

Abstract
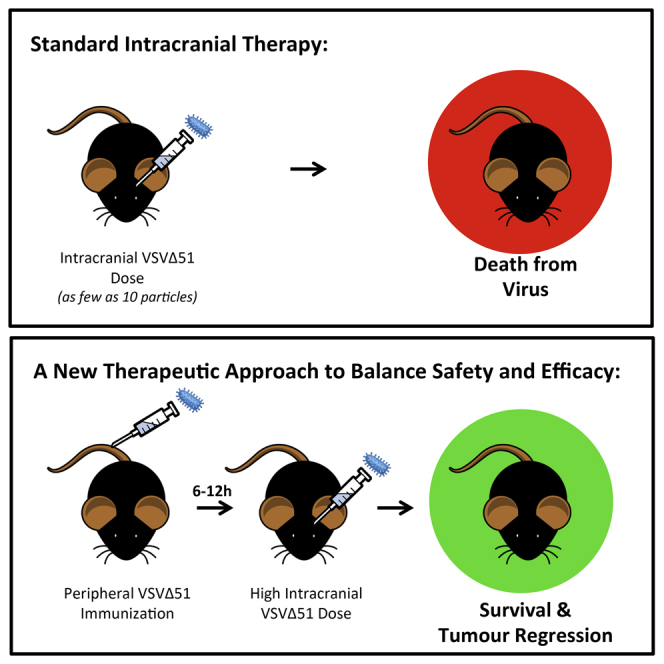
The oncolytic mutant vesicular stomatitis virus VSVΔ51 achieves robust efficacy in multiple extracranial tumor models. Yet for malignancies of the brain, direct intratumoral infusion of VSVΔ51 causes lethal virus-induced neuropathology. Here, we have developed a novel therapeutic regime that uses peripheral immunization with a single sub-lethal dose of VSVΔ51 to establish an acute anti-viral state that enables the safe intracranial (IC) infusion of an otherwise lethal dose of VSVΔ51 within just 6 hr. Although type I interferons alone appeared insufficient to explain this protective phenotype, serum isolated at early time points from primed animals conferred protection against an IC dose of virus. Adaptive immune populations had minimal contributions. Finally, the therapeutic utility of this novel strategy was demonstrated by peripherally priming and intracranially treating mice bearing aggressive CT2A syngeneic astrocytomas with VSVΔ51. Approximately 25% of animals achieved complete regression of established tumors, with no signs of virus-induced neurological impairment. This approach may harness an early warning system in the brain that has evolved to protect the host against otherwise lethal neurotropic viral infections. We have exploited this protective mechanism to safely and efficaciously treat brain tumors with an otherwise neurotoxic virus, potentially widening the available treatment options for oncolytic virotherapy in the brain.
Keywords: oncolytic, virus, brain, cancer, glioma, neuroimmunology, innate immunity
Graphical Abstract
Introduction
Brain cancer survival rates have not changed significantly over the last 35 years,1 and most forms of glioma remain refractory to current chemical, surgical, and radiation therapies.2 Although oncolytic virus (OV)-based approaches to brain cancer treatment have enormous potential for success, it has thus far proven difficult to balance potent anti-tumor activity with safety when treating via direct intracranial (IC) infusion. Although certain OVs, such as those based on herpes simplex virus (HSV), can be infused safely into the cancer-bearing brain, there is typically little evidence of a radiographic anti-tumor response.3, 4, 5 Conversely, other OVs, such as vesicular stomatitis virus (VSV), achieve robust anti-tumor efficacy in peripheral solid tumor models,6, 7 but are generally extremely neurotoxic when injected directly into the brain.8
We have previously generated the novel VSV mutant VSVΔ51 in an effort to improve VSV safety during oncolytic virotherapy.9 This virus has a single amino acid deletion at position 51 of the VSV M protein that solidifies its therapeutic index by preventing viral blockade of host cell transcription and nucleocytoplasmic transport, including interferon (IFN) production.9, 10 Following infection with VSVΔ51, non-malignant cells with intact IFN pathways are protected, whereas malignant cells, which have often lost the ability to mount an IFN response, remain largely susceptible. In mouse models of brain cancer, peripheral infusion of VSVΔ51 via the intravenous (i.v.) route allows the accumulation of virus at the brain tumor site in the absence of neurotoxic consequences, but few animals are successfully cured.11 However, direct IC delivery of VSVΔ51 remains lethal.12 This implies that VSVΔ51 can be safely tolerated in the brain if the initial location of virus sensing is in the periphery.
It has previously been shown that an extended period of repeat mucosal (intranasal [IN]) or systemic [i.v.] immunizations with wild-type VSV can protect mice from subsequent IC challenge with the same virus.12 This effect was predominantly mediated by adaptive cellular responses, including anti-viral antibodies and T cells.12, 13 In our present study, we show that introducing virus in the periphery can activate an acute anti-viral response that conditions the brain to enable IC therapeutic application of an oncolytic virus in the absence of otherwise lethal consequences. Further, we demonstrate that this therapeutic approach can be used to successfully treat aggressive, syngeneic brain tumors. This concept may widen the treatment options available for oncolytic virus therapy in the brain.
Results
A Short Priming Exposure to VSVΔ51 in the Periphery Prevents Lethal CNS Effects when Virus Is Introduced into the Brain
In order to establish if acute pre-exposure to a low dose of systemic virus could condition mice to tolerate an otherwise lethal IC dose, mice were “primed” in the periphery with VSVΔ51-GFP and treated with an IC dose of VSVΔ51 expressing firefly luciferase (VSVΔ51-fLuc) at different times following the prime. This IC dose was 106 higher than the lethal dose.12 The replication kinetics of the therapeutic (IC) virus were subsequently tracked by luciferase signal using IVIS imaging.
A CNS-protective effect established by the peripheral VSVΔ51-GFP prime was evident as early as 6 hr post-prime, with 67% of mice surviving a subsequent IC dose (Figure 1A). By 12 hr post-prime, 100% of VSVΔ51-primed mice were protected from the IC dose (Figure 1A). By 96 hr post-prime, the IC dose was suppressed very rapidly in the brain (Figure 1A).
Figure 1.

