

Abstract
Background
The authors characterized the γ-aminobutyric acid type A (GABAA) receptor pharmacology of the novel etomidate analogue naphthalene-etomidate, a potential lead compound for the development of anesthetic-selective competitive antagonists.
Methods
The positive modulatory potencies and efficacies of etomidate and naphthalene-etomidate were defined in oocyte-expressed α1β3γ2L GABAA receptors using voltage clamp electrophysiology. Using the same technique, the ability of naphthalene-etomidate to reduce currents evoked by GABA alone or GABA potentiated by etomidate, propofol, pentobarbital, and diazepam was quantified. The binding affinity of naphthalene-etomidate to the transmembrane anesthetic binding sites of the GABAA receptor was determined from its ability to inhibit receptor photoaffinity labeling by the site-selective photolabels 3[H]azi-etomidate and R-3[H]mTFD-MPAB.
Results
In contrast to etomidate, naphthalene-etomidate only weakly potentiated GABA-evoked currents and induced little direct activation even at a near-saturating aqueous concentration. It inhibited labeling of GABAA receptors by 3[H]azi-etomidate and R-3[H]mTFD-MPAB with similar half-maximal inhibitory concentrations of 48 μM (95% confidence interval, 28 to 81 μM) and 33 μM (95% confidence interval, 20 to 54 μM). It also reduced the positive modulatory actions of anesthetics (propofol > etomidate ~ pentobarbital), but not those of GABA or diazepam. At 300 μM, naphthalene-etomidate increased the half-maximal potentiating propofol concentration from 6.0 μM (95% confidence interval, 4.4 to 8.0 μM) to 36 μM (95% confidence interval, 17 to 78 μM) without affecting the maximal response obtained at high propofol concentrations.
Conclusion
Naphthalene-etomidate is a very low efficacy etomidate analog that exhibits the pharmacology of an anesthetic competitive antagonist at the GABAA receptor.
Introduction
Anesthesiologists often use competitive antagonists to pharmacologically reverse the actions of therapeutic drugs including opioids, muscle relaxants, benzodiazepines, and anticoagulants.1–6 Commonly, competitive antagonists are structurally similar to the therapeutic drugs they reverse (i.e. they are analogues) and bind to the same protein binding site(s). However, they possess very little or no intrinsic efficacy for altering the function of the target protein. They are highly valuable in clinical medicine because they allow the magnitude and duration of drug effects to be precisely controlled. They are also useful in scientific research as pharmacological tools to define the role that specific protein targets play in producing particular in vitro or in vivo drug effects.7–10 Unfortunately, competitive antagonists for general anesthetic agents have not yet been developed and the long-held (but now widely discredited) view that general anesthetics act non-specifically via lipid membranes implied that such antagonists could never be created.11–17
It is now generally accepted that, similar to most other therapeutic drugs, anesthetics act quite specifically by binding to discrete sites on proteins.18–20 In particular, the γ-aminobutyric acid type A (GABAA) receptor is now known to be the principle target for the hypnotic actions of propofol and etomidate, and an important target for anesthetic barbiturates.21–25 These drugs are highly efficacious positive allosteric modulators (PAMs) of GABAA receptor function, potentiating GABAA receptor-mediated currents that are evoked by low GABA concentrations and directly activating GABAA receptors in the absence of GABA.26–30
Recently, two classes of general anesthetic binding sites have been identified by photoaffinity labeling studies within the transmembrane domain of the GABAA receptor31,32. One class of sites is photolabeled by 3[H]azi-etomidate and located at the two β+−α− subunit interfaces whereas the other is photolabeled by R-3[H]mTFD-MPAB and located at the α+−β− and γ+−β− interfaces. Protection studies using these two photolabels have been used to define the selectivities of various anesthetics for these sites. They show that etomidate binds with >100-fold higher affinity to the β+–α− sites as compared to the α+–β−/γ+–β− sites whereas pentobarbital exhibits the reverse selectivity, and propofol exhibits essentially no selectivity at all.32 Photoaffinity labeling has also established that these transmembrane binding sites for general anesthetics (PAMs) can also bind negative allosteric modulators that are GABAA receptor inhibitors.33 These transmembrane anesthetic sites are distinct from those that bind GABA and benzodiazepines as the latter – while also located between subunits – are found within the extracellular domain.34,35 This highly specific receptor mechanism suggests the possibility of developing anesthetic analogues that bind selectively to the transmembrane anesthetic binding sites but possess little or no intrinsic efficacy for positively modulating GABAA receptor function. We hypothesized that such analogues would act as competitive anesthetic antagonists capable of selectively reversing the GABAA receptor actions of more efficacious anesthetic agents. In this manuscript, we describe the GABAA receptor pharmacology of naphthalene-etomidate, a novel etomidate analogue with very low intrinsic efficacy that selectively antagonizes anesthetic action.
