


Abstract
Since many synthetic targets in organic chemistry are larger, more functionalized, and more oxidized than the starting inputs used, methods that unite fragments with an increase in oxidation state possess inherent efficiencies. Thus, the potential for complexity building transforms by union of activated or unactivated C–H, N–H, or O–H centers is considerable, and the challenge lies in how to obtain selective reactions from any two of these centers.
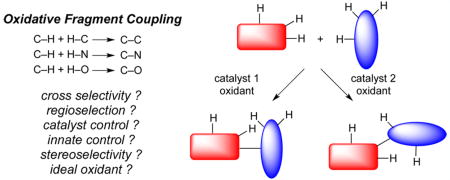
The construction of value-added organic molecules is built on a seemingly simple precept. Namely, small, inexpensive molecules, often referred to as building blocks, can be united by the formation of new bonds, such as C–C, C–N, C–O, and C═C bonds. Just as when building a house, some building blocks are easier to manipulate than others and some unions are easier to implement than others. In judging whether an overall synthesis is meritorious, many criteria can be used depending on the context. For a production scale synthesis, overall efficiency is paramount and factors in the cost of goods, waste, production facilities, time, etc. For a biological investigation, speed of access may be the most critical factor. Regardless, facile and cost-effective syntheses are both supported by synthetic methods that create complexity rapidly from the simplest possible starting materials. This concept is readily seen in the success of metal catalyzed cross coupling methods (Figure 1a) since it is often possible to unite two similarly sized units to create a much more complex ensemble. In terms of creating a perfect synthesis, one Holy Grail is to be able to perform such unions to create C–C, C–N, and C–O bonds from the corresponding C–H, O–H, and N–H bonds at will (Figure 1c).
Figure 1.

Redox neutral and oxidative fragment coupling reactions.
A perfect synthesis is elusive, but this Holy Grail would achieve several efficiencies that contribute to this goal. An ideal synthesis uses the minimal number of steps, each of which constructs the skeleton and requisite functionality while avoiding refunctionalizations and protecting group manipulations. Often, the starting inputs in a total synthesis are in a different oxidation state than the products. Thus, any oxidation or reduction operations need to be combined with skeleton forming operations to avoid unnecessary refunctionalizations and to enhance step economy. This idea of redox economy1 in total synthesis is particularly relevant to fragment couplings, as many common starting materials are in more reduced forms relative to the products.
The challenge herein lies in the large number of different C–H, O–H, and N–H bonds that could be present in any given molecule. How is it possible to selectively activate any two of these bonds to initiate an effective cross coupling event as shown in Figure 1c? The rapid explosion of successful C–H functionalization reactions (Figure 1b) is certainly a signpost in the right direction,2 and a previous Holy Grail Account3 highlighted alkanes as a frontier for which much progress has been made in the intervening years.4 An understanding has been achieved of some of the factors permitting selective C–H activation including innate reactivity (dictated by substrate reactivity) and guided reactivity (dictated by directing groups or external reagents).5 Indeed, the development of catalysts that can circumvent innate reactivity is extremely promising.4,6 However, the coupling partners employed in most C–H activation processes are already preactivated (e.g., ArBr, PhI(OAc)2, etc.). The next challenge is to activate two separate hydrogens on two different substrates to initiate an oxidative coupling, also referred to as a dehydrogenative coupling,7–9 as shown in Figure 1c.
Within this construct, there are a very large number of mechanistic possibilities. We advocate no specific transformation, metal, or mechanism here; rather, the field is in clear need of an outline of the limitations and applications of each type to allow users to select specific transformations for specific applications and to identify areas in need of development. Within this short Commentary, it would be impossible to give a full accounting given the numerous metals (Pd, Cu, Rh, Ru = common, Ni, Fe, Ag, Mn, Mo, Ir, Au, V, Cr = less common) and metal-free oxidative systems that have been investigated. Notably, methods that rely on different activation principles will exhibit different levels of selectivity for various C–H, O–H, and N–H bonds permitting selective intermolecular couplings of different substrates, as well as selection among these bond types within each substrate. A host of outer sphere and inner sphere mechanisms are possible depending on the substrate and catalyst.10,11 Possibilities where the metal may or may not be directly involved in the X–H activation include radical, metal-intercepted radical, metalloradical, and metal electron-transfer mechanisms. Even within inner sphere mechanisms, X–H bond activation can involve metal species that range from nucleophilic to electrophilic and a spectrum of mechanisms are possible including oxidative addition, σ bond metathesis, concerted metalation deprotonation, 1,2-addition, Heck-type, or electrophilic types. Each of these mechanisms relies on a different set of control factors, few of which are well understood. For example, the effects of metal nucleophilicity or electrophilicity, orbital frameworks, and attendant ligands on reactivity have been delineated in numerous systems. The controlling factors for activation of different X–H bonds, especially when a range of similar bond types are present remains, however, less well understood. With multiple independent and interdependent factors to consider such as acidity, bond strengths, hybridization, electronegativities, steric interactions, and directing moieties, deconvolution of these effects remains a premier challenge to the field.
Even so, a number of intriguing reports show the possibilities and the power of these types of transformations,7–9 and that Nature also has exploited the efficiency of oxidizing while constructing skeletal frameworks. For example, our syntheses of cercosporin12 and hypocrellin13 relied on a biomimetic asymmetric oxidative coupling of a monomeric naphthol to create a dimeric binaphthol, rapidly building up complexity (Figure 2). This transformation is hypothesized to involve ligand exchange to form a metal phenolate complex followed by electron transfer to generate the key reactive species, which either dimerizes or reacts with an additional molecule of substrate. Regardless of the exact steps, carbon–carbon bond formation must occur while the copper complex is associated to permit stereocontrol. In addition to generating all the carbons of the perylenequinone core in this step, formation of the chiral axis at this stage was key to allow control of the chiral helical elements in the final products.
Figure 2.