Peripheral Priming with VSVΔ51 Acutely Protects against an Otherwise Lethal Therapeutic Dose of Virus in the Brain
(A) BALB/c mice were treated with an intracranial dose of 2 × 107 PFU VSVΔ51-fLuc virus at 6, 12, 24, 72, or 96 hr post-prime with 1 × 108 PFU VSVΔ51-GFP or PBS (i.v.) (left). IC virus replication was tracked by IVIS imaging to capture luminescence (photons per second per cm2 per steridian [p/s/cm2/sr]) based on 3 mice per group. Dotted line indicates background luminescence (right). Example luminescence images are provided (bottom). Error bars refer to SD. (B) Outline of the prime/treat model. A peripheral i.v. prime with 1 × 108 PFU VSVΔ51-GFP or PBS was followed by an intracranial challenge 24 hr later with 2 × 107 PFU VSVΔ51-fLuc or PBS in female BALB/c mice, unless otherwise stated. (C) Correlation between virus titer and luciferase signal. Replication of IC-administered VSVΔ51-fLuc virus quantified over time by in vivo IVIS imaging based on two mice per group (left). Dotted line indicates background luminescence. Virus titers from brain homogenates determined by plaque assay at different time points post-IC treatment (right). Dotted line indicates limit of detection. Error bars refer to SD. (D) Survival curves for male versus female BALB/c mice based on 5 mice per group.
For many oncolytic virus platforms, a degree of virus replication appears to be a necessary requirement for efficacy.14, 15, 16, 17 Since IC treatment at 24 hr post-prime allowed virus replication but ultimately safely controlled IC viral loads, we further explored this priming schedule in subsequent experiments (Figure 1B). We established that luciferase activity from the replicating IC-delivered virus, as measured by total photon flux, was directly correlated with virus titers in the brain (Figure 1C; see also Figure S1). Ultimately, all VSVΔ51-primed animals survived an otherwise uniformly lethal IC dose, whereas all PBS-primed animals succumbed to encephalitis (Figure 1D). This effect was observed in both male and female animals and was maintained across multiple strains of mice, including C57BL/6, FVBN, and BALB/c (Figure 1D; see also Figure S2).
Route of Peripheral Priming Impacts Viral Clearance and Protection in the CNS
To establish if the route of peripheral priming had an impact on the clearance of IC virus from the brain and survival outcomes, mice were primed with VSVΔ51-GFP for 24 hr via intraperitoneal (i.p.), intramuscular (i.m.), i.v., IN, or subcutaneous (s.c.) routes. Subsequently, animals received an IC dose of VSVΔ51-fLuc.
Intraperitoneal priming resulted in the most efficient clearance of virus from the brain following IC treatment, with a more rapid reduction in viral loads in the brain compared to other routes (p < 0.001 at 48 hr) (Figure 2A). Other routes of administration, including i.m., IN, i.v., and s.c., achieved similar rates of VSVΔ51-fLuc clearance from the brain post-IC treatment. However, only priming through i.p., i.v., and i.m. routes provided complete protection from the otherwise lethal neurotoxic consequences following IC treatment (Figure 2B).
Figure 2.

Route of Priming Affects Viral Replication in the Brain
BALB/c mice were primed via the following routes: i.m., IN, i.p., s.c., or i.v., followed by intracranial treatment, based on 3 mice per group. (A) Clearance of the IC virus treatment dose in VSVΔ51-GFP- or PBS-primed mice was monitored by measuring the luminescent signal from the VSVΔ51-fLuc virus via IVIS imaging. Dotted line represents background luminescence. Statistical analysis performed by 2-way ANOVA. ***p < 0.001. Error bars refer to SD. (B) Survival curves for mice primed via each route. Statistical analysis performed by log-rank (Mantel-Cox) test. *p < 0.05.
Peripheral Priming Reduces but Does Not Prevent VSVΔ51 Infection in the Brain
In order to determine the impact of priming on the behavior of virus in the brain, we analyzed VSVΔ51-fLuc localization and infection following IC treatment in VSVΔ51-primed or control animals. Viral antigen was analyzed in the brain by immunohistochemistry or immunofluorescence at different time points following IC treatment.
Overall, less viral antigen was evident in the brains of VSVΔ51-primed animals compared to mock-primed animals following IC treatment. At 24 hr post-IC, viral antigen was detected around the ventricles and hippocampus in the brains of mock-primed mice but was largely confined to the ventricles in animals primed with VSVΔ51 (Figure 3A). By 72 hr post-IC treatment, virus had spread extensively throughout the hippocampus and midsection near the lateral ventricle in mock-primed animals, whereas in VSVΔ51-primed mice, virus remained restricted to the lateral ventricle, with relatively minimal viral antigen loads detected in the hippocampus (Figure 3A). By 96 hr, viral antigen had spread from the lateral ventricle (LV), third ventricle (3V), and dorsal third ventricle (D3V) into the hippocampus, thalamus, striatum, and fourth ventricle (4V) in mock-primed animals. However, in VSVΔ51-primed mice, virus continued to be contained within discrete areas at relatively low levels, with no spread into 4V (Figure 3A). No viral antigen was detected in the brains of mice receiving a VSVΔ51-GFP priming dose alone (no IC treatment) (data not shown).
Figure 3.

Peripheral Priming Reduces VSVΔ51 Infection in the Healthy Brain
(A) VSV antigen was detected by immunohistochemistry in the brains of BALB/c mice primed for 24 hr and treated IC for 24, 72, or 96 hr. Solid arrows highlight the choroid plexus; dashed arrows highlight the lateral ventricles. Scale bar, 3 mm. (B) Co-localization immunofluorescent analysis of VSV antigen (green) with different brain cell populations at 24 hr post-IC treatment. Sections were labeled with Texas Red secondary antibodies, including neurons (NeuN+), microglia/endothelia (RCA-1+), and astrocytes/ependymal cells (GFAP+). Hoescht (blue) was applied for nuclear staining. Images taken at 20x magnification.
At the cellular level, viral antigen co-localized with neuronal markers (NeuN+) in both VSVΔ51-primed and mock-primed mice (Figure 3B). However, in unprotected mock-primed animals, robust virus-associated fluorescence was observed in both the cell body and neurites, suggesting a fulminant infection. In protected VSVΔ51-primed animals, low levels of virus-associated fluorescence were confined to the cell body. Viral antigen could not be detected within microglia/endothelia (RCA-1+) or astrocyte (GFAP+) populations in the brains of either group of animals (Figure 3B), consistent with previous reports.18 Thus, peripheral priming with VSVΔ51-GFP did not alter viral cellular specificity, but had a profound impact on limiting VSVΔ51-fLuc viral replication and spread throughout the brain.
