

Abstract
The first chemical synthesis of the heptasaccharide repeating unit of type V Group B Streptococcus (GBS) capsular polysaccharide (CPS) by a preactivation-based one-pot [2+1+4] glycosylation strategy is described. The synthetic efficiency was further improved by using diol glycosyl acceptors for regio/stereoselective glycosylation reactions. The synthetic target contains a free amino group at the reducing end, facilitating additional regioselective elaboration. The results should be useful for synthetic and biological studies of related CPSs.
Graphical Abstract
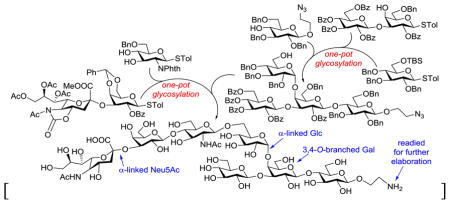
Group B Streptococcus (GBS) is the primary cause of neonatal sepsis and meningitis,1–3 which can be classified into at least ten serotypes based on their structurally unique capsular polysaccharides (CPSs).4,5 Type V GBS is one of the three predominant GBS serotypes and is responsible for 13% of early-onset diseases, but the mortality rate caused by type V GBS is higher than others.6 CPS is the major virulent factor of GBS that can assist bacteria to evade human innate immune responses.7–9
Thus, GBS CPSs are attractive targets for vaccine development.10,11 However, natural CPSs from bacteria are heterogeneous. As a result, synthetic CPS repeating units with defined structures are receiving growing attention. The synthetic molecules are not only useful for the development of semi- and fully synthetic glycoconjugate vaccines12,13 but also for detailed biological studies and structure-activity relationship (SAR) analysis to expand understanding of CPSs.
The structure of type V GBS CPS (1, Figure 1) was elucidated by Wessels et al. in 1991,14 but to date no synthesis of its repeating units has been reported. The main chain of 1 is composed of a repeating trisaccharide, 4)-α-D-Glc-(1→4)-β-D-Gal-(1→4)-β-D-Glc-(1→, which is further decorated with two branches: a trisaccharide, α-Neu5Ac-(2→3)-β-D-Gal-(1→4)-β-D-GlcNAc-(1→, attached to the 6-O-position of its non-reducing end Glc residue and a β-D-Glc linked to the Gal 3-O-position. Aiming at in-depth studies of 1, we investigated the chemical synthesis of its repeating unit 2 (Figure 1) with an aminoethyl group at the reducing end, which we felt would facilitate further elaboration. This heptasaccharide is chemically challenging because of the presence of a branch and several difficult glycosyl linkages such as that of α-D-Glc and α-NeuNAc.
Figure 1.

Structures of type V GBS CPS 1 and its repeating unit 2 as the synthetic target
Our synthetic plan for accessing 2 is outlined in Scheme 1. First, appropriate protecting groups were assigned to the various sugar units and positions to reach 3, the fully protected form of 2. In this design, the accessibility of protected monosaccharides, the overall stability of protecting groups, and the fact that participating groups at the donor C2-position could direct stereoselective trans-glycosylation were all taken into account. To reduce the risks of running into problems at a later stage, difficult glycosylation reactions, such as that of the Neu5Ac, and the branched structure would be achieved early in the synthesis. Accordingly, 3 was disconnected to afford 4 and 6 as key intermediates. In addition, thioglycosides would be used as glycosyl donors for most glycosylation reactions so that glycans could be assembled by Huang and Ye’s preactivation-based one-pot synthesis.15–18
Scheme 1.