Materials and Methods
Anesthetics and Anesthetic Photoaffinity Labels
Figure 1 shows the molecular structures of etomidate and naphthalene-etomidate. Etomidate was purchased from Bachem Americas (Torrance, CA). Propofol, pentobarbital, and diazepam were purchased from Sigma-Aldrich (St. Louis, MO). Azi-etomidate and R-TFD-MPAB were synthesized as previously described.36,37 Naphthalene-etomidate was synthesized by Aberjona Laboratories (Woburn, MA).
Figure 1.
Molecular structures of etomidate and naphthalene-etomidate.
GABAA Receptor Electrophysiology
Oocytes were harvested from Xenopus frogs with the approval of and in accordance with rules and regulations of our Institutional Animal Care and Use Committee, injected with messenger RNA encoding the α1, β3, and γ2L subunits of the human GABAA receptor, and the resulting expressed GABAA receptors studied using the whole cell two-electrode voltage-clamp technique as previously described.38 For all studies of GABA potentiation, a GABA concentration-peak current response curve was generated for each oocyte to define the GABA concentration that elicits either 5% or 50% of the current evoked by 1 mM GABA (i.e. EC5 GABA or EC50 GABA, respectively). Between electrophysiological recordings, oocytes where perfused with buffer for at least 3 min (washout period) to remove GABA and/or drugs and to allow receptors to recover from desensitization.
Electrophysiological Protocols to Study Modulation of GABAA Receptors by Etomidate and Naphthalene-etomidate
1) Potentiation of GABA-evoked Currents
The oocyte was first perfused with 1 mM GABA and the maximal peak current response was recorded. After a washout period, the oocyte was perfused with EC5 GABA alone for 15 – 20 s followed immediately by EC5 GABA plus drug (etomidate or naphthalene-etomidate) at the desired concentration for 20 – 60 s and the peak current response was recorded. After another washout period, the oocyte was again perfused with 1 mM GABA and the maximal peak current response was recorded. The current response recorded in the presence of EC5 GABA plus drug was then normalized to the average of the two current responses evoked by 1 mM GABA.
2) Direct Activation of GABAA Receptors
The oocyte was first perfused with 1 mM GABA and the maximal peak current response was recorded. After a washout period, the oocyte was perfused with the desired concentration of drug (etomidate or naphthalene-etomidate) for 15 – 20 s and the peak current response was recorded. After another washout period, the oocyte was again perfused with 1 mM GABA and the maximal peak current response was recorded. The peak current response in the presence of drug was then normalized to the average of the two current responses produced by 1 mM GABA.
Electrophysiology Protocols to Study Interactions Between Naphthalene-etomidate, GABA, and PAMs
To evaluate the ability of naphthalene-etomidate to modify current responses evoked by EC50 GABA or a combination of EC5 GABA plus a PAM (i.e. etomidate, propofol, pentobarbital, or diazepam), we utilized three drug administration protocols.