Oxidative homocoupling of naphthols en route to perylenequinone natural products.
An example that shows the overall potential of oxidative fragment coupling approaches can be found in the work of Porco who recognized that an oxidation of an alcohol to an aldehyde would initiate an electrocyclization leading to a pyran intermediate (Figure 3).14 This intermediate is poised to undergo a Diels–Alder reaction leading to the complete skeleton of torreyanic acid along with control of six new stereocenters. Although oxidation is not concomitant with dimerization, this example illustrates the power of oxidative complexity building transforms in molecular assembly.
Figure 3.

Biomimetic oxidative coupling.
In the systems described in Figures 2 and 3, the reactive site is largely dictated by the substrates, begging the question of whether oxidative channels to different products are viable. Our research has attempted to tackle this issue in transformations involving metal electron transfer, such as the oxidative coupling of phenols. By means of parallel microscale screening, a series of oxidizing catalysts were found that would allow site selectivity in oxidative dimerization of several phenolic substrates (Scheme 1a).15 For example, selective C–C coupling vs C–C and C–O bond formation could be achieved from the same substrate by simply altering the catalyst. Other reports, including a case from Lumb (Scheme 1b),16 confirm that selective control of C–O vs C–C bond formation is achievable. Clearly, control of different elements (e.g., nucleophilicity) can be exploited such that reactions do not occur only from the most oxidizable carbon site. These efforts show that either C–C or C–O bonds can be made via oxidative fragment unions when commencing from electron rich substrates.
Scheme 1.

Controlling Regioselectivity in Oxidative Homocoupling: C–C vs C–O Bond Formation
The next logical challenge in oxidative unions is bringing together two different substrates selectively, especially if both can be oxidized under the reactions conditions. We have found that such heterocouplings of phenols are indeed viable.15 Using an inexpensive chromium salen catalyst and oxygen as the terminal oxidant, para-ortho (Scheme 2a) and para–para cross couplings can be achieved with a range of substrates using typical halogenated (DCE) or nonpolar (toluene) solvents and oxygen as the oxidant. Notably, the two substrates are used at near parity (1.0–1.5:1.0 ratio) indicating that the corresponding homocouplings of either monomer are considerably slower. Along this vein, Pappo has identified a different system comprised of an iron trichloride catalyst with hexafluoroisopropanol as solvent and di-tert-butyl peroxide as the oxidant, which can perform phenol–phenol cross couplings as well as phenol–nucleophilic arene cross couplings and phenol–enol cross couplings.17 The example in Scheme 2c illustrates a two step sequence employing the latter two of these modalities; the rapid assembly of units and complexity is striking. This case also shows that oxidative couplings are not restricted to naphthol and phenol substrates, highlighting the use of arenes, albeit electron rich ones, and enolic substrates such as β-ketoesters.
Scheme 2.

Controlling Regioselectivity in Oxidative Cross-Coupling: C–O vs C–C Bond Formation
The oxidative coupling of similar enolizable substrates with stoichiometric bases and oxidants enjoys a long history8 with more recent efforts to effect cross couplings as illustrated in Scheme 2d.5 Such constructions allow access to more complex skeletons poised for additional bond constructions leading toward biologically interesting structures as seen in the case of hapalindole U. The downside of this protocol is the use of stoichiometric base and oxidant. The development of selective, catalytic systems with atom economical oxidants would be a significant advance.
The systems discussed to this point utilize easily oxidizable substrates such as phenols, enols, and enamines. Heteroatoms and carbon centers adjacent to nitrogen or oxygen are also easily oxidized usually through a series of electron transfer events,7,9 but the coupling of other positions (sp2 or sp3 hybridized carbons) only occurs readily if the site is acidic or the corresponding metalated species is readily generated. The recent advances in C–H activation have been recognized in this context, and much work has been devoted to double C–H activation including sites that are typically immune to oxidative processes. One of the earliest examples of regioselective cross-coupling of two different unactivated arenes can be found in the work of Lu and co-workers (Scheme 3a).18 With the absence of a kinetic isotope effect in the naphthalene partner and the presence of an isotope effect in the monoarene, C–H activation appears to proceed by two different steps. Subsequently, a huge amount of work in the field has shown that sp2–sp2 couplings can be effected across a range of systems and are most effective when the two substrates are differentiated electronically or by the presence of a directing group.7–9
Scheme 3.

