.1007/s00439-004-1142-6. https://doi.org/10.1007/s00439-004-1142-6. [DOI] [PubMed] [Google Scholar]
- 33.Open database: The National Center for Biotechnology Information. Available from: http://www.ncbi.nlm.nih.gov/snp/?term=rs111033186&SITE=NcbiHome&submit=Go. [Edited 2012]
- 34.Sırmacı A, Oztürkmen-Akay H, Erbek S, et al. A founder TMIE mutation is a frequent cause of hearing loss in southeastern Anatolia. Clin Genet. 2009;75:562–7. doi: 10.1111/j.1399-0004.2009.01183.x. https://doi.org/10.1111/j.1399-0004.2009.01183.x. [DOI] [PubMed] [Google Scholar]
- 35.Mitchem KL, Hibbard E, Beyer LA, et al. Mutation of the novel gene Tmie results in sensory cell defects in the inner ear of spinner, a mouse model of human hearing loss DFNB6. Hum Mol Genet. 2002;11:1887–98. doi: 10.1093/hmg/11.16.1887. https://doi.org/10.1093/hmg/11.16.1887. [DOI] [PubMed] [Google Scholar]
- 36.Yang JJ, Su MC, Chien KH, Hsin CH, Li SY. Identification of novel variants in the TMIE gene of patients with nonsyndromic hearing loss. Int J Pediatr Otorhinolaryngol. 2010;74:489–93. doi: 10.1016/j.ijporl.2010.02.001. https://doi.org/10.1016/j.ijporl.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet Test Mol Biomarkers. 2011;15:29–33. doi: 10.1089/gtmb.2010.0120. https://doi.org/10.1089/gtmb.2010.0120. [DOI] [PubMed] [Google Scholar]
- 38.Naz S, Giguere CM, Kohrman DC, et al. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet. 2002;71:632–6. doi: 10.1086/342193. https://doi.org/10.1086/342193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang DY, Wang YC, Weil D, et al. Screening mutations of OTOF gene in Chinese patients with auditory neuropathy, including a familial case of temperature-sensitive auditory neuropathy. BMC Med Genet. 2010;11:79. doi: 10.1186/1471-2350-11-79. https://doi.org/10.1186/1471-2350-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi BY, Ahmed ZM, Riazuddin S, et al. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin Genet. 2009;75:237–43. doi: 10.1111/j.1399-0004.2008.01128.x. https://doi.org/10.1111/j.1399-0004.2008.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiu YH, Wu CC, Lu YC, et al. Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol Neurootol. 2010;15:364–74. doi: 10.1159/000293992. https://doi.org/10.1159/000293992. [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez-Ballesteros M, Reynoso R, Olarte M, et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum Mutat. 2008;29:823–31. doi: 10.1002/humu.20708. https://doi.org/10.1002/humu.20708. [DOI] [PubMed] [Google Scholar]
- 43.Scott HS, Kudoh J, Wattenhofer M, et al. Insertion of betasatellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet. 2001;27:59–63. doi: 10.1038/83768. https://doi.org/10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 44.Lee K, Khan S, Islam A, et al. Novel TMPRSS3 variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin Genet. 2012;82:56–63. doi: 10.1111/j.1399-0004.2011.01695.x. https://doi.org/10.1111/j.1399-0004.2011.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guipponi M, Antonarakis SE, Scott HS. TMPRSS3, a type II transmembrane serine protease mutated in non-syndromic autosomal recessive deafness. Front Biosci. 2008;13:1557–67. doi: 10.2741/2780. https://doi.org/10.2741/2780. [DOI] [PubMed] [Google Scholar]
- 46.Wattenhofer M, Di Iorio MV, Rabionet R, et al. Mutations in the TMPRSS3 gene are a rare cause of childhood nonsyndromic deafness in Caucasian patients. J Mol Med (Berl) 2002;80:124–31. doi: 10.1007/s00109-001-0310-6. https://doi.org/10.1007/s00109-001-0310-6. [DOI] [PubMed] [Google Scholar]
- 47.Longo-Guess CM, Gagnon LH, Cook SA, Wu J, Zheng QY, Johnson KR. A missense mutation in the previously undescribed gene Tmhs underlies deafness in hurry-scurry (hscy) mice. Proc Natl Acad Sci USA. 2005;102:7894–9. doi: 10.1073/pnas.0500760102. https://doi.org/10.1073/pnas.0500760102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shabbir MI, Ahmed ZM, Khan SY, et al. Mutations of human TMHS cause recessively inherited non-syndromic hearing loss. J Med Genet. 2006;43:634–40. doi: 10.1136/jmg.2005.039834. https://doi.org/10.1136/jmg.2005.039834. [DOI] [PMC free article] [PubMed] [Google Scholar]