Adaptive Immune Responses Have a Limited Contribution to Peripheral Priming-Induced Protection in the Brain
We analyzed the contribution of adaptive immune responses to the protective benefits conferred by our peripheral prime. Neither T cells nor B cells were observed to infiltrate the brain at early time points following IC treatment (see Figure S3). T cells were detectable by 6 days post-IC treatment in the brains of both PBS- and VSVΔ51-primed mice (see Figure S3), whereas B cells remained absent at this time point (see Figure S3). Interestingly, a substantial infiltration of cells with a phenotype consistent with a granulocytic/monocytic origin (CD45+CD11b+Gr-1+ cells) was observed in the brain of PBS-primed animals at 12 hr following IC VSVΔ51-fLuc treatment. However, this population was absent in the brains of VSVΔ51-primed animals (see Figure S4). We also observed an increase in the expression of toll-like receptor 2 (TLR2) in a microglial cell population defined as CD45loCD11b+Gr-1− in the brains of mice receiving a peripheral VSVΔ51 prime compared to control animals (see Figure S4), consistent with the establishment of a pro-inflammatory state that can protect against a lytic virus infection.19 This upregulation of TLR2 was largely driven by the peripheral virus dose (see Figure S4), further supporting the concept that the peripheral prime conditioned the brain to facilitate viral control.
The contribution of T cells to the protective effect of the VSVΔ51 prime was further assessed in nude mice, which lack mature T cells. VSVΔ51-primed nude mice controlled viral titers in the brain following IC treatment (Figures 4A and 4B) and were fully protected from CNS lethality (Figure 4C). Since B cells have been reported to have a role in mediating anti-viral effects following VSV infection in the periphery,20 we further assessed their contribution in our model using u-chain-deficient mice, which lack mature B cells. Following a peripheral VSVΔ51-GFP prime, u-chain-deficient mice controlled VSVΔ51-fLuc viral replication after IC treatment, and all animals survived (Figure 4D and data not shown). Thus, neither T cells nor B cells were critical to allowing peripherally primed mice to clear IC-administered virus in the acute therapeutic time frames established in our model.
Figure 4.

Adaptive Factors Have Minimal Contributions to the CNS Protection Induced by VSVΔ51 Priming
Top: nude mice were primed i.p. with 2 × 108 PFU VSVΔ51-GFP or PBS for 48 hr, followed by IC treatment with 1 × 107 PFU VSVΔ51-fLuc, based on 3 mice per group. (A and B) IVIS imaging was used to measure luciferase signal and track IC-administered virus: (A) luminesce images; (B) graph of total flux. Dotted line indicates background luminescence. Error bars refer to SD. (C) Survival curves were assessed by log-rank (Mantel-Cox) test. *p < 0.05. (D) Bottom: luminescence from IC-applied virus was analyzed in primed u-chain-deficient mice, based on 3 mice per group. Error bars refer to SD. (E) Detection of anti-VSV neutralizing antibodies in the serum of BALB/c mice primed with 1 × 108 PFU VSVΔ51-GFP or PBS and analyzed at 24 or 48 hr post-prime by plaque reduction assay on Vero cells based on 3 mice per group. Dotted line represents limit of detection. Statistics were calculated by one-way ANOVA analysis. ns, not significant; ***p < 0.001. Negative control represents media alone. Positive control represents serum isolated from VSV immune mice (primed with 1 × 108 PFU VSVΔ51-GFP for 144 hr).
Finally, we explored the potential contribution of virus-neutralizing antibodies to the protective effect observed in our prime/treat model. Serum isolated from VSVΔ51-primed animals at 48 hr but not 24 hr post-prime was able to reduce VSV infectivity in susceptible target cells above the negative control (Figure 4E). Thus, at the time of IC treatment (24 hr post-prime), there was no evidence of VSV-neutralizing activity in the blood, suggesting that antibodies did not contribute to the priming effect in our model.
IFNs Are Beneficial, but Not Sufficient, to Mediate Protection during IC Virus Therapy
The rapid induction of protection in the brain within hours following a peripheral VSVΔ51 prime suggested the involvement of innate rather than adaptive immune mechanisms. Although wild-type VSV can block IFN production to evade immune responses during host cell infection, the VSVΔ51 mutant is much less potent in its ability to impact host immune pathways. Indeed, host cell soluble factors, including type I IFNs, appear in the serum shortly after VSVΔ51 infection9 and play an important role in limiting local and systemic spread.21, 22
In order to explore the possibility that innate factors in the serum had a role in the early brain-protective effects observed following a VSVΔ51 peripheral prime, we isolated serum, splenocytes, and lymph nodes from VSVΔ51-primed mice at 6 hr post-prime. We determined if i.p. passive transfer of these isolated tissue fractions into naive mice could establish protection against an IC dose of VSVΔ51-fLuc. Transfer of serum but not splenocytes or lymph nodes isolated from 6 hr VSVΔ51-primed mice could control virus in the brain following IC treatment and ultimately protected mice from otherwise lethal CNS consequences (Figure 5A and data not shown). Serum isolated from mice primed with VSVΔ51 for 24 hr failed to control IC virus loads and did not protect mice (Figure 5A). This suggested that a protective factor(s) was present in the serum during a short window (<24 hr) following a peripheral VSVΔ51 prime.
Figure 5.

Soluble Innate Factors Are Important for Establishing Protection against Intracranial VSV Infection
(A) BALB/c mice were primed i.p. with virus-depleted serum, bulk splenocytes, or bulk iliac lymph node populations isolated from mice at 6 hr (all tissues) or 24 hr (serum only) post-VSVΔ51-GFP prime or with 1 × 108 PFU VSVΔ51-GFP (positive control) or PBS (negative control). At 24 hr post-prime, mice received an IC dose of 2 × 107 PFU VSVΔ51-fLuc. Replication of the IC dose in the brain was monitored by luciferase signal via IVIS imaging. Dotted line represents background luminescence. Error bars refer to SD. (B) Levels of IFNα, IFNβ, and IFNγ were assessed by ELISA in triplicate samples in the serum or CSF of naive mice at 6 or 12 hr following a single peripheral (i.p.) or IC dose of 1 × 108 PFU VSVΔ51-GFP. Error bars refer to SD. (C) BALB/c mice were primed with IFNα, IFNβ, IFNγ, or 1 × 108 PFU VSVΔ51-GFP (i.v.) (positive control) or PBS (i.v.) (negative control). At 24 hr post-prime, mice received an IC dose of 2 × 107 PFU VSVΔ51-fLuc. IC virus replication was tracked by luminescence using IVIS imaging (left), and corresponding survival data were captured (right) based on 3 mice per group. Dotted line represents background luminescence. Statistical analysis of survival curves was performed using the log-rank (Mantel-Cox) test relative to PBS. *p < 0.05; **p < 0.01; ns, no significance. Error bars refer to SD.