1) Simultaneous Exposure Protocol
The oocyte was perfused with either (1) EC50 GABA or (2) EC5 GABA plus the desired PAM for 10 s and the control peak current response was recorded. After a washout period, the oocyte was again perfused with EC50 GABA or EC5 GABA plus the desired PAM but this time along with 300 μM naphthalene-etomidate and the test peak current response was recorded. After another washout period, the control peak response obtained without naphthalene-etomidate was again recorded. The percent current amplitude change produced by naphthalene-etomidate was then defined from the difference between the peak current response recorded during the test experiment and the average of the two control peak current responses.
2) Naphthalene-etomidate Pre-exposure Protocol
The oocyte was perfused with either (1) EC50 GABA or (2) EC5 GABA plus the desired PAM for 10 s and the control peak current response was recorded. After a washout period, the oocyte was pre-exposed to 300 μM naphthalene-etomidate for 10 s before co-application with either EC50 GABA or EC5 GABA plus the desired PAM for 10 s and the test peak current response was recorded. After another washout period, the control peak response obtained without naphthalene-etomidate was again recorded. The percent current amplitude change produced by naphthalene-etomidate was then defined from the difference between the peak current response recorded during the test experiment and the average of the two control peak current responses.
3) GABA Pre-exposure Protocol
The oocyte was perfused (i.e. activated) with either (1) EC50 GABA or (2) EC5 GABA plus the desired PAM for 30 s. Ten seconds into this activation period, 300 μM naphthalene-etomidate was added for 10 s. The effect of naphthalene-etomidate on currents was quantified as the maximum change in current amplitude recorded during naphthalene-etomidate administration. To correct for receptor desensitization (and current run up or run down) during naphthalene-etomidate administration, an interpolated straight line was fit between the pre- and post- naphthalene-etomidate phases of the current recording period. That line was then used as the baseline against which the effect of naphthalene-etomidate was quantified. The percent current amplitude change produced by naphthalene-etomidate was then defined from the maximal current difference between that interpolated line amplitude and the recorded current amplitude at the same time point.
Photoaffinity Label Competition Experiments
α1β3γ2L GABAA receptors containing a FLAG epitope on the N terminus of the α1 subunit were heterologously expressed in a tetracycline-inducible, stably transfected HEK 293S cell line and affinity purified on an anti-FLAG resin as previously described.32,39 Purified receptors were then photolabeled (for 30 min) with either 3[H]azi-etomidate (~2.6 μM; ~2.5 μCi per analytic sample) or R-3[H]mTFD-MPAB (~1.4 μM; ~2.7 μCi per analytic sample) using a 365 nm lamp in the presence of ranging concentrations of naphthalene-etomidate. Photolabel incorporation into each receptor subunit was then measured by running solubilized receptor membranes on a gel, cutting out the Coomasie blue-stained bands corresponding to each subunit, and measuring the radioactivity in the bands as previously described.32,40
Data Analysis
Concentration-response curves for potentiation of EC5 GABA-evoked currents and direct activation of GABAA receptor currents were fit using Prism 6.0h software (GraphPad, La Jolla, CA) using its built-in four-parameter equation for stimulation (equation 1):
where minimum is the normalized peak current amplitude in the absence of drug, maximum is the normalized peak current amplitude at high drug concentrations, [drug] is the drug concentration, EC50 is the drug concentration that evokes a peak current amplitude that is half way between the maximum and minimum values, and n is the slope of the relationship. The minimum value was constrained to the experimentally determined value in the absence of drug.