Retrosynthesis of 2
Tetrasaccharide 4 was initially assembled through stepwise introduction of monosaccharides based on a Gal derivative 13 (Scheme 2), rather than the corresponding lactose analog that was difficult to purify. We previously demonstrated that the proper sequence to install branches for oligosaccharides carrying a 3/4-O-branched Gal should be 3-O-glycosylation prior to 4-O-glycosylation.19 Therefore, the 3-O-position of 13, which is more reactive than its 2-O-position,20,21 was selectively glycosylated (<10% 2-O-glycosylation) with 12. The reaction was also β-specific probably due to the presence of a participating 2-O-Bz group in 12. The two regioisomers were more easily separated after benzoylation, to afford pure β-disaccharide 14 (JH1′,2′ = 7.8 Hz) in a 65% yield (for two steps). The structure of 14 was verified by the downfield shift of its H-2Gal signal from δ 3.78 to 5.37 ppm as a result of benzoylation. Thereafter, 14 was elongated with 9 to produce trisaccharide 15 (JH1′,2′ = 7.8 Hz) stereospecifically also due to neighboring group participation. Regioselective opening of the benzylidene ring in 16 with NaCNBH3 under acidic conditions was followed by glycosylation of the exposed hydroxyl group with 7 to secure the tetrasaccharide 17 in an excellent yield and stereoselectivity. In fact, only the α-product 17 was isolated from this reaction, whose structure was confirmed by 1H NMR (JH1″,2″ = 3.0 Hz). As proposed by Huang and his coworkers,22 this result may be attributed to the use of diethyl ether as solvent, which can act as a nucleophile to participate in the reaction, leading to double SN2 attacks at the anomeric carbon of the α-glycosyl triflate intermediate formed upon activation of 7. It is worth noting that only 1.0 equiv of AgOTf (relatively to the donor) should be used in the reaction, since excessive AgOTf would promote SN1 type of reaction to result in an α,β-mixture. Eventually, 17 was desilylated with Et3N·3HF to furnish 4, which was ready as a building block for the one-pot synthesis of 2.
Scheme 2.

Synthesis of 4 by a linear assembly strategy
After the potentially difficult α-glucosylation was proved to be highly stereoselective under above conditions, we moved on to execute the plan for preactivation-based one-pot synthesis of 17 (Scheme 3). Preactivation of thioglycoside 7 was achieved with stoichiometric p-toluenesulfenyl triflate, generated in situ from the reaction of p-toluenesulfenyl chloride (TolSCl) with silver triflate (AgOTf), in diethyl ether at −78 °C for 10 min. Next, disaccharide 8, obtained upon selective opening of the benzylidene ring in 14, was added at −78 °C, and the reaction mixture was allowed to warm to rt and stirred for another 1.5 h to accomplish the first glycosylation. Then, 9 was added directly to the mixture to perform the second glycosylation under the promotion of TolSCl and AgOTf. Finally, the glycosylation product was isolated in a 68% overall yield, which was proved to be 17.
Scheme 3.

Preactivation-based one-pot synthesis of 17
In the synthesis of 2 (Scheme 4), sialodisaccharide 6 was prepared first and then used as one of the three building blocks for preactivation-based one-pot oligosaccharide assembly. Dibutyl phosphate sialoside 10, which was revealed to be an excellent sialyl donor for α-glycosylation,23 was employed to react with 11 under the promotion of TMSOTf to produce 6 in a 79% yield, whose α-sialyl linkage was verified by its H-3ax/C-1 coupling constant (J = 6.2 Hz). Thereafter, 6 was sequentially coupled with monosaccharide 5 and tetrasaccharide 4 by the preactivation-based one-pot glycosylation protocol described above to provide the fully protected heptasaccharide 3 (59% yield). The regioselectivity of the first glycosylation reaction was confirmed by the HMBC spectrum of its product isolated from a portion of the reaction mixture, which showed the C-1Gal/H-4GlcN correlation. Glycosylation reactions using both 4 and 5 were stereospecific probably because of neighboring group participation, and the new β-glycosidic bonds were confirmed by the 1H NMR spectrum of 3 (Gal: JH1,2 = 8.4 Hz; β-GlcN: JH1,2 = 7.8 Hz).
Scheme 4.

