

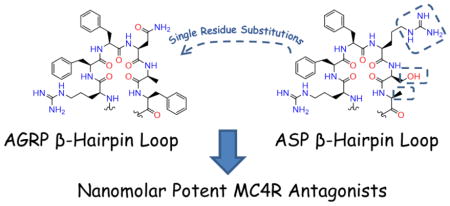
Abstract
The melanocortin system consists of five reported receptors, agonists from the proopiomelanocortin gene transcript, and two antagonists, agouti-signaling protein (ASP) and agouti-related protein (AGRP). For both ASP and AGRP, the hypothesized Arg-Phe-Phe pharmacophores are on exposed β-hairpin loops. In this study, the Asn and Ala positions of a reported AGRP macrocyclic scaffold (c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro]) were explored with 14-compound and 8-compound libraries, respectively, to generate more potent, selective melanocortin receptor antagonists. Substituting diaminopropionic acid (Dap), DDap, and His at the Asn position yielded potent MC4R ligands, while replacing Ala with Ser maintained MC4R potency. Since these substitutions correlate to ASP loop residues, an additional Phe to Ala substitution was synthesized and observed to maintain MC4R potency. Seventeen compounds also possessed inverse agonist activity at the MC5R, the first report of this pharmacology. These findings are useful in developing molecular probes to study negative energy balance conditions and unidentified functions of the MC5R.
Keywords: AGRP, agouti, structure-activity relationships, melanocortin receptors, inverse agonist
Graphical Abstract
Introduction
The melanocortin system has been implicated in many biological pathways. To date, five melanocortin G protein-coupled receptors (GPCRs) have been identified. The melanocortin-1 receptor (MC1R) plays an important role in pigmentation.1,2 The melanocortin-2 receptor (MC2R) is linked to steroidogenesis,2 and is only stimulated by the endogenous adrenocorticotropic hormone (ACTH).3 The centrally expressed melanocortin-3 and -4 receptors (MC3R and MC4R) have been associated with food intake and energy homeostasis.4–11 Dysregulation or single nucleotide polymorphisms (SNPs) of the MC3R may predispose an individual to obesity,12 while SNPs in the MC4R have been demonstrated to result in an obese phenotype.13 The melanocortin-5 receptor (MC5R) is expressed widely throughout the body with its physiological role less characterized,14,15 although it has been linked to exocrine function in mice.16 These melanocortin receptors (MCRs) are stimulated by endogenous agonists derived from the proopiomelanocortin gene transcript,17 which is processed into α-MSH, β-MSH, γ-MSH, and ACTH among other peptide products, as previously reviewed.18,19 The melanocortin system also possesses naturally occurring antagonists, agouti-signaling protein (ASP)20,21 and agouti-related protein (AGRP).22–24 Overexpression of ASP or AGRP in transgenic mice results in an obese phenotype, with the expression level of either protein correlating to weight gain.25,26 Transgenic mice overexpressing ASP recapitulate the lethal yellow (Ay) mouse strain, characterized by ectopic ASP expression,21,27 a yellow coat color (presumably due to MC1R antagonism),28 and an obese phenotype (originally hypothesized to be due to MC4R antagonism, but may also involve MC3R antagonism).28,29 While a yellow coat color is not observed (implying a lack of MC1R antagonism), ectopic expression of AGRP increases weight gain,26 similar to the dose-dependent increase in food intake with icv administration of AGRP.10,30 The AGRP-mediated increased consumption is observed in both MC3R and MC4R knockout mice, implying AGRP antagonism of both receptors may be responsible for the observed phenotype.10 Developing probes from ASP and AGRP may therefore be important in investigating the origins and potential therapeutics for obesity. Additionally, as potent orexigenic (appetite-inducing) compounds, ligands developed from ASP and AGRP may be useful in the treatment of negative energy balance disorders, including cachexia, aneroxia, and failure to thrive in infants.31–33
Full length ASP is 132 amino acids (Figure 1A), with the active form ASP(23-132) resulting from the cleavage of a 22-residue signal sequence.34–36 Recombinant mouse (m)ASP was reported to impair α-MSH-mediated cAMP production at the mMC1R28,37,38 and the human (h)MC4R,28 but did not at the rat (r)MC3R.28 The C-terminal domain of recombinant mASP is equipotent to ASP(23-132) in Xenopus melanophores35 and in murine B16F10 cells (both presumably express the MC1R),38 indicating this region possessing 5 disulfide bonds is responsible for the observed antagonist activity. Although recombinant mASP was purportedly not able to antagonize the rMC3R,28 later work with the recombinant hASP indicated this homolog was an antagonist at the hMC3R, with decreased potency relative to the hMC1R and hMC4R.39 The authors reported seeing a similar trend for recombinant mASP at the human receptors, but did not show these data.39 This functional activity correlated with a subsequent binding study which demonstrated that mASP was able to displace radiolabeled NDP-MSH at the mMC1R, mMC3R, and mMC4R with Ki app of 2.6, 190, and 54 nM, respectively.40 In the same report, an alanine positional scan of the C-terminal domain of mASP indicated that replacing the Arg116 and Phe118 residues of mASP decreased affinity at the mMC1R, mMC3R, and mMC4R; additionally, substituting Ala at the Phe117 of mASP decreased affinity at the mMC4R.40 To generate sufficient quantities of the hASP C-terminal domain for NMR analysis, a synthetic approach was explored.29 Although synthesis of the 53 residue C-terminal domain of hASP resulted in poor yields due to misfolded products, substitution of two residues (Q115Y, S124Y, corresponding to equivalent residues in AGRP) resulted in one major observed product that possessed the correct molecular mass for the properly folded protein.29 The synthetic hASP-YY possessed nanomolar antagonist potency at the hMC1R and hMC3R, and sub-nanomolar potency at the hMC4R.29 Within the C-terminal domain of ASP-YY, the C-terminal loop (residues Arg126-Asn131) has been implicated as necessary for potent MC1R binding.41
Figure 1.

A) Sequence alignment of human ASP and AGRP. The hypothesized Arg-Phe-Phe pharmacophore region is highlighted in red. The disulfide pairing is also indicated.
B) Sequence alignment of the postulated active loop of human and mouse ASP and AGRP. The conserved Arg-Phe-Phe is highlighted in red.
C) NMR solution structures of human AGRP(87-132) (PDB = 1HYK)50 and human ASP(80-132: Q115Y, S124Y) (PDB = 1Y7K).29 The β-hairpin loops are colored red and highlighted in the red box. The Arg-Phe-Phe tripeptide side-chains are drawn to illustrate their similar positions within the structures.
Agouti-related protein, AGRP, was first identified due to sequence homology and the Cys-spacing pattern with ASP (Figure 1A).22–24 Described as agouti-related protein,23 agouti-related peptide,42,43 or agouti-related transcript,22,24,26 AGRP possesses nanomolar antagonist pharmacology at the centrally expressed MC3R and MC4R,22,23 as well as inverse agonist activity at the MC4R.44,45 The biologically active form of AGRP has been hypothesized to be AGRP(83-132) following processing by proprotein convertase 1.46 The C-terminal domain of synthetic47,48 and recombinant AGRP23 has been demonstrated to be equipotent to longer AGRP variants.23,48 Like ASP, the C-terminal domain of AGRP possesses an Arg-Phe-Phe tripeptide sequence important for binding and antagonist activity (Figure 1B).49 Solution NMR structural studies of ASP-YY,29 the C-terminal domain of AGRP,50,51 and a shortened “mini-AGRP”52 have all indicated that the hypothesized Arg-Phe-Phe pharmacophores are located on similar solvent-exposed β-hairpin loops (Figure 1C). Truncation studies of hAGRP, mASP, and hASP indicated that cyclic octapeptide fragments based upon the active loops of these molecules (c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys], c[Cys-Arg-Phe-Phe-Gly-Ser-Ala-Cys], and c[Cys-Arg-Phe-Phe-Arg-Ser-Ala-Cys], respectively) were the minimal required sequences for MC4R binding affinity, with the hASP octapeptide also possessing affinity at the MC3R.49 While mASP was not further explored, elongation of hAGRP by two Tyr residues, H-Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-OH, was necessary for binding affinity at the MC3R and sub-micromolar functional activity at the MC4R.49 An additional four residues were required for functional antagonism at the MC3R, indicating the importance of amino acids outside the β-hairpin loop for MC3R potency.53 Although dodecapeptide and tetradecapeptides possess antagonist activity at the MC3R and MC4R, the potencies of truncated AGRP peptides are lower compared to AGRP.