Naphthalene-etomidate concentration-response curves for inhibition of 10 μM propofol potentiated currents and for inhibition of photoaffinity labeling were fit using Prism 6.0h software (GraphPad, La Jolla, CA) using its built-in four-parameter equation for inhibition (equation 2):
where minimum is the normalized peak current amplitude (or normalized specific Counts Per Minute for photoaffinity labeling studies) in the presence of high naphthalene-etomidate concentrations, maximum is the normalized peak current amplitude (or normalized specific Counts Per Minute for photoaffinity labeling studies) in the absence of naphthalene-etomidate, [NE] is the naphthalene-etomidate concentration, IC50 is the naphthalene-etomidate concentration that produces a current amplitude (or normalized specific Counts Per Minute for photoaffinity labeling studies) that is half way between the maximum and minimum values, and n is the slope of the relationship. The maximum value was constrained to the experimentally determined value in the absence of naphthalene-etomidate.
Allosteric Modeling of Direct Activation Data
Allosteric receptor modeling was used to derive the microscopic dissociation constants for etomidate and naphthalene-etomidate to the open and closed conformational states of the GABAA receptor. Etomidate and naphthalene-etomidate direct activation data were transformed to Popen values by assuming that Popen in the presence of a maximally activating GABA concentration (1 mM) is approximately 0.85.41,42 The relationship between Popen and the drug concentration was then fit to the allosteric equation (equation 3):41
where Popen is the fraction of receptors that are open in the presence of etomidate or naphthalene-etomidate, [drug] is the concentration of either etomidate or naphthalene-etomidate, Kdclosed and Kdopen respectively are the microscopic dissociation constants of the drug in the closed and open states, and n is the number of drug binding sites. L0, the closed:open receptor ratio in the absence of drug, was fixed at a median literature value of 40,000.41,43–46
Statistical Analysis
At each drug concentration, individual electrophysiological data points were obtained using different oocytes. Errors bars on mean electrophysiological data are reported as ± SD whereas those on mean photoincorporation data are reported as the range of two experiments obtained using two different receptor preparations. Sample sizes (4 – 6 points per drug concentration for electrophysiological experiments) were defined based on our previous experience.29,30,47,48 A one-sample t test (two tailed) was used to statistically assess whether 300 μM naphthalene-etomidate significantly changed peak currents evoked by EC50 GABA or EC5 GABA potentiated by each of the PAMs. The statistical comparisons between the EC50s for propofol potentiation of EC5 GABA-evoked currents in the presence versus absence of 300 μM naphthalene-etomidate, and those between naphthalene-etomidate potency for inhibiting photolabeling by [3H]azi- etomidate versus R-[3H]mTFD-MPAB were made using the extra sum-of-squares F test. The uncertainties in fitted parameters are reported as confidence intervals (CI). There was no lost or missing data. To avoid output saturation, oocytes producing 1 mM GABA-evoked peak currents greater than 5 μA were discarded. All fitting and statistical tests were performed with GraphPad Prism 6.0h (La Jolla, CA). Statistical significance was assumed for p < 0.05.
Results
Potentiation of EC5 GABA-evoked Currents by Etomidate and Naphthalene-etomidate