We interrogated serum and cerebrospinal fluid (CSF) at 6 or 12 hr in animals who had received a single dose of VSVΔ51-GFP in the periphery (i.p.) versus directly in the brain (IC) for the presence of IFNα, IFNβ, or IFNγ by ELISA. At 6 hr, all three IFNs were detectable in the serum of mice receiving an i.p. VSVΔ51-GFP prime, with levels declining by 12 hr (Figure 5B). In the CSF, only IFNα was detectable at 6 hr post-i.p. prime, declining below the limit of detection by 12 hr (Figure 5B). In mice receiving VSVΔ51-GFP directly into the brain, low or undetectable levels of all three cytokines were evident at 6 hr in the serum or CSF (Figure 5B). However, levels of IFNγ increased in the serum at 12 hr, whereas levels of IFNβ increased in the CSF at 12 hr, consistent with the previously defined role of IFNβ as a key protective type I IFN produced locally in the brain.23, 24, 25 Taken together, this suggested that the kinetics of IFN production during a primary IC infection may have been too slow to adequately control virus in the brain; however, the administration of virus via the i.p. route achieved an early peak in IFNs.
Since all three detected IFNs play a role in modulating blood-brain barrier (BBB) permeability26, 27, 28 and contribute to the anti-VSV response in intranasal infection models,25, 29 we proceeded to further investigate their contribution to CNS protection in the prime/treat setting. To do this, we introduced each individual cytokine exogenously as a “prime” (in the absence of virus), followed by IC application of VSVΔ51-fLuc. IFNβ or IFNγ were administered IC because they are typically produced locally in the brain during infection,23 whereas IFNα, typically considered a peripheral cytokine with limited penetrance across the BBB,30 was administered i.p. at doses known to block peripheral VSV infection.31
Mice primed with IFNα had no benefit in terms of viral control or survival following IC treatment (Figure 5C). Similarly, administering IFNβ as an exogenous prime had no protective effect (Figure 5C). Priming with exogenous IFNγ recapitulated the protection conferred by the VSVΔ51 prime, both in terms of replication control and survival (p < 0.05) (Figure 5C). However, VSVΔ51-priming in IFNγ−/− transgenic knockout mice controlled IC virus loads (see Figure S5) and provided complete protection, whereas all mock PBS-primed mice succumbed to lethal encephalitis (see Figure S5). This suggested that IFNγ could be protective, but did not appear to be absolutely required within the context of our prime/treat model. As an extended experimental approach, we aimed to knock out type I IFNs in the prime/treat setting by using either antibody-mediated blockade in wild-type mice or transgenic IFNAR−/− mice. However, the priming dose of virus alone was lethal in both settings, rendering these experiments untenable. This included priming with VSVΔ51-GFP or VSVΔ51-Gless (which can undergo VSV-G-mediated cell entry but is only capable of a single-round of infection and is protective in the prime/treat setting in wild-type mice); UV-inactivated VSVΔ51-GFP was also considered as a prime but failed to protect in wild-type mice and was thus discarded. Finally, priming animals with other viruses capable of inducing type I IFN responses, such as reovirus,32 or innate immune stimulators, such as lipopolysaccharide (LPS) or the TLR7 agonist loxoribine,33 also failed to control IC VSVΔ51 loads and failed to protect mice (see Figure S6). Thus, IFNs appeared to be necessary but not sufficient as standalone mediators of protection in our model.
Peripheral VSVΔ51 Priming Allows Effective IC Oncolytic Treatment in Syngeneic Gliomas
We evaluated the therapeutic relevance of the VSVΔ51 prime/treat regime in the aggressive orthotopic syngeneic CT2A glioma model. CT2A astrocytoma cells expressing fLuc were seeded intracranially in C57BL/6 mice. Following priming and IC treatment, approximately 20% of CT2A tumor-bearing mice responded with complete tumor regression, as monitored through fLuc expression (Figures 6A and 6B). Mice that initially responded to the prime/treat regime survived and remained tumor free (Figures 6A and 6B). Mice that did not respond to the prime/treat regime succumbed due to tumor complications, including weight loss and neurological symptoms. Priming with VSVΔ51 alone in the absence of a subsequent therapeutic IC dose did not have a significant impact on survival compared to PBS controls (Figures 6A and 6B).
Figure 6.

Peripheral Priming Enables Efficacious Loco-regional Therapy against Aggressive Brain Tumors
(A) C57BL/6 mice bearing CT2A-fLuc tumors were primed i.p. with 5 × 108 VSVΔ51-GFP at 6 days post-tumor implantation (day 0) and treated IC with a single dose of 5 × 107 VSVΔ51-NR 18 hr later (n = 23). Control mice received an intracranial infusion of PBS following a VSVΔ51-GFP prime (n = 9) or dual PBS prime/PBS treat (n = 9). (B) Survival curves for different treatment groups. *p < 0.05 (VSV prime/PBS treat versus VSV prime/VSV treat) and ***p < 0.0001 (PBS prime/PBS treat versus VSV prime/VSV treat) assessed by log-rank (Mantel-Cox) test.
Discussion
We have developed a novel strategy for oncolytic virus therapy in the brain that appears to harness an early warning system that can protect from the otherwise lethal encephalitic effects of a rhabdovirus penetrating into the CNS. From a therapeutic perspective, this provides a window of opportunity in which peripheral host responses can be sufficiently engaged to protect against neurotoxicity by rendering the normal brain resistant to an IC therapeutic dose of virus while simultaneously maintaining tumor susceptibility to viral oncolytic effects. Ultimately, this prime/treat approach creates a therapeutic index that allows the application of a loco-regional viral therapy to achieve cures in an aggressive syngeneic glioma model.
VSV induces a myriad of host immune responses in the first 24 hr following infection.34 This includes the long-distance activation of an IFN-independent anti-viral state in uninfected cells of the brain following systemic infection.25 Extended (6 week) peripheral immunization regimes have previously been shown to enable the direct IC infusion of attenuated VSV mutants without lethal consequences, highlighting a role for adaptive immunity and neutralizing antibodies in the control of VSV over longer therapeutic time frames.12 Yet the rapid ability of a priming dose to control IC VSVΔ51 replication in our model was fully established by 12 hr and was maintained in the absence of mature T cells (in nude mice) or mature B cells (in u-chain-deficient mice). We were unable to detect virus-neutralizing antibodies earlier than 48 hr following the peripheral VSVΔ51 prime, similar to previous reports.29, 35, 36, 37 Further, our observations that an IC challenge at 96 hr post-prime severely limited virus replication in the brain suggested that sterilizing immunity was established around 96 hr following the initial priming exposure, which is in line with previous reports.38, 39, 40 Collectively, this implied that B cells (including neutralizing antibodies) and T cells were not overtly involved in the early and rapid protective effects conferred by the peripheral VSVΔ51 prime. This does not exclude their contribution at later stages of virus replication in solidifying CNS protection, as previously reported.41, 42
Peripheral priming with VSVΔ51 reduced the spread of IC virus through the brain and did not alter viral tropism following IC treatment, with VSV antigen co-localizing with neuronal populations, as previously observed.43 Importantly, there was no evidence of neurological dysfunction in the prime/treat model, suggesting that this approach was clinically tractable.