Previously, it was hypothesized that inducing a β-hairpin structure in small AGRP-derived peptides containing the postulated Arg-Phe-Phe pharmacophore may result in more potent antagonists.54 A macrocyclic scaffold containing the active hexapeptide loop of ARGP cyclized through a DPro-Pro motif, c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro], was 50-fold less potent at the MC4R compared to AGRP.54 Further structure-activity relationship studies at the Asn position indicated basic residues (diaminopropionic acid [Dap], diaminobutyric acid [Dab], Orn, Lys, Arg) or Gly increased MC4R potency. The most potent substitution, the Asn to Dap replacement, resulted in a sub-nanomolar potent MC4R antagonist that was 160-fold selective for the MC4R over the MC3R, induced partial MC1R activation at 100 μM concentrations, and did not possess agonist activity at the MC5R.54 To further investigate this scaffold, a series of truncated macrocycles were synthesized containing either the Asn or Dap residues and replacing the Arg-Phe-Phe tripeptide antagonist sequence with the melanocortin agonist tetrapeptide His-DPhe-Arg-Trp or tripeptide DPhe-Arg-Trp sequences.55 This study examined if the AGRP DPro-Pro loop scaffold was amendable to substitutions that may result in melanocortin agonist activity as weight-management therapeutic leads. Two macrocycles were identified that possessed nanomolar agonist potencies at the MC4R, nanomolar to sub-nanomolar potencies at the MC1R and MC5R, and 30–40 nanomolar potencies at the MC3R, c[Pro-His-DPhe-Arg-Trp-Asn-Ala-Phe-DPro] and c[Pro-His-DPhe-Arg-Trp-Dap-Ala-DPro].55
Due to the increased ligand potency for basic and Gly substitutions at the Asn position in the octapeptide macrocyclic scaffold, an additional SAR study was performed at the Asn position. To explore the ligand-receptor interface, substitutions at the Asn position with basic, acidic, polar, non-polar, and aromatic groups were assayed at the mouse MCRs. A similar series of substitutions explored the Ala position. From these two series, a substitution pattern was observed that substitution of the equivalent residue type in the exposed loop of ASP maintained MC4R potency in this AGRP scaffold while other substitutions decreased potency. Therefore, the non-pharmacophore Phe residue was replaced with an Ala found in the active loops of both hASP and mASP to examine if insertion of ASP residues into an AGRP scaffold would maintain MC4R potency.
Results & Discussion
Peptide Synthesis and Characterization
Peptides were synthesized manually or using an automated peptide synthesizer with standard fluorenylmethoxycarbonyl (Fmoc) chemistry.56,57 The macrocyclization chemistry to form the amide bond between the Arg and Pro residues using BOP and HOBt has previously been reported.54,55 Following cyclization and side-chain deprotection, peptides were purified by semi-preparative reverse-phase high pressure liquid chromatography (RP-HPLC). Peptides were assessed for purity (>95%) by analytical RP-HPLC in two solvent systems (Table 1), and the correct molecular mass was confirmed through matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS, University of Minnesota Mass Spectrometry Laboratory).
Table 1.
Analytical Data for Peptides Synthesized in this Study.
| Peptide | Sequence | Retention Time (min)a
|
M (calc) | mass spectral analysis (M+1) | purity % | |
|---|---|---|---|---|---|---|
| system 1 | system 2 | |||||
| 1 | c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] | 18.1 | 26.8 | 976.5 | 977.5 | >99 |
| 2 | c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro] | 18.0 | 29.3 | 948.5 | 949.4 | >98 |
| 3 | c[Pro-Arg-Phe-Phe-Ala-Ala-Phe-DPro] | 18.9 | 28.7 | 933.5 | 934.3 | >99 |
| 4 | c[Pro-Arg-Phe-Phe-Abu-Ala-Phe-DPro] | 19.5 | 29.4 | 947.5 | 948.5 | >98 |
| 5 | c[Pro-Arg-Phe-Phe-Ser-Ala-Phe-DPro] | 18.8 | 29.2 | 949.5 | 950.3 | >99 |
| 6 | c[Pro-Arg-Phe-Phe-Thr-Ala-Phe-DPro] | 19.5 | 29.9 | 963.5 | 964.2 | >99 |
| 7 | c[Pro-Arg-Phe-Phe-Asp-Ala-Phe-DPro] | 18.4 | 28.5 | 977.5 | 978.5 | >99 |
| 8 | c[Pro-Arg-Phe-Phe-Glu-Ala-Phe-DPro] | 18.3 | 28.1 | 991.5 | 992.2 | >99 |
| 9 | c[Pro-Arg-Phe-Phe-DDap-Ala-Phe-DPro] | 18.1 | 29.4 | 948.5 | 949.4 | >95 |
| 10 | c[Pro-Arg-Phe-Phe-His-Ala-Phe-DPro] | 18.0 | 29.1 | 999.5 | 1000.2 | >96 |
| 11 | c[Pro-Arg-Phe-Phe-Nle-Ala-Phe-DPro] | 21.2 | 31.0 | 975.5 | 976.3 | >99 |
| 12 | c[Pro-Arg-Phe-Phe-Leu-Ala-Phe-DPro] | 21.0 | 30.8 | 975.5 | 976.3 | >99 |
| 13 | c[Pro-Arg-Phe-Phe-Val-Ala-Phe-DPro] | 20.4 | 30.8 | 961.5 | 962.2 | >99 |
| 14 | c[Pro-Arg-Phe-Phe-Phe-Ala-Phe-DPro] | 21.6 | 31.6 | 1009.5 | 1010.3 | >98 |
| 15 | c[Pro-Arg-Phe-Phe-Trp-Ala-Phe-DPro] | 23.2 | 30.7 | 1048.5 | 1049.2 | >97 |
| 16 | c[Pro-Arg-Phe-Phe-Asn-Asp-Phe-DPro] | 17.7 | 26.6 | 1020.5 | 1021.6 | >99 |
| 17 | c[Pro-Arg-Phe-Phe-Asn-Glu-Phe-DPro] | 17.4 | 26.7 | 1034.5 | 1035.6 | >98 |
| 18 | c[Pro-Arg-Phe-Phe-Asn-Lys-Phe-DPro] | 16.0 | 25.2 | 1033.6 | 1034.6 | >99 |
| 19 | c[Pro-Arg-Phe-Phe-Asn-His-Phe-DPro] | 16.2 | 25.4 | 1042.5 | 1043.6 | >99 |
| 20 | c[Pro-Arg-Phe-Phe-Asn-Phe-Phe-DPro] | 21.4 | 32.4 | 1052.5 | 1053.7 | >97 |
| 21 | c[Pro-Arg-Phe-Phe-Asn-Ser-Phe-DPro] | 17.7 | 27.2 | 992.5 | 993.6 | >96 |
| 22 | c[Pro-Arg-Phe-Phe-Asn-Leu-Phe-DPro] | 21.2 | 32.8 | 1018.5 | 1019.6 | >98 |
| 23 | c[Pro-Arg-Phe-Phe-Asn-Gly-Phe-DPro] | 17.6 | 26.5 | 962.5 | 963.5 | >96 |
| 24 | c[Pro-Arg-Phe-Phe-Asn-Ala-Ala-DPro] | 15.4 | 25.0 | 900.5 | 901.8 | >97 |
Peptide retention times (min) are reported for solvent system 1 (10% acetonitrile in 0.1% trifluoroacetic acid/water and a gradient to 90% acetonitrile over 35 min) and solvent system 2 (10% methanol in 0.1% trifluoroacetic acid/water and a gradient to 90% methanol over 35 min). An analytical Vydac C18 column (Vydac 218TP104) was used with a flow rate of 1.5 mL/min. The peptide purity was determined by HPLC at a wavelength of 214 nm.
In vitro AlphaScreen cAMP Assay
The compounds were assayed using the AlphaScreen cAMP assay in HEK293 cells stably expressing the mouse melanocortin 1, 3, 4, and 5 receptors according to the manufacturer’s instructions and as previously reported.58–60 The MC2R is only stimulated by ACTH and was therefore excluded from this study. Compounds were first assayed for agonist activity at the MCRs; ligands that did not possess full agonist activity at the MC3R and MC4R were then assayed for antagonist activity using a Schild paradigm61 and NDP-MSH as the agonist. Since the AlphaScreen assay is a competition assay (higher concentrations of ligand result in lower signal), concentration-activity curves were normalized to baseline and maximal NDP-MSH signal for illustrative purposes as previously described.58,59 Due to the inherent error associated with the assay in our laboratory, compounds that were within a 3-fold potency range were considered equipotent.