We characterized the effects of etomidate and naphthalene-etomidate over a wide range of concentrations on currents evoked by EC5 GABA and mediated by α1β3γ2L GABAA receptors expressed in Xenopus oocytes using the two-electrode voltage clamp technique. Figure 2A (top) shows representative electrophysiological traces obtained using a single oocyte and demonstrates that etomidate potentiated EC5 GABA-evoked currents in a concentration-dependent manner. At the highest etomidate concentration shown in that figure (100 μM), the current was potentiated by 14-fold, reaching a magnitude that was similar to that evoked by 1 mM GABA (GABA trace not shown). Figure 2A (bottom) shows representative electrophysiological traces obtained using a single oocyte and demonstrates that naphthalene-etomidate also potentiated EC5 GABA-evoked currents in a concentration-dependent manner. However, the magnitude of potentiation produced by naphthalene-etomidate was relatively small, ≤ 1/10th that produced by the same concentration of etomidate. Figure 2B plots the concentration-mean peak response relationship for potentiation of EC5 GABA-evoked currents by etomidate (n = 6 oocytes per concentration) and naphthalene-etomidate (n = 4 oocytes per concentration). It shows that etomidate increased peak currents evoked by EC5 GABA in a manner that was not only potent but also highly efficacious; in the presence of 300 μM etomidate, EC5-evoked currents that were 90 ± 7% of those evoked by 1 mM GABA. We fit the data to equation 1 with the minimum constrained to 5% (by definition for EC5 GABA-evoked currents). It yielded an EC50 for etomidate potentiation of 3.4 μM (95% CI, 2.5 to 4.5 μM), a maximum peak current amplitude at high etomidate concentrations that was 95% (95% CI, 89 to 102%) of that produced by 1 mM GABA, and a slope of 1.2 (95% CI, 0.8 to 1.5). Figure 2B also shows that naphthalene-etomidate was significantly less potent and efficacious than etomidate; in the presence of 300 μM naphthalene-etomidate (a near aqueous-saturating concentration), EC5-evoked currents were only 11 ± 2.7% of those evoked by 1 mM GABA. A fit of the naphthalene-etomidate data to equation 1 yielded an EC50 for naphthalene-etomidate potentiation of 38 μM (95% CI, 16 to 93 μM), a maximum peak current value at high concentrations of only 11% (95% CI, 8.7 to 13%), and a slope of 3 (95% CI, −5 to 11).
Figure 2.
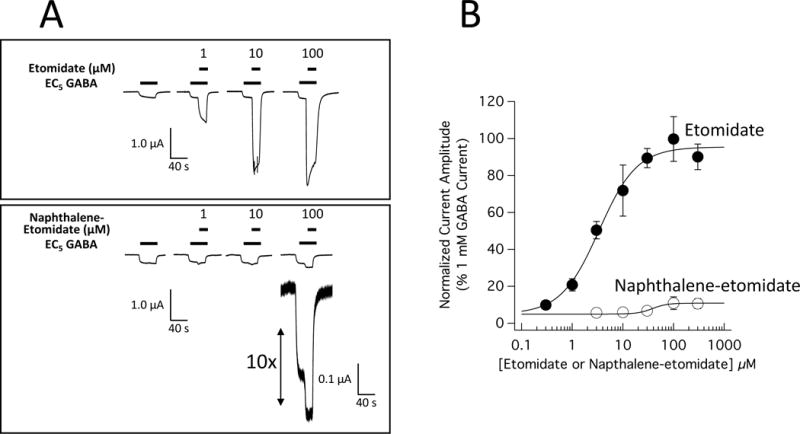
Potentiation of α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor currents by etomidate and naphthalene-etomidate. (A) Electrophysiological traces showing the potentiating effect of etomidate (top) or naphthalene-etomidate (bottom) on currents evoked by a GABA concentration that elicits 5% of the current evoked by 1 mM GABA (EC5 GABA). For each dataset, currents at all drug concentrations were obtained using the same oocyte. (B) Etomidate and naphthalene-etomidate concentration-response curves for potentiation of EC5 GABA-evoked currents. Each symbol is the mean ± SD derived from 6 (etomidate) or 4 (naphthalene-etomidate) different oocytes. The curves are fits of the datasets to equation 1. For etomidate, the fit yielded a half-maximal potentiating concentration of 3.4 μM (95% CI, 2.5 to 4.5 μM), a maximum peak current amplitude at high etomidate concentrations of 95% (95% CI, 89 to 102%) of that produced by 1 mM GABA, and a slope of 1.2 (95% CI, 0.8 to 1.5). For naphthalene-etomidate, the fit yielded a half-maximal potentiating concentration of 38 μM (95% CI, 16 to 93 μM), a maximum peak current value at high concentrations of 11% (95% CI, 8.7 to 13%), and a slope of 3 (95% CI, −5 to 11).