Serum isolated from 6 hr VSVΔ51-primed mice conferred protection against IC virus in naive animals, whereas 24 hr primed serum was not protective. This suggested that a soluble factor(s) in the serum may be mediating the protective effect. Interestingly, the protection conferred by passive transfer was transient, whereas the protection achieved by natural infection in the periphery appeared to be relatively long lasting. We hypothesize that in vivo, the natural course of peripheral infection likely establishes depots of viral antigen that persist following the resolution of VSVΔ51 infection and contribute to ongoing, low-level immune system stimulation, as previously described.44, 45 This likely sustains the protective effect over an extended period of time. However, in the case of the passive transfer of serum, which has been treated to remove infectious virus, protective factor(s) are undoubtedly present in a finite amount, and these immune resources are likely rapidly consumed. Elevated levels of IFNα, IFNβ, and IFNγ were present at 6 hr in the serum of i.p. VSVΔ51-primed mice, a time point when we observed the majority of mice (∼67%) to be protected from a subsequent IC challenge, with concentrations of these cytokines declining by 12 hr post-prime. Indeed, the rapid production of IFNβ we observed following the peripheral VSVΔ51 prime, which has previously been shown for other rhabdoviruses,46 may have been responsible for the block in granulocyte infiltration into the brain that we observed following peripheral priming.47 Such a reduction in granulocytic infiltration can dampen pathological inflammation and promote oncolytic safety as granulocytes contribute to the development of viral encephalitis but do not affect viral clearance.48
After testing the contribution of individual IFNs to the protective effect by administering them as an exogenous prime prior to IC virus administration, we concluded that neither IFNα nor IFNβ alone was sufficient to orchestrate protection because i.p. or IC delivery of these cytokines, respectively, failed to recapitulate the benefits of a VSVΔ51 prime. Of note, IFNγ was capable of achieving protection in the brain because its exogenous administration as a local IC “prime” 6 hr prior to VSVΔ51 IC treatment controlled viral loads in the brain and achieved complete protection, similar to the VSVΔ51 peripheral prime itself. Indeed, other groups have demonstrated that IFNγ can protect neurons from VSV infection.49, 50, 51 Yet because IFNγ knockout mice could be protected with a VSVΔ51 prime to the same degree as wild-type mice, this suggested a redundant role for IFNγ in our model. Other groups have also shown that systemic IFNγ contributes to but is not essential for local clearance of VSV in the brain.52 Interestingly, our attempts to utilize other viruses or “virus-like” danger signals by priming with reovirus or innate immune stimulators, such as LPS or loxoribine, were similarly unable to confer protection against IC VSVΔ51 treatment.
Here, we have described a mechanism that allows VSVΔ51 to be tolerated in both the normal and tumor-bearing brain. This mechanism can be exploited to improve the safety of oncolytic virus therapy by initiating an anti-viral state through a short peripheral exposure to virus, thereby enabling the IC administration of therapeutically relevant doses (over 10,000 times more virus particles than have been previously reported12) of a normally neurotoxic virus to treat brain cancer. Importantly, this approach achieved successful cures in an aggressive, syngeneic glioma model with a single IC dose of VSVΔ51. These findings have clear translational relevance because work in our laboratory suggests that the outcome of oncolytic virotherapy in the brain may be enhanced when virus is delivered by dual i.v. and IC routes (unpublished observation). The approach we have described may guide the development of similarly clinically tractable strategies to treat CNS malignancies with other extremely potent oncolytic viruses, such as Maraba virus.6, 7 Future studies that determine the precise combination of innate factors that contribute to CNS protection will prove useful for guiding new interventions aimed at preventing diseases affecting the brain.
Materials and Methods
Mice
6- to 8-week-old BALB/c, C57BL/6, FVBN, and nude mice were purchased from Charles River Canada (Constant, QC, Canada). IFNγ knockout (BALB/c-Ifgtml), IFNAR knockout (C57BL/6-129S2-Ifnar1tm1Agt/Mmjax), and mu-chain deficient (C57BL/6-129S2-Ighmtm1Cgn/J) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were kept in sterile isolation cages and maintained on a 12-hr dark-light cycle. To establish the CT2A astrocytoma model,53, 54 C57BL/6 mice were injected sterotactically with 105 CT2A-fLuc cells. All animal procedures were performed in accordance with the institutional guidelines of the University of Ottawa committee on the Use of Live Animals in Teaching and Research.
Viruses and Infections
The VSVΔ51-GFP (Indiana serotype), VSVΔ51-GFP-fLuc (herein referred to as VSVΔ51-fLuc), and VSVΔ51 no reporter (VSVΔ51-NR) viruses were provided courtesy of Dr. John Bell (OHRI, Canada). Unless otherwise stated, mice were primed intravenously with 1 × 108 plaque forming unit (PFU) (BALB/c) or 5 × 108 PFU (C57BL/6) of VSVΔ51-GFP or PBS as a control. Unless otherwise stated, mice were treated intracranially with 2 × 107 PFU VSVΔ51-fLuc or VSVΔ51-NR using stereotactic injection into the striatum. Mice were monitored daily by weight and euthanized at endpoint (neurological symptoms or loss of >20% of initial body weight). Reovirus (type 3 Dearing) was administered i.p. at a sub-lethal dose of 108 PFU.
IVIS Imaging
Luciferase-expressing virus or CT2A-Luc astrocytomas were detected by optical in vivo imaging using the Xenogen IVIS-200 system. Mice were anesthetized by isofluorane and injected i.p. with 4 mg of luciferin (Molecular Imaging Products) 5 min prior to imaging. Images were processed and analyzed using Living Image 3.1 software (Xenogen). Flux curves were plotted as mean with SD, unless otherwise indicated.
ELISA
CSF was collected from the cisterna magna. Serum was isolated from whole blood by centrifugation at 10,000 × g. IFNα and -β were assayed in CSF and serum by ELISA (PBL Biomedical Laboratories) using a Spectramax 340 PC automated plate reader at 450 nm. Data were acquired using Softmax Pro 4.7.1 (Molecular Devices).
Soluble Priming Treatments
BALB/c mice were injected i.p. with 20,000 U IFNα or PBS 24 hr prior to receiving an IC dose of VSVΔ51. Two additional doses of 10,000 U IFNα were administered i.p. just prior to IC dosing and at 24 hr post-IC dosing. Exogenous IFNβ was administered at 10,000 U IC 6 hr prior to IC treatment. Exogenous IFNγ was administered at 50,000 U IC 6 hr prior to IC treatment. Loxoribine was administered i.p. at 2 mg per mouse, and LPS was administered i.p. at 1 mg/kg.