Asn Position Substitutions
Previously, the AGRP-derived macrocyclic octapeptide c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] was found to possess sub-micromolar antagonist potency at the MC4R (pA2 = 7.7), and substitution at the Asn position with basic residues or Gly further increased potency (pA2 values ranged from 8.3 to 9.1).54 The most potent substitution, Asn to diaminopropionic acid (Dap), resulted in a ligand that was equipotent to the C-terminal domain of AGRP at the MC4R.54 It was previously hypothesized that the active loop Asn residue in AGRP may be in close proximity to an Asp residue in the MC4R (Asp189), and that incorporation of basic residues into AGRP analogues at this position might form a novel salt bridge with the MC4R.47 While the increased potency observed for basic substitutions supported the postulated salt bridge, the increased potency for Gly suggested other residues might also be incorporated without diminishing potency. Therefore, an additional SAR study was performed at the Asn position to explore the side-chain requirements for antagonist potency and to further examine the hypothesized salt-bridge interaction. Short aliphatic residues (Ala and Abu) were utilized to mimic the steric space of Dap without the charge. Residues were included that could mimic the hydrogen bond donor and acceptor potential of Dap without the positive charge (Ser and Thr). The negatively charged Asp and Glu were incorporated to reverse the charge while varying side-chain length. Additional basic residues were also explored by inverting the stereochemistry (DDap) and incorporating a small charged heterocycle (His). Longer and/or branched aliphatic side chains Nle, Leu, and Val were integrated to examine the functional consequence of expanding the uncharged side chain, as were the aromatic side chains in Phe and Trp. The Dap substitution was synthesized as a control, and the C-terminal domain of AGRP [AGRP(86-132)] was run in parallel as an antagonist at the MC3R and MC4R using the AlphaScreen assay as an additional known ligand. All amino acid structures can found in Figure 2.
Figure 2.

Structures of the amino acids used in this study.
In the present study, AGRP(86-132) possessed identical pA2 values of 8.7 at both the MC3R and MC4R, similar to previous reported nanomolar potencies at these receptors (Table 2, Figure 3).22,23,41,46,48,53 The native sequence of the exposed β-hairpin loop (1) partially stimulated the MC1R at 100 μM concentrations (25% of the maximal NDP-MSH signal) and did not possess agonist activity at the MC3R or MC4R up to 100 μM. This peptide possessed sub-micromolar antagonist potency at the MC3R (pA2 = 6.3) and nanomolar potency at the MC4R (pA2 = 8.2; Figure 3). These values are comparable to a previous report where this peptide possessed micromolar agonist activity at the MC1R, and was an antagonist at the MC3R (pA2 = 6.4) and MC4R (pA2 = 7.7) using a β-galactatosidase reporter gene assay.54 Insertion of the Dap residue (2) resulted in partial activation of the MC1R (30% NDP-MSH maximal signal) and did not stimulate cAMP production at the MC3R or MC4R up to 100 μM concentrations. Peptide 2 possessed antagonist activity at the MC3R (pA2 = 6.5) and MC4R (pA2 = 8.7; Figure 3), similar to the previously reported pA2 values of 6.9 and 9.1, respectively (2 also was reported to partially activate the MC1R to 75% NDP-MSH at 100 μM).54 In both the previous and present study, 2 was an equipotent antagonist to AGRP(86-132) at the MC4R, and was over 100-fold selective for the MC4R over the MC3R despite using two different cAMP reporter assays.54
Table 2.
Pharmacology of AGRP Loop Analogues at the Mouse Melanocortin Receptors.a
| Peptide | Sequence | mMC1R
|
mMC3R
|
mMC4R
|
mMC5R
|
||
|---|---|---|---|---|---|---|---|
| EC50 (nM) | EC50 (nM) | pA2 | EC50 (nM) | pA2 | EC50 (nM) | ||
| NDP-MSH | 0.021±0.002 | 0.19±0.02 | 0.41±0.03 | 0.18±0.02 | |||
| hAGRP(86-132) | N.D. | 8.7±0.1 | 8.7±0.2 | N.D. | |||
| 1 | c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] | 25% @ 100μM | >100,000 | 6.3±0.1 | >100,000 | 8.2±0.1 | Inverse Agonist −10%, 130 nM |
| 2 | c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro] | 30% @ 100μM | >100,000 | 6.52±0.09 | >100,000 | 8.7±0.1 | Inverse Agonist −15%, 60 nM |
| 3 | c[Pro-Arg-Phe-Phe-Ala-Ala-Phe-DPro] | >100,000 | >100,000 | <5.5 | >100,000 | 7.32±0.09 | Inverse Agonist −20%, 250 nM |
| 4 | c[Pro-Arg-Phe-Phe-Abu-Ala-Phe-DPro] | >100,000 | >100,000 | <5.5 | >100,000 | 7.3±0.2 | Inverse Agonist −20%, 160 nM |
| 5 | c[Pro-Arg-Phe-Phe-Ser-Ala-Phe-DPro] | >100,000 | >100,000 | 5.9±0.2 | >100,000 | 7.7±0.3 | Inverse Agonist −25%, 140 nM |
| 6 | c[Pro-Arg-Phe-Phe-Thr-Ala-Phe-DPro] | >100,000 | >100,000 | 6.4±0.2 | >100,000 | 7.76±0.07 | Inverse Agonist −20%, 50 nM |
| 7 | c[Pro-Arg-Phe-Phe-Asp-Ala-Phe-DPro] | >100,000 | >100,000 | <5.5 | >100,000 | 6.79±0.07 | Inverse Agonist −25% @ 100 μM |
| 8 | c[Pro-Arg-Phe-Phe-Glu-Ala-Phe-DPro] | 20,500±500 | >100,000 | <5.5 | >100,000 | 7.00±0.09 | Inverse Agonist −10%, 700 nM |
| 9 | c[Pro-Arg-Phe-Phe-DDap-Ala-Phe-DPro] | >100,000 | >100,000 | 6.7±0.2 | >100,000 | 8.58±0.04 | Inverse Agonist −10%, 110 nM |
| 10 | c[Pro-Arg-Phe-Phe-His-Ala-Phe-DPro] | >100,000 | >100,000 | 6.46±0.07 | >100,000 | 8.3±0.1 | Inverse Agonist −15%, 10 nM |
| 11 | c[Pro-Arg-Phe-Phe-Nle-Ala-Phe-DPro] | >100,000 | >100,000 | <5.5 | >100,000 | 6.9±0.1 | >100,000 |
| 12 | c[Pro-Arg-Phe-Phe-Leu-Ala-Phe-DPro] | >100,000 | >100,000 | 5.7±0.2 | >100,000 | 7.34±0.04 | Inverse Agonist −20%, 110 nM |
| 13 | c[Pro-Arg-Phe-Phe-Val-Ala-Phe-DPro] | 55% @ 100μM | >100,000 | <5.5 | >100,000 | 5.9±0.1 | Inverse Agonist −25%, 510 nM |
| 14 | c[Pro-Arg-Phe-Phe-Phe-Ala-Phe-DPro] | >100,000 | >100,000 | <5.5 | >100,000 | 6.63±0.09 | >100,000 |
| 15 | c[Pro-Arg-Phe-Phe-Trp-Ala-Phe-DPro] | 80% @ 100μM | 40% @ 100μM | 5.70±0.08 | 50% @ 100μM | 6.81±0.08 | Inverse Agonist −20%, 150 nM |
| 16 | c[Pro-Arg-Phe-Phe-Asn-Asp-Phe-DPro] | 65% @ 100μM | >100,000 | <5.5 | >100,000 | <5.5 | Inverse Agonist −25% @ 100 μM |
| 17 | c[Pro-Arg-Phe-Phe-Asn-Glu-Phe-DPro] | 60% @ 100μM | >100,000 | <5.5 | >100,000 | <5.5 | >100,000 |
| 18 | c[Pro-Arg-Phe-Phe-Asn-Lys-Phe-DPro] | 60% @ 100μM | 45% @ 100μM | <5.5 | >100,000 | 6.11±0.01 | 45% @ 100μM |
| 19 | c[Pro-Arg-Phe-Phe-Asn-His-Phe-DPro] | Partial Agonist 40% NDP (2,600±1,200) | >100,000 | <5.5 | >100,000 | 6.07±0.09 | >100,000 |
| 20 | c[Pro-Arg-Phe-Phe-Asn-Phe-Phe-DPro] | 70% @ 100μM | 35% @ 100μM | 5.72±0.08 | 35% @ 100μM | 5.71±0.03 | 25% @ 100μM |
| 21 | c[Pro-Arg-Phe-Phe-Asn-Ser-Phe-DPro] | >100,000 | >100,000 | 6.2±0.1 | >100,000 | 8.2±0.2 | Inverse Agonist −30% @ 100 μM |
| 22 | c[Pro-Arg-Phe-Phe-Asn-Leu-Phe-DPro] | 80% @ 100μM | 25% @ 100μM | <5.5 | 25% @ 100μM | 6.18±0.05 | >100,000 |
| 23 | c[Pro-Arg-Phe-Phe-Asn-Gly-Phe-DPro] | >100,000 | >100,000 | <5.5 | >100,000 | 6.1±0.3 | Inverse Agonist −35% @ 100 μM |
| 24 | c[Pro-Arg-Phe-Phe-Asn-Ala-Ala-DPro] | >100,000 | >100,000 | 6.1±0.2 | >100,000 | 8.16±0.08 | Inverse Agonist −25% @100 μM |
The indicated errors represent the standard error of the mean determined from at least three independent experiments. The antagonistic pA2 values were determined using the Schild analysis and the agonist NDP-MSH. The use of >100,000 indicates that the compound was examined but lacked agonist activity at up to 100 μM concentrations. A percentage denotes the percent maximal stimulatory response observed at 100 μM concentrations but not enough stimulation was observed to determine an EC50 value. N.D. indicates values not determined. The use of <5.5 indicates that no antagonist potency was observed in the highest concentration ranged assayed (10,000, 5,000, 1,000, and 500 nM). Partial agonist indicates partial agonist activity was observed, along with the percentage of activation relative to NDP-MSH and the EC50. Inverse agonist indicates that inverse agonist pharmacology was observed with the percent decrease from basal indicated. For inverse agonists, an apparent potency is indicated for the inflection point on sigmoidal dose-response curves; if a decrease in cAMP signal was observed without a sigmoidal dose-response curve, the percent change from basal at 100 μM concentrations is indicated.