Direct Activation of GABAA Receptor Currents by Etomidate and Naphthalene-etomidate
We then compared the abilities of etomidate and naphthalene-etomidate to directly activate α1β3γ2L GABAA receptor currents in the absence of GABA. Using the same oocyte for etomidate and naphthalene-etomidate, figure 3A shows that currents directly activated by naphthalene-etomidate (bottom) are orders of magnitude smaller than those activated by etomidate (top). Figure 3B plots the concentration-mean peak response relationship for direct activation by etomidate (n = 4 oocytes/concentration) and naphthalene-etomidate (n = 4 oocytes/concentration). A fit of the etomidate data to equation 1 with the minimum constrained to 0% (i.e. no current in the absence of drug) yielded an EC50 for direct activation, maximum current amplitude at high concentrations, and slope of 65 μM (95% CI, 41 to 103 μM) and 54% (95% CI, 46 to 63%), and 1.2 (95% CI, 0.7 to 1.8) respectively. An analogous fit of the naphthalene-etomidate dataset failed to converge.
Figure 3.
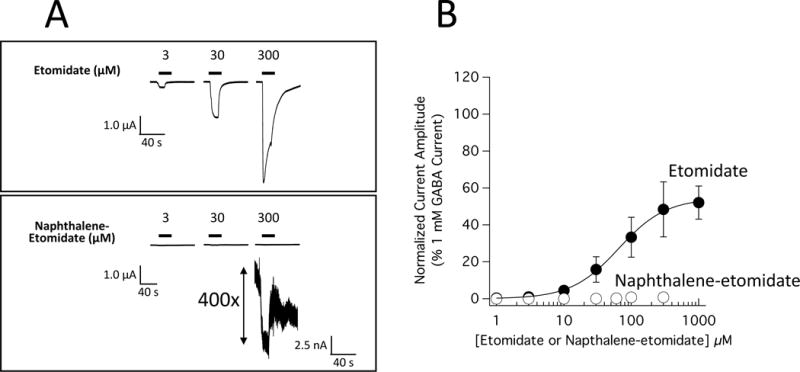
Direct activation of α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor currents by etomidate and naphthalene-etomidate. (A) Electrophysiological traces showing the direct activation by etomidate (top) or naphthalene-etomidate (bottom). To allow a direct comparison between drugs, a single oocyte was used to obtain both datasets. (B) Etomidate and naphthalene-etomidate concentration-response curves for direct activation. Each symbol is the mean ± SD derived from 4 different oocytes. The curve is a fit of the etomidate dataset to equation 1 yielding a half-maximal direct activating concentration, maximum current amplitude at high etomidate concentrations, and slope of 65 μM (95% CI, 41 to 103 μM) and 54% (95% CI, 46 to 63%), and 1.2 (95% CI, 0.7 to 1.8) respectively. A fit of the naphthalene-etomidate dataset to equation 1 did not converge.
Naphthalene-etomidate Inhibits GABAA Receptor Photolabeling by 3[H]Azi-etomidate and R-3[H]mTFD-MPAB
To test whether naphthalene-etomidate bound to either (or both) classes of transmembrane anesthetic binding sites, we quantified its ability to inhibit photoaffinity labeling of α1β3γ2L GABAA receptors by 3[H]azi-etomidate and the barbiturate R-3[H]mTFD-MPAB. Figure 4 shows that naphthalene-etomidate reduced photoincorporation of both 3[H]azi-etomidate and R-3[H]mTFD-MPAB in a concentration-dependent manner. At the highest concentrations studied, naphthalene-etomidate inhibited specific (i.e. etomidate-displaceable) 3[H]azi-etomidate photoincorporation and specific (i.e. R-mTFD-MPAB-displaceable) R-3[H]mTFD-MPAB photoincorporation by ≥90%. Naphthalene-etomidate IC50s for inhibiting specific photoincorporation of the two photolabels were 48 μM (95% CI, 28 to 81 μM) for 3[H]azi-etomidate and 33 μM (95% CI, 20 to 54 μM) for R-3[H]mTFD-MPAB with respective slopes of −1.2 (95% CI, −2.0 to −0.6) and −2.0 (95% CI, −3.5 to −0.5). These IC50 values were not significantly different from one another, strongly suggesting that naphthalene-etomidate binds to both classes of transmembrane anesthetic binding sites on the GABAA receptor with similar affinities.