For serum, spleen, and lymph node fraction adoptive transfers, organs were harvested from animals primed with 1 × 108 PFU VSVΔ51-GFP at 6 or 24 hr (serum only) post-prime. Serum was isolated as above. Spleens or lymph nodes were manually homogenized, dissociated, and passed through a 2 μm filter to prepare pooled bulk splenocytes or lymph node homogenates, respectively. Each dissociated organ preparation was incubated with anti-VSV(G) (a gift from Dr. E. Brown, University of Ottawa) and run through a Protein G column to precipitate antibody-associated virus. Plaque assays were performed to verify that no infectious virus was present before adoptive transfer.
Virus Neutralization Assay
Serum samples were isolated as described above, diluted 1:100, and incubated with serial dilutions of VSVΔ51 for 1 hr at 37°C prior to plating on confluent Vero cells. After 1 hr, VSV-infected Vero cells were overlaid with a 1:1 mixture of 2×αMEM containing 20% fetal calf serum (FCS) and agarose. Plaques were counted after 24 hr.
Immunohistochemistry
Mice were anesthetized with a lethal dose of sodium pentobarbital and trans-cardially perfused with PBS. For frozen sections, mice were subsequently perfused and brains were isolated in 4% paraformaldehyde (PFA) followed by 20% sucrose and then flash frozen in liquid nitrogen. 10 μm horizontal or sagittal sections were cut and mounted on Superfrost Plus (Fisher Scientific) glass slides. For paraffin sections, fresh brains were transferred into 10% phosphate-buffered formalin followed by 70% EtOH and paraffin embedding. 5 μm sections were mounted onto glass slides. Sections were de-paraffinized and antigen retrieval was performed in sodium citrate buffer. Sections were incubated with 3% hydrogen peroxide and blocked in 5% serum for 1 hr at room temperature (RT). Slides were incubated with primary antibody (anti-CD3ε [BD Biosciences], anti-CD45R/B220 [BD Biosciences], or anti-VSV(G) [a gift from Dr. E. Brown, University of Ottawa]) for 2 hr, followed by a biotinylated secondary antibody (BD Biosciences) for 30 min at RT. Avidin-Biotin-Peroxidase (ABC Kit, Vector Laboratories) was applied as per the manufacturer’s protocol. 3,3'-Diaminobenzidine (DAB; Sigma) was added for 2 min before immersing in ddH2O and counterstaining with hematoxylin, followed by slide mounting in xylene medium. Sections were scanned using a UMAX Astra 1220S scanner.
Immunofluorescence Microscopy
Slides were incubated with primary antibody (anti-GFAP [Abcam], anti-RCA-1-biotin [Vector Laboratories], anti-NeuN [clone A60, Chemicon International], and anti-VSV(G) [as above]) for 2 hr followed by secondary antibodies (streptavidin-Texas Red [Vector Laboratories], goat anti-chicken immunoglobulin Y [IgY]-Texas Red [Abcam], goat anti-mouse rhodamine [Abcam], goat anti-rabbit Cy3 [BD Biosciences], or goat anti-rabbit AF488 [BD Biosciences]). Sections were viewed using Zeiss Axioskop 2 mot (Carl Zeiss Canada) fitted with a QICAM digital CCD camera (Qimaging). Northern Eclipse software (EMPIX Imaging) was used to acquire all images.
Flow Cytometry
Brains were isolated and homogenized, and single-cell suspensions were prepared by filtration through a 100 μm mesh. Cellular infiltrates were stained with anti-CD45.2 (clone 104, BD Biosciences), anti-CD11b (clone M1/70, BD Biosciences), anti-Ly-6G/Ly-6C (clone RB6-8C5, BD Biosciences), and/or TLR2 (clone 6C2, eBioscience), and acquired on a FACS Canto (BD Biosciences) running DIVA software.
Author Contributions
L.B., V.A.T., B.Y., M.L., C.L., J.B., S.L.S., and D.F.S. conducted the experiments. L.B., V.A.T., S.L.S., and D.F.S. designed the experiments. V.A.T., S.L.S., and D.F.S. wrote the manuscript. D.F.S. secured funding for the project.
Conflicts of Interest
D.F.S. owns shares and patents in several rhabdovirus-based oncolytic technologies and is a founding scientist of the oncolytic virus immunotherapy company Turnstone Biologics.
Acknowledgments
The authors would like to thank Teresa Falls and Jennifer Sparling for supporting this project. This research was funded by the National Cancer Institute of Canada (to D.F.S.). The funders had no role in the study design, data collection, data interpretation, or decision to submit the work for publication.
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.omto.2017.09.004.
Supplemental Information
References
- 1.Lwin Z., MacFadden D., Al-Zahrani A., Atenafu E., Miller B.A., Sahgal A., Menard C., Laperriere N., Mason W.P. Glioblastoma management in the temozolomide era: have we improved outcome? J. Neurooncol. 2013;115:303–310. doi: 10.1007/s11060-013-1230-3. [DOI] [PubMed] [Google Scholar]
- 2.Lim S.K., Llaguno S.R., McKay R.M., Parada L.F. Glioblastoma multiforme: a perspective on recent findings in human cancer and mouse models. BMB Rep. 2011;44:158–164. doi: 10.5483/BMBRep.2011.44.3.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrow S., Papanastassiou V., Harland J., Mabbs R., Petty R., Fraser M., Hadley D., Patterson J., Brown S.M., Rampling R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004;11:1648–1658. doi: 10.1038/sj.gt.3302289. [DOI] [PubMed] [Google Scholar]
- 4.Markert J.M., Liechty P.G., Wang W., Gaston S., Braz E., Karrasch M., Nabors L.B., Markiewicz M., Lakeman A.D., Palmer C.A. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 2009;17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markert J.M., Razdan S.N., Kuo H.C., Cantor A., Knoll A., Karrasch M., Nabors L.B., Markiewicz M., Agee B.S., Coleman J.M. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014;22:1048–1055. doi: 10.1038/mt.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brun J., McManus D., Lefebvre C., Hu K., Falls T., Atkins H., Bell J.C., McCart J.A., Mahoney D., Stojdl D.F. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol. Ther. 2010;18:1440–1449. doi: 10.1038/mt.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pol J.G., Zhang L., Bridle B.W., Stephenson K.B., Resseguier J., Hanson S., Chen L., Kazdhan N., Bramson J.L., Stojdl D.F. Maraba virus as a potent oncolytic vaccine vector. Mol. Ther. 2014;22:420–429. doi: 10.1038/mt.2013.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knudson D.L. Rhabdoviruses. J. Gen. Virol. 1973;20(Suppl):105–130. doi: 10.1099/0022-1317-20-Supplement-105. [DOI] [PubMed] [Google Scholar]