Figure 3.

Illustration of the antagonist pharmacology at the mMC3R and mMC4R for AGRP, 1, 2, 21, and 24.
While similar activity was observed for 1 and 2 at the MC1R, MC3R, and MC4R, a difference was observed at the MC5R. Previously, these compounds were reported to possess no activity at the MC5R at 100 μM concentrations using a β-galactosidase assay.54 In the present study, a decrease in normalized signal from basal activity was observed (Table 2, Figure 4), correlating to decreased levels of cAMP and suggesting an inverse agonist response for these ligands at the MC5R. From basal activity, a 10% and 15% dose-response difference in signal were observed for 1 (Figure 4) and 2, respectively. Since a sigmoidal curve was observed, 1 and 2 possessed apparent potencies (the inflection point on the curve) of 130 nM and 60 nM, respectively. This activity was only observed at the MC5R, and not the other MCRs in the present study. Since the MCRs are all stably expressed in HEK293 cells, if another factor such as cellular toxicity or signaling through an alternative receptor was responsible, the decreased signal would be expected at additional melanocortin receptors. It therefore appears that the observed decreased levels of cAMP may be the result of inverse agonist activity at the MC5R. While many ligands displayed a similar sigmoidal dose-response compared to 1 and 2 (Figure 4, 4, 5, and 8 as examples), some ligands decreased cAMP levels at the MC5R without plateauing at 100 μM concentrations (Figure 4, 21 and 24 as examples). The inverse agonist response of these compounds is reported as the percent decreased from basal at 100 μM concentrations.
Figure 4.

Illustration of the pharmacology at the MC5R for 1, 4, 5, 8, 18, 19, 21, and 24.
Substitution of the short aliphatic Ala (3) or Abu (4) residues at the Asn position resulted in ligands that were unable to stimulate any receptor tested. At the MC5R, 3 and 4 (Figure 4) decreased from basal signal 20% and had apparent potencies of 250 nM and 160 nM, respectively. Both peptides did not possess antagonist activity at the MC3R, and were 8-fold less potent at the MC4R compared to 1 (pA2 = 7.3 for both). No agonist activity was observed when Asn was replaced with the polar amino acids Ser (5) or Thr (6) at the MC1R, MC3R, or MC4R. These peptides produced inverse agonist activity of 25% (with an apparent potency of 140 nM) and 20% (50 nM) for 5 (Figure 4) and 6, respectively. Peptide 5 possessed antagonist potency at the MC3R (pA2 = 5.9) and MC4R (pA2 = 7.7), similar to the response of 6 (pA2 = 6.4 and 7.8 at the MC3R and MC4R). When Asn was substituted with the acidic Asp residue (7), no agonist activity was observed for the MC1R, MC3R, and MC4R, while a 25% inverse agonist response was observed at 100 μM concentrations at the MC5R. This peptide did not possess antagonist activity at the MC3R, and was 25-fold less potent at the MC4R compared to 1. Elongating the acidic side chain by one methylene unit, 8, resulted in the only peptide in this study with full MC1R agonist activity (EC50 = 20.5 μM). This Glu substitution did not result in measurable activity at the MC3R, had a 10% inverse agonist response (apparent potency = 700 nM) at the MC5R, and was a 16-fold less potent antagonist at the MC4R compared to 1. When substituting the basic residues DDap or His (9 or 10, respectively), no agonist activity was observed at the MC1R, MC3R, and MC4R up to 100 μM concentrations. Similar 10% inverse agonist responses were observed at the MC5R, with apparent potencies of 110 nM (9) and 10 nM (10). Both substitutions possessed sub-micromolar antagonist potencies at the MC3R (pA2 values of 6.7 and 6.5) and nanomolar antagonist potencies at the MC4R (pA2 values of 8.6, and 8.3). For the longer and/or branched aliphatic substitutions Nle, Leu, and Val (11, 12, and 13), the Val substituted 13 stimulated the MC1R to 55% the maximal signal of NDP-MSH at 100 μM. These three ligands did not produce an agonist response at any other MCR. At the MC5R, 12 and 13 decreased the basal signal 20% and 25%, respectively, with apparent potencies of 110 nM and 510 nM. Peptide 12 was the only ligand of the three to possess micromolar antagonist potency at the MC3R (pA2 = 5.7). These aliphatic substitutions possessed a range of potencies at the MC4R (pA2 = 6.9, 7.3, and 5.9 for 11, 12, and 13, respectively), and were at least 8-fold less potent than 1. When substituting the aromatic amino acid Phe (14), only antagonist activity at the MC4R was observed (pA2 = 6.6). Incorporation of Trp, 15, resulted in a ligand that partially stimulated the MC1R, MC3R, and MC4R (80%, 40%, and 50% maximal NDP-MSH signal) at 100 μM concentrations. This substitution resulted in a 20% inverse agonist response with an apparent potency of 150 nM at the MC5R, and antagonist activity at the MC3R and MC4R (pA2 =5.7 and 6.8, respectively).
Certain potency trends at the MC4R were observed when grouping these AGRP-derived macrocycles varied at the Asn position by side-chain type. Residues containing basic side chains (Dap, DDap, and His) possessed the highest MC4R potency, similar to the previous report.54 These results support a potential salt-bridge between this position and the acidic residue Asp189 of the MC4R, as previously hypothesized.47 Interestingly, the stereochemistry of the basic charge did not appear significant, as the Dap (pA2 = 8.7) and DDap (pA2 = 8.6) were equipotent at the MC4R. Amino acids with side chains capable of donating and accepting hydrogen bonds (Ser, Thr, and Asn) possessed slightly lower potency at the MC4R compared to basic substitutions. Aliphatic substitutions of similar size to Dap (Ala and Abu) or Asn (Leu) possessed similar potencies, less than the hydrogen bond donor capable side-chains. Substitution of Asn with Asp, replacing an amide with a carboxylic acid, decreased potency more than 10-fold, indicating a negative charge adversely impacted activity. The remaining acidic, aliphatic, and aromatic substitutions all possessed similar potency at the MC4R, with pA2 ≤ 7. The Val substituted ligand (13) was the only compound with micromolar or higher potency at the MC4R (pA2 = 5.9). Although Val is similar in structure to Thr (both possess a branched side chain and differ by a methyl versus hydroxyl substitution), the Thr substituted 6 was 70-fold more potent compared to 13. At the MC5R, no trends in percent inverse agonist activity of apparent potency were observed.
Ala Position Substitutions
While SAR studies for the Asn and two pharmacophore Phe positions have been reported on this scaffold,54 the Ala position has not previously been studied. One structural model of AGRP interacting with the MC4R indicated that this Ala position of AGRP may be in close proximity to the His264 residue of the MC4R.62 Therefore, it was hypothesized that incorporating an acidic amino acid into the corresponding Ala position in the truncated AGRP macrocycle may result in the formation of a new salt bridge and increase potency. A library of 8 compounds varied at the Ala position was synthesized and assayed at the mMCRs. To probe for a potential salt bridge with His264, the acidic residues Asp and Glu were included. Additional residues were incorporated to begin to probe the ligand requirements at this position for potent melanocortin receptor activity including the basic Lys and His, aromatic Phe, hydrogen bond donor and acceptor Ser, aliphatic Leu, and small Gly.