Oxidative Coupling at Oxidatively Resistant Sites
An example that highlights the use of oxidative cross coupling in sequenced transformations on unactivated substrates can be found in a report from Shi and co-workers. A directed metalation combined with a concerted metalation–deprotonation of an unactivated arene was found to form a biaryl (Scheme 3b).19 Conspicuously, no homocoupling was observed, presumably due to an initial N–Ac directed insertion followed by an second insertion into the less hindered benzene. Further treatment of the biaryl product with the palladium catalyst and oxygen gave rise to a second oxidative coupling resulting in selective C–N formation. It is notable that these two oxidative couplings permit a rapid buildup of complexity to form the core of deoxycarbazomyicn B from simple precursors.
Overall, oxidative couplings to form sp–sp, sp–sp2, and sp2–sp2 bonds are well advanced, but the corresponding reactions with sp3 centers remain a major challenge, although exploration of reactions employing acidic species, allylic centers, and radicals have resulted in important advances.7–9 In our work, we have found an unexpected C–H insertion into the benzylic, rather than arene, bonds under concerted metalation deprotonation conditions allowing selective cross-coupling with an acidic species (Scheme 3c).20 Interestingly, the mechanism and rate-determining step of the alkyl C–H insertion differs significantly from concerted metalation deprotonation processes in that alkyl C–H bonds are selectively activated relative to aryl C–H bonds and that migration (via a β-hydride elimination/migratory insertion mechanism) is competitive (Scheme 3d) with the rapid reductive elimination typically observed in such systems. In this transformation, the azlactone substrate appears to play an additional role modulating the reactivity of the palladium center to allow for selective alkyl C–H insertion rather than the typically observed aryl C–H insertion. This observation has considerable import in that an understanding of the process and reengineering of the palladium coordination sphere may allow insertion into many other alkyl C–H bonds and their selective coupling to a range of partners.
In summary, the potential for complexity building transforms by union of activated or unactivated C–H, N–H, or O–H centers is considerable. Recent results from our laboratories highlight that preconceptions regarding oxidative activation modalities need to be treated with caution. Rather, we should set our sights on the most desirable transformations possible with the goal of achieving processes such as those illustrated in Figure 1c using catalyst control, ideally with base metal catalysts, as well as with the most efficient oxidants possible such as O2 or H2O2. Critical areas in need of development include couplings involving sp3 centers, enantioselective couplings for the formation of stereocenters, and selective couplings compatible with a broader range of functional groups and involving much more complex substrates. To do so, a greater understanding of the mechanisms driving selectivity in such processes needs to be achieved together with a greater understanding of how to modulate catalysts to achieve the desired reactivity. In particular, detailed analyses and correlations of selectivities with readily measured or calculated parameters (acidity, bond strengths, oxidation potentials, steric accessibility, etc.) is needed to provide useful guides for those wishing to use the methods (see ref 6b and references cited therein).
Acknowledgments
Thanks are extended to the many co-workers who have contributed both materially and intellectually to the work described herein and to the funding agencies supporting these efforts including, most recently, NSF (CHE1464778) and NIH (R01GM112684).
Footnotes
The author declares no competing financial interest.
References
- 1.Burns NZ, Baran PS, Hoffmann RW. Redox Economy in Organic Synthesis. Angew. Chem., Int. Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 2.Davies HML, Morton D. Recent Advances in C–H Functionalization. J. Org. Chem. 2016;81:343–350. doi: 10.1021/acs.joc.5b02818. [DOI] [PubMed] [Google Scholar]
- 3.Arndtsen BA, Bergman RG, Mobley TA, Peterson TH. Selective Intermolecular Carbon-Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc. Chem. Res. 1995;28:154–162. [Google Scholar]