- 9.Stojdl D.F., Lichty B.D., tenOever B.R., Paterson J.M., Power A.T., Knowles S., Marius R., Reynard J., Poliquin L., Atkins H. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 10.Black B.L., Lyles D.S. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J. Virol. 1992;66:4058–4064. doi: 10.1128/jvi.66.7.4058-4064.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lun X., Senger D.L., Alain T., Oprea A., Parato K., Stojdl D., Lichty B., Power A., Johnston R.N., Hamilton M. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J. Natl. Cancer Inst. 2006;98:1546–1557. doi: 10.1093/jnci/djj413. [DOI] [PubMed] [Google Scholar]
- 12.Ozduman K., Wollmann G., Ahmadi S.A., van den Pol A.N. Peripheral immunization blocks lethal actions of vesicular stomatitis virus within the brain. J. Virol. 2009;83:11540–11549. doi: 10.1128/JVI.02558-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed M., Marino T.R., Puckett S., Kock N.D., Lyles D.S. Immune response in the absence of neurovirulence in mice infected with m protein mutant vesicular stomatitis virus. J. Virol. 2008;82:9273–9277. doi: 10.1128/JVI.00915-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breitbach C.J., Paterson J.M., Lemay C.G., Falls T.J., McGuire A., Parato K.A., Stojdl D.F., Daneshmand M., Speth K., Kirn D. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007;15:1686–1693. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- 15.Galivo F., Diaz R.M., Wongthida P., Thompson J., Kottke T., Barber G., Melcher A., Vile R. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17:158–170. doi: 10.1038/gt.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parrish C., Scott G.B., Migneco G., Scott K.J., Steele L.P., Ilett E., West E.J., Hall K., Selby P.J., Buchanan D. Oncolytic reovirus enhances rituximab-mediated antibody-dependent cellular cytotoxicity against chronic lymphocytic leukaemia. Leukemia. 2015;29:1799–1810. doi: 10.1038/leu.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guse K., Sloniecka M., Diaconu I., Ottolino-Perry K., Tang N., Ng C., Le Boeuf F., Bell J.C., McCart J.A., Ristimäki A. Antiangiogenic arming of an oncolytic vaccinia virus enhances antitumor efficacy in renal cell cancer models. J. Virol. 2010;84:856–866. doi: 10.1128/JVI.00692-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huneycutt B.S., Plakhov I.V., Shusterman Z., Bartido S.M., Huang A., Reiss C.S., Aoki C. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 1994;635:81–95. doi: 10.1016/0006-8993(94)91426-5. [DOI] [PubMed] [Google Scholar]
- 19.Perkins D.J., Polumuri S.K., Pennini M.E., Lai W., Xie P., Vogel S.N. Reprogramming of murine macrophages through TLR2 confers viral resistance via TRAF3-mediated, enhanced interferon production. PLoS Pathog. 2013;9:e1003479. doi: 10.1371/journal.ppat.1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moseman E.A., Iannacone M., Bosurgi L., Tonti E., Chevrier N., Tumanov A., Fu Y.X., Hacohen N., von Andrian U.H. B cell maintenance of subcapsular sinus macrophages protects against a fatal viral infection independent of adaptive immunity. Immunity. 2012;36:415–426. doi: 10.1016/j.immuni.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bründler M.A., Aichele P., Bachmann M., Kitamura D., Rajewsky K., Zinkernagel R.M. Immunity to viruses in B cell-deficient mice: influence of antibodies on virus persistence and on T cell memory. Eur. J. Immunol. 1996;26:2257–2262. doi: 10.1002/eji.1830260943. [DOI] [PubMed] [Google Scholar]
- 22.Gobet R., Cerny A., Rüedi E., Hengartner H., Zinkernagel R.M. The role of antibodies in natural and acquired resistance of mice to vesicular stomatitis virus. Exp. Cell Biol. 1988;56:175–180. doi: 10.1159/000163477. [DOI] [PubMed] [Google Scholar]
- 23.Detje C.N., Lienenklaus S., Chhatbar C., Spanier J., Prajeeth C.K., Soldner C., Tovey M.G., Schlüter D., Weiss S., Stangel M. Upon intranasal vesicular stomatitis virus infection, astrocytes in the olfactory bulb are important interferon beta producers that protect from lethal encephalitis. J. Virol. 2015;89:2731–2738. doi: 10.1128/JVI.02044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandberg K., Eloranta M.L., Campbell I.L. Expression of alpha/beta interferons (IFN-alpha/beta) and their relationship to IFN-alpha/beta-induced genes in lymphocytic choriomeningitis. J. Virol. 1994;68:7358–7366. doi: 10.1128/jvi.68.11.7358-7366.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Pol A.N., Ding S., Robek M.D. Long-distance interferon signaling within the brain blocks virus spread. J. Virol. 2014;88:3695–3704. doi: 10.1128/JVI.03509-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitagami T., Yamada K., Miura H., Hashimoto R., Nabeshima T., Ohta T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003;978:104–114. doi: 10.1016/s0006-8993(03)02776-8. [DOI] [PubMed] [Google Scholar]
- 27.Kraus J., Ling A.K., Hamm S., Voigt K., Oschmann P., Engelhardt B. Interferon-beta stabilizes barrier characteristics of brain endothelial cells in vitro. Ann. Neurol. 2004;56:192–205. doi: 10.1002/ana.20161. [DOI] [PubMed] [Google Scholar]
- 28.Minagar A., Long A., Ma T., Jackson T.H., Kelley R.E., Ostanin D.V., Sasaki M., Warren A.C., Jawahar A., Cappell B. Interferon (IFN)-beta 1a and IFN-beta 1b block IFN-gamma-induced disintegration of endothelial junction integrity and barrier. Endothelium. 2003;10:299–307. doi: 10.1080/10623320390272299. [DOI] [PubMed] [Google Scholar]
- 29.Detje C.N., Meyer T., Schmidt H., Kreuz D., Rose J.K., Bechmann I., Prinz M., Kalinke U. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 2009;182:2297–2304. doi: 10.4049/jimmunol.0800596. [DOI] [PubMed] [Google Scholar]
- 30.Spanier J., Lienenklaus S., Paijo J., Kessler A., Borst K., Heindorf S., Baker D.P., Kröger A., Weiss S., Detje C.N. Concomitant TLR/RLH signaling of radioresistant and radiosensitive cells is essential for protection against vesicular stomatitis virus infection. J. Immunol. 2014;193:3045–3054. doi: 10.4049/jimmunol.1400959. [DOI] [PubMed] [Google Scholar]