While the Ala position in the active loop of AGRP was previously hypothesized to be in close proximity to His264, substitution of Asp (16) or Glu (17) did not result in observable agonist or antagonist activity at the MC3R and MC4R. Both peptides partially stimulated the MC1R at 100 μM concentrations (65% and 60% for 16 and 17, respectively) and 16 produced an inverse agonist response at the MC5R (25% at 100 μM). Incorporation of a basic Lys (18) resulted in a ligand that partially stimulated the MC1R (60%), MC3R (45%), and MC5R (45%; Figure 4), and possessed sub-micromolar antagonist potency at the MC4R (pA2 = 6.1). Identical antagonist potency at the MC4R was observed for a His substitution (19, pA2 = 6.1) compared to 18, while a partial agonist response was observed at the MC1R (40% NDP-MSH efficacy with an EC50 = 3,000 nM). This compound did not possess activity at the MC5R (Figure 4). Substitution of an aromatic Phe (20) resulted in partial stimulation of all the MCRs assayed at 100 μM concentrations (70%, 35%, 35%, and 25% of the maximal NDP-MSH signal at the MC1R, MC3R, MC4R, and MC5R, respectively), as well as micromolar antagonist potencies at the MC3R and MC4R (pA2 = 5.7 for both receptors). Incorporating the polar Ser capable of donating and accepting hydrogen bonds resulted in peptide 21, which did not possess agonist activity at the MC1R, MC3R, and MC4R up to 100 μM concentrations, and generated an inverse agonist response at the MC5R (Figure 4, 30% at 100 μM). Peptide 21 possessed sub-micromolar antagonist potency at the MC3R (pA2 = 6.2), and was the only ligand in the Ala substitution series that was equipotent to 1 at the MC4R (pA2 = 8.2; Figure 3). The aliphatic Leu substitution (22) induced a partial agonist response at the MC1R, MC3R, and MC4R (80%, 25%, and 25%, respectively), and was a sub-micromolar antagonist at the MC4R (pA2 = 6.2). Incorporating the small Gly residue (23) resulted in a ligand that possessed inverse agonist activity at the MC5R (35% at 100 μM concentrations) and sub-micromolar antagonist potency at the MC4R (pA2 = 6.1).
The Ala position had previously been postulated to be near the His264 residue in the MC4R,62 and it was therefore hypothesized that incorporating a negatively charged residue may result in the formation of a novel salt bridge. If correct, replacement of the Ala with two acidic amino acids (Asp and Glu) might be expected to increase antagonist potency. However, both 16 and 17 did not possess antagonist activity at the MC3R or MC4R. While the current study does not support the postulated Ala-His264 proximity, it is possible that a longer form of AGRP orients this position in a different conformation that may result in a productive interaction. Altering the side chain stereochemistry, elongating/shortening a side chain possessing a negative charge, or utilizing longer AGRP substituted derivatives could be utilized to further examine this hypothesis.
Phe Position Substitution
Unlike substitutions at the Asn position, which resulted in a range of MC4R potencies, only one ligand substituted at the Ala position (Ser, 21) was equipotent to the parent macrocycle 1 (pA2 = 8.2 for both compounds). All other substitutions decreased potency more than 100-fold (pA2 < 6.2) relative to 1, perhaps indicating a more stringent structural requirement for ligand-receptor interactions at this position. While attempting to rationalize why Ser might be tolerated at the Ala position, it was observed that the equivalent loop position in both mouse and human ASP is a Ser (Figure 1B). Another compelling observation was that the equivalent position of Asn in the active loop of AGRP was either a basic Arg (human) or Gly (mouse) residue in ASP, a substitution pattern described herein and previously as possessing potent MC4R antagonism.54 To extend these observations, it was hypothesized that substitution of the non-pharmacophore Phe position in the AGRP loop with Ala (found in the equivalent ASP position) would maintain MC4R antagonist potency. The resulting Phe to Ala substitution, 24, possessed equipotent MC4R antagonist potency (pA2 = 8.2; Figure 3) compared to the parent macrocycle 1, supporting the observations that ASP loop residues substituted onto an AGRP macrocyclic template maintain MC4R antagonist potency. This peptide possessed sub-micromolar antagonist potency at the MC3R (pA2 = 6.1), no agonist activity at the MC1R up to 100 μM concentrations, and resulted in inverse agonist activity at the MC5R (25% at 100 μM, Figure 4).
Thus, single substitutions of ASP active loop residues into a macrocyclic AGRP-derived peptide result in the maintenance of MC4R antagonist potency and approximately 100-fold decrease in MC3R antagonist potency, a similar decrease to the parent ligand. In the current study, AGRP was shown to be equipotent at the MC3R and MC4R (pA2 = 8.7 at both receptors), in agreement with prior reports of nanomolar potencies at both receptors.22,23,53 The decreased MC3R potency has been observed in many AGRP truncated analogs, and was described by Tota et al. who found the minimal fragment needed to bind to the MC4R (H-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-OH) was unable to bind the MC3R.49 Acetylating the N-terminal and amidating the C-terminal of this octapeptide was sufficient to achieve micromolar MC4R antagonist potency.49 Further elongation by adding Tyr residues to both terminals was required to achieve MC3R binding and resulted in sub-micromolar potency at the MC4R.49,53 Two residues added to both the N- and C-terminals (Thr-Ala-Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-Ala-Arg-NH2) or four residues added to the C-terminal (Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-Ala-Arg-Lys-Leu-NH2) were required for sub-micromolar potency at the MC3R.53 The prior study with DPro-Pro cyclized AGRP macrocycles also resulted in more potent antagonism at the MC4R relative to the MC3R.54 Therefore, a common trend of truncated AGRP analogs is decreased potency at the MC3R, perhaps indicating that additional residues outside of the active loop are necessary for potent interaction at the MC3R.
While decreased MC3R potency is observed in many AGRP truncation studies, ASP was first described to interfere with α-MSH stimulation of the MC1R and MC4R, and not affect the MC3R and MC5R.28 One interpretation of the current data, assuming ASP is an antagonist at the MC1R and MC4R while AGRP antagonizes the MC3R and MC4R, is that replacement of AGRP residues in the active loop sequence with ASP residues would be expected to maintain MC4R antagonism while decreasing and/or minimizing MC3R potency. However, such an interpretation would ignore subsequent publications indicating ASP was capable of binding40 and functionally antagonizing39 the MC3R. Throughout all of these conflicting reports, a recombinant ASP (expressed using a baculovirus construct in insect cells) was utilized, potentially resulting in varying degrees of ASP purity and difficulty in accurately assessing the concentration of ASP.
To generate a pure ASP for functional characterization and NMR studies, McNulty et al. attempted to chemically synthesize the C-terminal domain of ASP,29 previously shown to be as active as the full-length construct.35 However, when attempting to cyclize the 53-residue fragment, less than 10% of the linear peptide folded with the correct disulfide bond pairing, with greater than 90% of the observed products resulting from incorrect disulfide bond formation.29 This is unlike the chemical synthesis of the C-terminal domain of AGRP, which correctly folds to the proper disulfide bonding when put under oxidative conditions.48,50 In order to generate sufficient quantities of chemically synthesized ASP, two Tyr residues of AGRP were incorporated into the ASP synthesis (Gln115Tyr and Ser124Tyr) to generate a double-substituted form of ASP that properly folded under oxidative conditions.29 This ASP-YY ligand was shown to possess potent antagonism at the MC3R (2.6 nM) as well as the MC1R (3.9 nM) and MC4R (0.5 nM), and incorporation of the two Tyr residues was crucial in yielding sufficient product for NMR studies. However, the addition of these Tyr amino acids confounds the activity of ASP at the MC3R, since it is impossible to discern if ASP-YY without the Tyr would retain the same activity, if the Tyr residues induce an altered conformation of the ligand, or if the Tyr amino acids result in novel ligand-receptor interactions. Despite these limitations, the in vitro pharmacological profile of ASP-YY at the MC1R, MC3R, and MC4R is similar to purified forms of recombinant ASP,39,40 supporting the hypothesis that ASP can interact at the MC3R.