- 4.Hartwig JF. Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc. 2016;138:2–24. doi: 10.1021/jacs.5b08707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brückl T, Baxter RD, Ishihara Y, Baran PS. Innate and Guided C–H Functionalization Logic. Acc. Chem. Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) White MC. Adding Aliphatic C–H Bond Oxidations to Synthesis. Science. 2012;335:807–808. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]; (b) Gormisky PE, White MC. Catalyst-Controlled Aliphatic C–H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc. 2013;135:14052–14055. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- 7.(a) Li C-J. Cross-Dehydrogenative Coupling (CDC): Exploring C-C Bond Formations Beyond Functional Group Transformations. Acc. Chem. Res. 2009;42:335–344. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]; (b) Girard SA, Knauber T, Li C-J. The Cross-Dehydrogenative Coupling of Csp3–H Bonds: A Versatile Strategy for C–C Bond Formations. Angew. Chem., Int. Ed. 2014;53:74–100. doi: 10.1002/anie.201304268. [DOI] [PubMed] [Google Scholar]
- 8.(a) Yeung CS, Dong VM. Catalytic Dehydrogenative Cross-Coupling: Forming Carbon-Carbon Bonds by Oxidizing Two Carbon-Hydrogen Bonds. Chem. Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; (b) Liu C, Zhang H, Shi W, Lei A. Bond Formations between Two Nucleophiles: Transition MetalCatalyzed Oxidative Cross-Coupling Reactions. Chem. Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]; (c) Liu C, Yuan J, Gao M, Tang S, Li W, Shi R, Lei A. Oxidative Coupling between Two Hydrocarbons: An Update of Recent C–H Functionalizations. Chem. Rev. 2015;115:12138–12204. doi: 10.1021/cr500431s. [DOI] [PubMed] [Google Scholar]
- 9.Girard SA, Knauber T, Li C-J. In: From C-H to C-C Bonds: Cross-Dehydrogenative-Coupling. Li C-J, editor. RSC Green Chemistry; 2014. pp. 1–32. [Google Scholar]
- 10.Labinger JA, Bercaw JE. Understanding and exploiting C–H bond activation. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- 11.Roudesly F, Oble J, Poli G. Metal-Catalyzed C H Activation/Functionalization: The Fundamentals. J. Mol. Catal. A: Chem. 2017;426:275–296. [Google Scholar]
- 12.Morgan BJ, Dey S, Johnson SW, Kozlowski MC. Design, Synthesis, and Investigation of Protein Kinase C Inhibitors: Total Syntheses of (+)-Calphostin D, (+)-Phleichrome, Cercosporin, and New Photoactive Perylenequinones. J. Am. Chem. Soc. 2009;131:9413–9425. doi: 10.1021/ja902324j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Brien EM, Morgan BJ, Kozlowski MC. Dynamic Stereochemistry Transfer in a Transannular Aldol Reaction: Total Synthesis of Hypocrellin A. Angew. Chem., Int. Ed. 2008;47:6877–6880. doi: 10.1002/anie.200800734. [DOI] [PubMed] [Google Scholar]
- 14.Li C, Johnson RP, Porco JA., Jr Total Synthesis of the Quinone Epoxide Dimer (+)-Torreyanic Acid: Application of a Biomimetic Oxidation/Electrocyclization/Diels-Alder Dimerization Cascade. J. Am. Chem. Soc. 2003;125:5095–5106. doi: 10.1021/ja021396c. [DOI] [PubMed] [Google Scholar]
- 15.Lee YE, Cao T, Torruellas C, Kozlowski MC. Selective Oxidative Homo- and Cross-Coupling of Phenols with Aerobic Catalysts. J. Am. Chem. Soc. 2014;136:6782–6785. doi: 10.1021/ja500183z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esguerra KVN, Lumb J-P. Adapting Melanogenesis to a Regioselective C–H Functionalization of Phenols. Synlett. 2015;26:2731–2738. [Google Scholar]
- 17.Gaster E, Vainer Y, Regev A, Narute S, Sudheendran K, Werbeloff A, Shalit H, Pappo D. Significant Enhancement in the Efficiency and Selectivity of Iron-Catalyzed Oxidative Cross-Coupling of Phenols by Fluoroalcohols. Angew. Chem., Int. Ed. 2015;54:4198–4202. doi: 10.1002/anie.201409694. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Jiang L, Lu W. Intermolecular Cross-Coupling of Simple Arenes via C-H Activation by Tuning Concentrations of Arenes and TFA. Organometallics. 2006;25:5973–5975. [Google Scholar]
- 19.Li B-J, Tian Z-L, Fang Z, Shi Z-J. Multiple C-H Activations To Construct Biologically Active Molecules in a Process Completely Free of Organohalogen and Organometallic Components. Angew. Chem., Int. Ed. 2008;47:1115–1118. doi: 10.1002/anie.200704092. [DOI] [PubMed] [Google Scholar]
- 20.Curto JM, Kozlowski MC. Chemoselective Activation of sp3 vs sp2 C–H Bonds with Pd(II) J. Am. Chem. Soc. 2015;137:18–21. doi: 10.1021/ja5093166. [DOI] [PMC free article] [PubMed] [Google Scholar]