Preactivation-based [2+1+4] one-pot assembly of 2
It is commonly acknowledged that global deprotection of complex oligosaccharides can be intricate. This is especially accurate for 3 that contained distinct protecting groups, including carboxylate, carbamate, amido, benzylidene, benzyl, azido and acyl groups, which need distinct and typically incompatible deprotection conditions. After screening and optimizing a series of reactions and conditions, we eventually established a five-step deprotection protocol. First, the methyl ester of sialic acid was selectively deprotected on treatment of 3 with LiI in pyridine at reflux.24 Next, the resultant product was heated with hydrazine in EtOH at reflux to remove the carbamate and acyl groups concurrently. It is noteworthy that performing the two reactions in such an order was critical, as removal of the Phth group in the presence of an ester can lead to complex results.25 Subsequently, selective N-acetylation was realized in two steps, including acetylation with acetic anhydride and then NaOMe treatment to remove O-acetyl groups. After the N-acetylated product was purified on a Sephadex LH-20 column with MeOH as eluent, it was subjected to Pd/C-catalyzed hydrogenolysis in MeOH and H2O (4:1) to remove all benzyl and benzylidene groups and reduce the azide concomitantly. The final product was purified on a Sephadex G-25 column with H2O as eluent to afford 2 after lyophilization in a 54% overall yield. Its structure was verified by 1D and 2D NMR and HR MS. It is worth noting that to facilitate final product purification it was crucial to monitor the reactions closely in the deprotection process and make sure that every step was complete.
In summary, we have achieved the first chemical synthesis of the heptasaccharide repeating unit of type V GBS CPS, which had several challenging features. The synthesis is highlighted by the usage of preactivation-based one-pot glycosylation for rapid and efficient assembly of the title compound and the key tetrasaccharide intermediate. Compared to conventional synthetic strategies, preactivation-based one-pot synthesis has saved multiple steps of glycosyl donor manipulation and intermediate purification and thus improved the overall synthetic efficiency. For example, 17 was obtained from 14 in a 62% overall yield by the conventional method, while its one-pot synthesis gave a 68% overall yield from 7, not mentioning the purification steps that were saved. Moreover, after optimization, all glycosylations afforded excellent stereoselectivity. Another highlight of this work is the use of diol acceptors, such as 5 and 13, for regioselective glycosylations, based on the different reactivities of the free hydroxyl groups in the structures. This had not only saved multiple protection-deprotection manipulations but also significantly simplified the overall design of protecting tactics. Finally, despite the presence of a variety of functional groups in heptasaccharide 3, its global deprotection was accomplished effectively following careful planning and optimization of the deprotection sequence and reaction conditions. The discoveries of this work should be useful for future synthetic studies on other GBS CPSs. Furthermore, since the synthetic target 2 carried a free amino group at its reducing end, it should be feasible to regioselectively couple 2 with other molecules to create glycoconjugates that would facilitate more in-depth studies on type V GBS CPS and its medical applications, such as in the development of new antibacterial vaccines.
Supplementary Material
Acknowledgments
The authors appreciate NIH/NCI support (R01CA095142) for this research work.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors, and all authors have given approval to the final version of this manuscript.
The Supporting Information, including synthetic procedures, analytical data and NMR and MS spectra of all new compounds, is available free of charge on the ACS Publications website.