In addition to being found in the active loop of ASP, select residue types in the current study are also found in species variants of AGRP. A PubMed BLASTp search of the sequence Cys-Arg-Phe-Phe-Lys-Ala-Phe-Cys (Asn to Lys) found this sequence to be in the AGRP precursor peptide of Carassius auratus (goldfish) and Danio rerio (zebrafish), indicating some species possess a basic residue in this position of the active loop of AGRP. Similarly, searching Cys-Arg-Phe-Phe-Asn-Ser-Phe-Cys (Ala to Ser) and Cys-Arg-Phe-Phe-Asn-Thr-Phe-Cys (Ala to Thr) found these sequences to be in the predicted AGRP of Sarcophilus harrisii (Tasmanian devil) and the precursor AGRP of Rattus norvegicus (rat), respectively. The observed residues types in the current study that maintained or increased MC4R potency (replacing Asn with basic residues, replacing Ala with hydroxyl-containing Ser) can also be found in naturally occurring AGRP orthologs, and may explain why these substitutions are tolerated at the MC4R.
An additional discovery from these structure-activity relationship studies was the apparent inverse agonism of several ligands at the MC5R. While the MC5R was previously reported to be constitutively active,45 to the best of the authors’ knowledge, no ligand to date has previously been shown to decrease cAMP through interaction with the MC5R. Twelve compounds (1, 2, 3, 4, 5, 6, 8, 9, 10, 12, 13, and 15) decreased the basal-normalized signal in a sigmoidal dose-response curve. This set of compounds decreased the observed response 10–25% that of the basal response, with apparent potencies (the inflection point of the sigmoidal dose-response) ranging from 10 to 700 nM. All compounds with the sigmoidal dose-response MC5R inverse agonist activity were substituted at the Asn position within the scaffold. An additional 5 ligands (7, 16, 21, 23, and 24) decreased the response from basal 25–35%, although this activity did not plateau at high concentrations. This scaffold did not uniformly result in MC5R inverse agonism, as five ligands possessed no activity at the MC5R (11, 14, 17, 19, and 22), while two more compounds partially stimulated the MC5R (18 and 20). The variable MC5R activities suggest this is not an artifact due to the ligand scaffold, since a uniform response to all ligands was not observed. Additional structure-activity relationship studies on this scaffold at the MC5R, coupled with mutagenesis and docking studies, may help elucidate the putative ligand-receptor interactions between this class of ligands and the MC5R to explain the variable inverse agonist activity and how to improve the apparent potency and percent change from basal activity.
Conclusions
The present SAR study investigated the Asn and Ala positions within an AGRP-derived macrocyclic scaffold, c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro], in attempts to generate more potent and/or selective MCR antagonists. Similar to a prior SAR study, basic substitutions at the Asn position increased MC4R potency. Substitution of Ser at the Ala position maintained MC4R potency, while all other substitutions at the Ala position decreased MC4R antagonist potency. The same trends were observed at the MC3R, though ligands were approximately 100-fold less potent at this receptor compared to the MC4R. Observing that the potent MC4R substitutions were also found in the active loop of ASP, a final Phe to Ala substitution was synthesized and assayed, which was equipotent at the MC4R compared to the scaffold lead. These results indicated that the equivalent β-hairpin active loop positions of ASP could be inserted into this AGRP scaffold and resulted in similar or increased MC4R potency. From the increased knowledge of structural requirements for AGRP-derived ligand at the MC4R, this study may be important in the development of MC4R selective probes and lead molecules for potential weight gain therapeutics. Additionally, the observed MC5R inverse agonism from these SAR studies are useful in designing ligands and probes to clarify the role of the ubiquitously expressed MC5R in vivo.
Methods
Peptide Synthesis
All peptides were synthesized using standard Fmoc chemistry.56,57 The coupling reagents 2-(1-H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), benzotriazol-1-yl-oxy-tris(dimethylamino) phosphonium hexafluorophosphate (BOP) and 1-hydroxybenzotriazole (HOBt), the H-Pro-2-chlorotrityl resin, amino acids Fmoc-DPro, Fmoc-Phe, Fmoc-Ala, Fmoc-Abu (aminobutyric acid), Fmoc-Thr(tBu), Fmoc-Asp(tBu), Fmoc-Glu(tBu), Fmoc-Dap(Boc) (Diaminopropionic acid), Fmoc-Nle, Fmoc-Val, Fmoc-Leu, Fmoc-Trp(Boc), Fmoc-His(Trt), Fmoc-Gly, Fmoc-Ser(tBu), Fmoc-Lys(Boc), and Fmoc-Asn(Trt), and the agouti-related protein (AGRP86-132) were purchased from Peptides International (Louisville, KY). Amino acid Fmoc-D-Dap(Boc) was purchased from Bachem (Torrance, CA). Dichloromethane (DCM), methanol (MeOH), acetonitrile (ACN), dimethylformamide (DMF) and anhydrous ethyl ether were purchased from Fisher (Fairlawn, NJ). Trifluoroacetic acid (TFA), dimethyl sulfoxide (DMSO), piperidine, triisopropylsilane (TIS), and N,N-diisopropylethylamine (DIEA) were purchased from Sigma-Aldrich (St. Louis, MO). All reagents and chemicals were ACS grade or better and were used without further purification.
Peptides were synthesized on a 0.05 mmol scale using H-Pro-2-chlorotrityl resin (0.68 to 0.76 meq/g substitution) in a 96-well block with an automated (Vantage Automated Parallel Peptide Synthesizer; Advanced ChemTech, Louisville, KY) or a semi-automated (LabTech I; Advanced ChemTech, Louisville, KY) instrument. Parallel syntheses consisted of two repeated steps separated by DMF washes: (i) removal of the Fmoc group with 20% piperidine in DMF (1x at rt for 5 min, 1x at rt for 20 min), and (ii) double coupling of the incoming Fmoc-protected amino acid (3.1 eq) with HBTU (3 eq) and DIEA (5 eq) in DMF at rt for 45 min. After completion of the syntheses, peptides were cleaved with a 99:1 DCM:TFA solution for 6 min. The cleavage solutions were concentrated and the side-chain protected linear peptides were precipitated using ice-cold ethyl ether. Peptides were cyclized in DCM with BOP (3 eq) and HOBt (3 eq) overnight using a peptide concentration of 1 mg/mL, and the DCM was removed under vacuum. Without further purification, the cyclized peptides were side-chain deprotected using a 95:2.5:2.5 TFA:TIS:H2O solution for 2 h. The solution was concentrated and cyclic peptides precipitated with ice-cold ethyl ether.
Crude peptides were purified by RP-HPLC using a Shimadzu system with a UV detector and a semi-preparative RP-HPLC C18 bonded silica column (Vydac 218TP1010, 1 × 25 cm). Assayed peptides were at least 95% pure as assessed by analytical RP-HPLC utilizing a Shimadzu system with a photodiode array detector and an analytical C18 silica column (Vydac 218TP104, 0.46 × 25 cm) in two diverse solvent systems and had the correct average molecular mass by MALDI-MS (Applied Biosystems-Sciex 5800 MALDI/TOF/TOF-MS, University of Minnesota Mass Spectrometry Lab).
cAMP AlphaScreen® Bioassay
Cyclized peptides were dissolved in DMSO (NDP-MSH and AGRP in H2O) at a stock concentration of 10−2M and were characterized pharmacologically using HEK293 cells stably expressing the mouse MC1R, MC3-5R by the cAMP AlphaScreen® assay (PerkinElmer) according to the manufacturer’s instructions and as previously described.58,60
Briefly, cells 70–90% confluent were dislodged with Versene (Gibco®) at 37 °C and plated 10,000 cells/well in a 384-well plate (Optiplate™) with 10 μL freshly prepared stimulation buffer (1X HBSS, 5 mM HEPES, 0.5 mM IBMX, 0.1% BSA, pH = 7.4) with 0.5 μg anti-cAMP acceptor beads per well. The cells were stimulated with the addition of 5 μL stimulation buffer containing peptide (concentrations from 10−4 to 10−13 M, determined by ligand potency) or forskolin (10−4 M) and incubated in the dark at room temperature for 2 hr.
Following stimulation, streptavidin donor beads (0.5 μg) and biotinylated-cAMP (0.62 μmol) were added to the wells in a green light environment with 10 μL lysis buffer (5 mM HEPES, 0.3% Tween-20, 0.1% BSA, pH = 7.4) and the plates were incubated in the dark at room temperature for an additional 2 hr. Plates were read on a Enspire (PerkinElmer) Alpha-plate reader using a pre-normalized assay protocol (set by the manufacturer).
Data Analysis
The EC50 and pA2 values represent the mean of duplicate replicates performed in at least three independent experiments. The EC50 and pA2 estimates and associated standard errors (SEM) were determined by fitting the data to a nonlinear least-squares analysis using the PRISM program (v4.0, GraphPad Inc.). When analyzing the inverse agonist activity at the MC5R, each replicate was normalized to the replicate signal at 10−10 M to observe change from basal activity. The percent inverse agonist activity was calculated from the normalized signal of three independent experiments. When a sigmoidal dose-response was observed, the percent inverse agonist activity reported was the change from basal to the plateau signal at high ligand concentrations; in these instances, the apparent potency was reported to be the inflection point of the sigmoidal curve. When inverse agonist activity was observed without a plateau at high concentrations, the percent inverse activity reported was the percent change from basal to signal at 100 μM concentrations. The ligands were assayed as TFA salts and not corrected for peptide context.