- 31.Stojdl D.F., Abraham N., Knowles S., Marius R., Brasey A., Lichty B.D., Brown E.G., Sonenberg N., Bell J.C. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 2000;74:9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dionne K.R., Galvin J.M., Schittone S.A., Clarke P., Tyler K.L. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J. Neurovirol. 2011;17:314–326. doi: 10.1007/s13365-011-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heil F., Ahmad-Nejad P., Hemmi H., Hochrein H., Ampenberger F., Gellert T., Dietrich H., Lipford G., Takeda K., Akira S. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur. J. Immunol. 2003;33:2987–2997. doi: 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
- 34.Hastie E., Grdzelishvili V.Z. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 2012;93:2529–2545. doi: 10.1099/vir.0.046672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iannacone M., Moseman E.A., Tonti E., Bosurgi L., Junt T., Henrickson S.E., Whelan S.P., Guidotti L.G., von Andrian U.H. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. 2010;465:1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang K.S., Navarini A.A., Recher M., Lang P.A., Heikenwalder M., Stecher B., Bergthaler A., Odermatt B., Akira S., Honda K. MyD88 protects from lethal encephalitis during infection with vesicular stomatitis virus. Eur. J. Immunol. 2007;37:2434–2440. doi: 10.1002/eji.200737310. [DOI] [PubMed] [Google Scholar]
- 37.Pinschewer D.D., Perez M., Jeetendra E., Bächi T., Horvath E., Hengartner H., Whitt M.A., de la Torre J.C., Zinkernagel R.M. Kinetics of protective antibodies are determined by the viral surface antigen. J. Clin. Invest. 2004;114:988–993. doi: 10.1172/JCI22374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seiler P., Bründler M.A., Zimmermann C., Weibel D., Bruns M., Hengartner H., Zinkernagel R.M. Induction of protective cytotoxic T cell responses in the presence of high titers of virus-neutralizing antibodies: implications for passive and active immunization. J. Exp. Med. 1998;187:649–654. doi: 10.1084/jem.187.4.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinhoff U., Müller U., Schertler A., Hengartner H., Aguet M., Zinkernagel R.M. Antiviral protection by vesicular stomatitis virus-specific antibodies in alpha/beta interferon receptor-deficient mice. J. Virol. 1995;69:2153–2158. doi: 10.1128/jvi.69.4.2153-2158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomsen A.R., Nansen A., Andersen C., Johansen J., Marker O., Christensen J.P. Cooperation of B cells and T cells is required for survival of mice infected with vesicular stomatitis virus. Int. Immunol. 1997;9:1757–1766. doi: 10.1093/intimm/9.11.1757. [DOI] [PubMed] [Google Scholar]
- 41.Bi Z., Barna M., Komatsu T., Reiss C.S. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J. Virol. 1995;69:6466–6472. doi: 10.1128/jvi.69.10.6466-6472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huneycutt B.S., Bi Z., Aoki C.J., Reiss C.S. Central neuropathogenesis of vesicular stomatitis virus infection of immunodeficient mice. J. Virol. 1993;67:6698–6706. doi: 10.1128/jvi.67.11.6698-6706.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sur J.H., Allende R., Doster A.R. Vesicular stomatitis virus infection and neuropathogenesis in the murine model are associated with apoptosis. Vet. Pathol. 2003;40:512–520. doi: 10.1354/vp.40-5-512. [DOI] [PubMed] [Google Scholar]
- 44.Simon I.D., Publicover J., Rose J.K. Replication and propagation of attenuated vesicular stomatitis virus vectors in vivo: vector spread correlates with induction of immune responses and persistence of genomic RNA. J. Virol. 2007;81:2078–2082. doi: 10.1128/JVI.02525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turner D.L., Cauley L.S., Khanna K.M., Lefrançois L. Persistent antigen presentation after acute vesicular stomatitis virus infection. J. Virol. 2007;81:2039–2046. doi: 10.1128/JVI.02167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson N., McKimmie C.S., Mansfield K.L., Wakeley P.R., Brookes S.M., Fazakerley J.K., Fooks A.R. Lyssavirus infection activates interferon gene expression in the brain. J. Gen. Virol. 2006;87:2663–2667. doi: 10.1099/vir.0.82024-0. [DOI] [PubMed] [Google Scholar]
- 47.Veldhuis W.B., Floris S., van der Meide P.H., Vos I.M., de Vries H.E., Dijkstra C.D., Bär P.R., Nicolay K. Interferon-beta prevents cytokine-induced neutrophil infiltration and attenuates blood-brain barrier disruption. J. Cereb. Blood Flow Metab. 2003;23:1060–1069. doi: 10.1097/01.WCB.0000080701.47016.24. [DOI] [PubMed] [Google Scholar]
- 48.Steel C.D., Kim W.K., Sanford L.D., Wellman L.L., Burnett S., Van Rooijen N., Ciavarra R.P. Distinct macrophage subpopulations regulate viral encephalitis but not viral clearance in the CNS. J. Neuroimmunol. 2010;226:81–92. doi: 10.1016/j.jneuroim.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takeuchi H., Wang J., Kawanokuchi J., Mitsuma N., Mizuno T., Suzumura A. Interferon-gamma induces microglial-activation-induced cell death: a hypothetical mechanism of relapse and remission in multiple sclerosis. Neurobiol. Dis. 2006;22:33–39. doi: 10.1016/j.nbd.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 50.Komatsu T., Bi Z., Reiss C.S. Interferon-gamma induced type I nitric oxide synthase activity inhibits viral replication in neurons. J. Neuroimmunol. 1996;68:101–108. doi: 10.1016/0165-5728(96)00083-5. [DOI] [PubMed] [Google Scholar]
- 51.Reiss C.S., Komatsu T., Barna M., Bi Z. Interleukin-12 promotes enhanced recovery from viral infection of neurons in the central nervous system. Ann. N Y Acad. Sci. 1996;795:257–265. doi: 10.1111/j.1749-6632.1996.tb52675.x. [DOI] [PubMed] [Google Scholar]
- 52.Komatsu T., Reiss C.S. IFN-gamma is not required in the IL-12 response to vesicular stomatitis virus infection of the olfactory bulb. J. Immunol. 1997;159:3444–3452. [PubMed] [Google Scholar]
- 53.Martínez-Murillo R., Martínez A. Standardization of an orthotopic mouse brain tumor model following transplantation of CT-2A astrocytoma cells. Histol. Histopathol. 2007;22:1309–1326. doi: 10.14670/HH-22.1309. [DOI] [PubMed] [Google Scholar]
- 54.Seyfried T.N., el-Abbadi M., Roy M.L. Ganglioside distribution in murine neural tumors. Mol. Chem. Neuropathol. 1992;17:147–167. doi: 10.1007/BF03159989. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.