References
- 1.Le Doare K, Heath PT. Vaccine. 2013;31(Suppl 4):D7. doi: 10.1016/j.vaccine.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 2.MMWR Recomm Rep. 1996;45:1. [Google Scholar]
- 3.Eickhoff TC, Daly AK, Ingall D, Finland M, Klein JO. New Engl J Med. 1964;271:1221. doi: 10.1056/NEJM196412102712401. [DOI] [PubMed] [Google Scholar]
- 4.Cieslewicz MJ, Chaffin D, Glusman G, Kasper D, Madan A, Rodrigues S, Fahey J, Wessels MR, Rubens CE. Infect Immun. 2005;73:3096. doi: 10.1128/IAI.73.5.3096-3103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berti F, Campisi E, Toniolo C, Morelli L, Crotti S, Rosini R, Romano MR, Pinto V, Brogioni B, Torricelli G, Janulczyk R, Grandi G, Margarit I. J Biol Chem. 2014;289:23437. doi: 10.1074/jbc.M114.567974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weisner AM, Johnson AP, Lamagni TL, Arnold E, Warner M, Heath PT, Efstratiou A. Clin Infect Dis. 2004;38:1203. doi: 10.1086/382881. [DOI] [PubMed] [Google Scholar]
- 7.Howard CJ, Glynn AA. Immunology. 1971;20:767. [PMC free article] [PubMed] [Google Scholar]
- 8.Albanyan EA, Edwards MS. Infect Immun. 2000;68:5794. doi: 10.1128/iai.68.10.5794-5802.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards MS, Kasper DL, Jennings HJ, Baker CJ, Nicholson-Weller A. J Immunol. 1982;128:1278. [PubMed] [Google Scholar]
- 10.Wessels MR, Paoletti LC, Pinel J, Kasper DL. J Infect Dis. 1995;171:879. doi: 10.1093/infdis/171.4.879. [DOI] [PubMed] [Google Scholar]
- 11.Nilo A, Passalacqua I, Fabbrini M, Allan M, Usera A, Carboni F, Brogioni B, Pezzicoli A, Cobb J, Romano MR, Margarit I, Hu Q, Berti F, Adamo R. Bioconjugate Chem. 2015;26:1839. doi: 10.1021/acs.bioconjchem.5b00365. [DOI] [PubMed] [Google Scholar]
- 12.Hevey R, Ling CC. Future Med Chem. 2012;4:545. doi: 10.4155/fmc.11.193. [DOI] [PubMed] [Google Scholar]
- 13.Hecht ML, Stallforth P, Silva DV, Adibekian A, Seeberger PH. Curr Opin Chem Biol. 2009;13:354. doi: 10.1016/j.cbpa.2009.05.127. [DOI] [PubMed] [Google Scholar]
- 14.Wessels MR, DiFabio JL, Benedi VJ, Kasper DL, Michon F, Brisson JR, Jelinkova J, Jennings HJ. J Biol Chem. 1991;266:6714. [PubMed] [Google Scholar]
- 15.Huang X, Huang L, Wang H, Ye XS. Angew Chem Int Ed Engl. 2004;43:5221. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 16.Sun B, Srinivasan B, Huang X. Chem Eur J. 2008;14:7072. doi: 10.1002/chem.200800757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao J, Guo Z. J Org Chem. 2013;78:12717. doi: 10.1021/jo4021979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Liao G, Wang L, Guo Z. Org Lett. 2014;16:988. doi: 10.1021/ol4036903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mondal PK, Liao G, Mondal MA, Guo Z. Org Lett. 2015;17:1102. doi: 10.1021/ol5036563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu G, Du Y, Linhardt RJ. J Org Chem. 2004;69:5497. doi: 10.1021/jo0493929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao JA, Li X, Gu G, Sun B, Cui M, Ji M, Lou HX. Bioorg Med Chem Lett. 2011;21:622. doi: 10.1016/j.bmcl.2010.12.046. [DOI] [PubMed] [Google Scholar]
- 22.Gilbert W, Youlin Z, Xuefei H. Sci China Chem. 2011;54:66. doi: 10.1007/s11426-010-4186-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu CH, Chu KC, Lin YS, Han JL, Peng YS, Ren CT, Wu CY, Wong CH. Chem Eur J. 2010;16:1754. doi: 10.1002/chem.200903035. [DOI] [PubMed] [Google Scholar]
- 24.Xia J, Alderfer JL, Piskorz CF, Matta KL. Chem Eur J. 2000;6:3442. doi: 10.1002/1521-3765(20000915)6:18<3442::aid-chem3442>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 25.Seifert J, Lergenmuller M, Ito Y. Angew Chem Int Ed Engl. 2000;39:531. doi: 10.1002/(sici)1521-3773(20000204)39:3<531::aid-anie531>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.