Acknowledgments
Funding Sources: This work has been supported by NIH Grant R01DK091906 (C.H.-L.). M.D.E. is a recipient of an NIH F32 Postdoctoral Fellowship (F32DK108402).
Figure 1C was generated using the PDB files 1Y7K29 and 1HYK,50 and PyMOL Molecular Graphics System, version 1.7.0.5, Schrödinger, LLC, with the computing resources of University of Minnesota Supercomputing Institute.
Abbreviations
- CTH
adrenocorticotropin hormone
- Fmoc
9-fluorenylmethoxycarbonyl
- AGRP
agouti-related protein
- GPCR
G protein-coupled receptor
- cAMP
cyclic 5′-adenosine monophosphate
- MC1R
melanocortin-1 receptor
- MC2R
melanocortin-2 receptor
- MC3R
melanocortin-3 receptor
- MC4R
melanocortin-4 receptor
- MC5R
melanocortin-5 receptor
- MCR
melanocortin receptor
- MSH
melanocyte stimulating hormone
- POMC
proopiomelanocortin
- α-MSH
alpha-melanocyte stimulating hormone
- β-MSH
beta-melanocyte stimulating hormone
- γ-MSH
gamma-melanocyte stimulating hormone
- μM
micromolar
- NDP-MSH (4-Norleucine-7-D-Phenylalanine)
Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2
- Nle
norleucine
- Dap
diaminopropioinic acid
- RP-HPLC
reverse-phase high-pressure liquid chromatography
- SAR
structure-activity relationships
- SNPs
single nucleotide polymorphisms
Footnotes
Author Contributions: M.D.E. and C.H.-L. designed the research. M.D.E., K.T.F., S.M.S. and K.A.F. performed the experiments. M.D.E. and C.H.-L. analyzed the data. M.D.E. wrote the manuscript with the help of C.H.-L.
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Chhajlani V, Wikberg JE. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 2.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 3.Schioth HB, Chhajlani V, Muceniece R, Klusa V, Wikberg JE. Major pharmacological distinction of the ACTH receptor from other melanocortin receptors. Life Sci. 1996;59:797–801. doi: 10.1016/0024-3205(96)00370-0. [DOI] [PubMed] [Google Scholar]
- 4.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 5.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 6.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 7.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 8.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for γ melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci US A. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 10.Irani BG, Xiang ZM, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the melanocortin-3 receptor in the regulation of food intake. Eur J Pharmacol. 2011;660:80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z, Tao YX. Mutations in melanocortin-3 receptor gene and human obesity. Prog Mol Biol Transl Sci. 2016;140:97–129. doi: 10.1016/bs.pmbts.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 14.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochem Biophys Res Commun. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 15.Griffon N, Mignon V, Facchinetti P, Diaz J, Schwartz JC, Sokoloff P. Molecular cloning and characterization of the rat fifth melanocortin receptor. Biochem Biophys Res Commun. 1994;200:1007–1014. doi: 10.1006/bbrc.1994.1550. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Kelly MA, Opitz-Araya X, Thomas RE, Low MJ, Cone RD. Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of exocrine gland function by melanocortin peptides. Cell. 1997;91:789–798. doi: 10.1016/s0092-8674(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 17.Nakanishi S, Inoue A, Kita T, Nakamura M, Chang AC, Cohen SN, Numa S. Nucleotide sequence of cloned cDNA for bovine corticotropin-beta-lipotropin precursor. Nature. 1979;278:423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- 18.Eipper BA, Mains RE. Structure and biosynthesis of pro- adrenocorticotropin/endorphin and related peptides. Endocr Rev. 1980;1:1–27. doi: 10.1210/edrv-1-1-1. [DOI] [PubMed] [Google Scholar]
- 19.Smith AI, Funder JW. Proopiomelanocortin processing in the pituitary, central nervous system, and peripheral tissues. Endocr Rev. 1988;9:159–179. doi: 10.1210/edrv-9-1-159. [DOI] [PubMed] [Google Scholar]
- 20.Miller MW, Duhl DM, Vrieling H, Cordes SP, Ollmann MM, Winkes BM, Barsh GS. Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the lethal yellow mutation. Genes Dev. 1993;7:454–467. doi: 10.1101/gad.7.3.454. [DOI] [PubMed] [Google Scholar]
- 21.Bultman SJ, Michaud EJ, Woychik RP. Molecular characterization of the mouse agouti locus. Cell. 1992;71:1195–1204. doi: 10.1016/s0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- 22.Fong TM, Mao C, MacNeil T, Kalyani R, Smith T, Weinberg D, Tota MR, Van der Ploeg LHT. ART (protein product of agouti-related transcript) as an antagonist of MC-3 and MC-4 receptors. Biochem Biophys Res Commun. 1997;237:629–631. doi: 10.1006/bbrc.1997.7200. [DOI] [PubMed] [Google Scholar]
- 23.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen YR, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 24.Shutter JR, Graham M, Kinsey AC, Scully S, Luthy R, Stark KL. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes Dev. 1997;11:593–602. doi: 10.1101/gad.11.5.593. [DOI] [PubMed] [Google Scholar]
- 25.Klebig ML, Wilkinson JE, Geisler JG, Woychik RP. Ectopic expression of the agouti gene in transgenic mice causes obesity, features of type II diabetes, and yellow fur. Proc Natl Acad Sci US A. 1995;92:4728–4732. doi: 10.1073/pnas.92.11.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham M, Shutter JR, Sarmiento U, Sarosi I, Stark KL. Overexpression of AGRT leads to obesity in transgenic mice. Nat Genet. 1997;17:273–274. doi: 10.1038/ng1197-273. [DOI] [PubMed] [Google Scholar]
- 27.Duhl DM, Vrieling H, Miller KA, Wolff GL, Barsh GS. Neomorphic agouti mutations in obese yellow mice. Nat Genet. 1994;8:59–65. doi: 10.1038/ng0994-59. [DOI] [PubMed] [Google Scholar]
- 28.Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, Cone RD. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 29.McNulty JC, Jackson PJ, Thompson DA, Chai B, Gantz I, Barsh GS, Dawson PE, Millhauser GL. Structures of the agouti signaling protein. J Mol Biol. 2005;346:1059–1070. doi: 10.1016/j.jmb.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 30.Ebihara K, Ogawa Y, Katsuura G, Numata Y, Masuzaki H, Satoh N, Tamaki M, Yoshioka T, Hayase M, Matsuoka N, Aizawa-Abe M, Yoshimasa Y, Nakao K. Involvement of agouti-related protein, an endogenous antagonist of hypothalamic melanocortin receptor, in leptin action. Diabetes. 1999;48:2028–2033. doi: 10.2337/diabetes.48.10.2028. [DOI] [PubMed] [Google Scholar]
- 31.Adan RA, Hillebrand JJ, De Rijke C, Nijenhuis W, Vink T, Garner KM, Kas MJ. Melanocortin system and eating disorders. Ann NY Acad Sci. 2003;994:267–274. doi: 10.1111/j.1749-6632.2003.tb03189.x. [DOI] [PubMed] [Google Scholar]
- 32.Ge Y, Ohta T, Driscoll DJ, Nicholls RD, Kalra SP. Anorexigenic melanocortin signaling in the hypothalamus is augmented in association with failure-to-thrive in a transgenic mouse model for Prader-Willi syndrome. Brain Res. 2002;957:42–45. doi: 10.1016/s0006-8993(02)03583-7. [DOI] [PubMed] [Google Scholar]
- 33.Kas MJ, van Dijk G, Scheurink AJ, Adan RA. Agouti-related protein prevents self-starvation. Mol Psychiatry. 2003;8:235–240. doi: 10.1038/sj.mp.4001206. [DOI] [PubMed] [Google Scholar]
- 34.He L, Gunn TM, Bouley DM, Lu XY, Watson SJ, Schlossman SF, Duke-Cohan JS, Barsh GS. A biochemical function for attractin in agouti-induced pigmentation and obesity. Nat Genet. 2001;27:40–47. doi: 10.1038/83741. [DOI] [PubMed] [Google Scholar]
- 35.Ollmann MM, Barsh GS. Down-regulation of melanocortin receptor signaling mediated by the amino terminus of Agouti protein in Xenopus melanophores. J Biol Chem. 1999;274:15837–15846. doi: 10.1074/jbc.274.22.15837. [DOI] [PubMed] [Google Scholar]
- 36.Ollmann MM, Lamoreux ML, Wilson BD, Barsh GS. Interaction of agouti protein with the melanocortin 1 receptor in vitro and in vivo. Genes Dev. 1998;12:316–330. doi: 10.1101/gad.12.3.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanchard SG, Harris CO, Ittoop OR, Nichols JS, Parks DJ, Truesdale AT, Wilkison WO. Agouti antagonism of melanocortin binding and action in the B16F10 murine melanoma cell line. Biochemistry. 1995;34:10406–10411. doi: 10.1021/bi00033a012. [DOI] [PubMed] [Google Scholar]
- 38.Willard DH, Bodnar W, Harris C, Kiefer L, Nichols JS, Blanchard S, Hoffman C, Moyer M, Burkhart W, Weiel J, Luther MA, Wilkison WO, Rocque WJ. Agouti structure and function: characterization of a potent alpha-melanocyte stimulating hormone receptor antagonist. Biochemistry. 1995;34:12341–12346. doi: 10.1021/bi00038a030. [DOI] [PubMed] [Google Scholar]
- 39.Yang YK, Ollmann MM, Wilson BD, Dickinson C, Yamada T, Barsh GS, Gantz I. Effects of recombinant agouti-signaling protein on melanocortin action. Mol Endocrinol. 1997;11:274–280. doi: 10.1210/mend.11.3.9898. [DOI] [PubMed] [Google Scholar]
- 40.Kiefer LL, Veal JM, Mountjoy KG, Wilkinson WO. Melanocortin receptor binding determinants in the agouti protein. Biochemistry. 1998;37:991–997. doi: 10.1021/bi971913h. [DOI] [PubMed] [Google Scholar]
- 41.Patel MP, Cribb Fabersunne CS, Yang YK, Kaelin CB, Barsh GS, Millhauser GL. Loop-swapped chimeras of the agouti-related protein and the agouti signaling protein identify contacts required for melanocortin 1 receptor selectivity and antagonism. J Mol Biol. 2010;404:45–55. doi: 10.1016/j.jmb.2010.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stanley SA, Small CJ, Kim MS, Heath MM, Seal LJ, Russell SH, Ghatei MA, Bloom SR. Agouti related peptide (Agrp) stimulates the hypothalamo pituitary gonadal axis in vivo & in vitro in male rats. Endocrinology. 1999;140:5459–5462. doi: 10.1210/endo.140.11.7248. [DOI] [PubMed] [Google Scholar]
- 43.Mizuno TM, Makimura H, Silverstein J, Roberts JL, Lopingco T, Mobbs CV. Fasting regulates hypothalamic neuropeptide Y, agouti-related peptide, and proopiomelanocortin in diabetic mice independent of changes in leptin or insulin. Endocrinology. 1999;140:4551–4557. doi: 10.1210/endo.140.10.6966. [DOI] [PubMed] [Google Scholar]
- 44.Haskell-Luevano C, Monck EK. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul Pept. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 45.Nijenhuis WAJ, Oosterom J, Adan RAH. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 46.Creemers JW, Pritchard LE, Gyte A, Le Rouzic P, Meulemans S, Wardlaw SL, Zhu X, Steiner DF, Davies N, Armstrong D, Lawrence CB, Luckman SM, Schmitz CA, Davies RA, Brennand JC, White A. Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83-132: Interaction between AGRP83-132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology. 2006;147:1621–1631. doi: 10.1210/en.2005-1373. [DOI] [PubMed] [Google Scholar]
- 47.Wilczynski A, Wang XS, Joseph CG, Xiang Z, Bauzo RM, Scott JW, Sorensen NB, Shaw AM, Millard WJ, Richards NG, Haskell-Luevano C. Identification of putative agouti-related protein(87-132)-melanocortin-4 receptor interactions by homology molecular modeling and validation using chimeric peptide ligands. J Med Chem. 2004;47:2194–2207. doi: 10.1021/jm0303608. [DOI] [PubMed] [Google Scholar]
- 48.Yang YK, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SB, Gantz I. Characterization of agouti-related protein binding to melanocortin receptors. Mol Endocrinol. 1999;13:148–155. doi: 10.1210/mend.13.1.0223. [DOI] [PubMed] [Google Scholar]
- 49.Tota MR, Smith TS, Mao C, MacNeil T, Mosley RT, Van der Ploeg LH, Fong TM. Molecular interaction of agouti protein and agouti-related protein with human melanocortin receptors. Biochemistry. 1999;38:897–904. doi: 10.1021/bi9815602. [DOI] [PubMed] [Google Scholar]
- 50.Bolin KA, Anderson DJ, Trulson JA, Thompson DA, Wilken J, Kent SBH, Gantz I, Millhauser GL. NMR structure of a minimized human agouti related protein prepared by total chemical synthesis. FEBS Lett. 1999;451:125–131. doi: 10.1016/s0014-5793(99)00553-0. [DOI] [PubMed] [Google Scholar]
- 51.McNulty JC, Thompson DA, Bolin KA, Wilken J, Barsh GS, Millhauser GL. High-resolution NMR structure of the chemically-synthesized melanocortin receptor binding domain AGRP(87-132) of the agouti-related protein. Biochemistry. 2001;40:15520–15527. doi: 10.1021/bi0117192. [DOI] [PubMed] [Google Scholar]
- 52.Jackson PJ, McNulty JC, Yang YK, Thompson DA, Chai B, Gantz I, Barsh GS, Millhauser GL. Design, pharmacology, and NMR structure of a minimized cystine knot with agouti-related protein activity. Biochemistry. 2002;41:7565–7572. doi: 10.1021/bi012000x. [DOI] [PubMed] [Google Scholar]
- 53.Joseph CG, Bauzo RM, Xiang ZM, Shaw AM, Millard WJ, Haskell-Luevano C. Elongation studies of the human agouti-related protein (AGRP) core decapeptide (Yc[CRFFNAFC]Y) results in antagonism at the mouse melanocortin-3 receptor. Peptides. 2003;24:263–270. doi: 10.1016/s0196-9781(03)00030-5. [DOI] [PubMed] [Google Scholar]
- 54.Ericson MD, Wilczynski A, Sorensen NB, Xiang ZM, Haskell-Luevano C. Discovery of a β-hairpin octapeptide, c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro], mimetic of agouti-related protein(87-132) [AGRP(87-132)] with equipotent mouse melanocortin-4 receptor (mMC4R) antagonist pharmacology. J Med Chem. 2015;58:4638–4647. doi: 10.1021/acs.jmedchem.5b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ericson MD, Freeman KT, Schnell SM, Haskell-Luevano C. A macrocyclic agouti-related protein/[Nle4,DPhe7]α-melanocyte stimulating hormone chimeric scaffold produces subnanomolar melanocortin receptor ligands. J Med Chem. 2017;60:805–813. doi: 10.1021/acs.jmedchem.6b01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J Am Chem Soc. 1970;92:5748–5749. [Google Scholar]
- 57.Carpino LA, Han GY. The 9-fluorenylmethoxycarbonyl amino-protecting group. J Org Chem. 1972;37:3404–3409. [Google Scholar]
- 58.Ericson MD, Schnell SM, Freeman KT, Haskell-Luevano C. A fragment of the Escherichia coli ClpB heat-shock protein is a micromolar melanocortin 1 receptor agonist. Bioorg Med Chem Lett. 2015;25:5306–5308. doi: 10.1016/j.bmcl.2015.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lensing CJ, Freeman KT, Schnell SM, Adank DN, Speth RC, Haskell-Luevano C. An in vitro and in vivo investigation of bivalent ligands that display preferential binding and functional activity for different melanocortin receptor homodimers. J Med Chem. 2016;59:3112–3128. doi: 10.1021/acs.jmedchem.5b01894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh A, Tala SR, Flores V, Freeman K, Haskell-Luevano C. Synthesis and pharmacology of α/β3-peptides based on the melanocortin agonist Ac-His-DPhe-Arg-Trp-NH2 sequence. ACS Med Chem Lett. 2015;6:568–572. doi: 10.1021/acsmedchemlett.5b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schild HO. pA, a new scale for the measurement of drug antagonism. Br J Pharmacol Chemother. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chai BX, Pogozheva ID, Lai YM, Li JY, Neubig RR, Mosberg HI, Gantz I. Receptor-antagonist interactions in the complexes of agouti and agouti-related protein with human melanocortin 1 and 4 receptors. Biochemistry. 2005;44:3418–3431. doi: 10.1021/bi0478704. [DOI] [PubMed] [Google Scholar]