Figure 4.
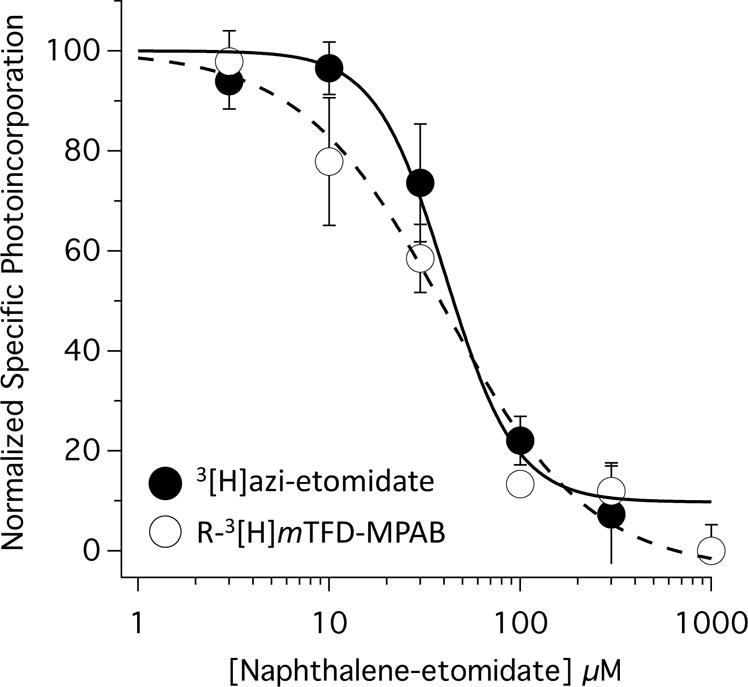
Naphthalene-etomidate concentration-response curves for inhibition of specific [3H]azi- etomidate and R-[3H]mTFD-MPAB photolabeling of α1β3γ2L GABAA receptors. For the two photolabels, the half-maximal inhibitory concentrations of naphthalene-etomidate were 48 μM (95% CI, 28 to 81 μM) and 33 μM (95% CI, 20 to 54 μM), respectively. The slopes were −2.0 (95% CI, −3.5 to −0.5) and −1.3 (95% CI, −2.0 to −0.6), respectively. Data was normalized to Counts Per Minute measured in the absence of naphthalene-etomidate. Non-specific photolabeling was defined in the presence of 300 μM etomidate (for [3H]azi- etomidate photolabeling experiments) or 100 μM R-mTFD-MPAB (for R-[3H]mTFD-MPAB photolabeling experiments). All photolabeling was done in the presence of 300 μM GABA.
Naphthalene-etomidate Antagonizes Anesthetic Potentiated GABAA Receptor Currents
The observation that naphthalene-etomidate inhibits photoaffinity labeling of the transmembrane anesthetic binding sites of the GABAA receptor (most likely because it binds to these sites) but has very low efficacy for positively modulating GABAA receptor function suggested to us that it might be capable of acting as a competitive antagonist of anesthetics that also bind to these sites. An analogous competitive mechanism at the classical benzodiazepine binding site of the GABAA receptor (located at the extracellular α+ – γ− subunit interface) accounts for the ability of flumazenil to reverse the GABAA receptor actions of benzodiazepines.35 To test this possibility, we assessed whether naphthalene-etomidate would reduce the agonist potentiating effects of etomidate, propofol, and pentobarbital. As control experiments, we also assessed the actions of naphthalene-etomidate on receptors similarly potentiated by diazepam or activated with an EC50 GABA concentration.
In one set of studies, we simultaneously applied naphthalene-etomidate (300 μM) along with either (1) EC50 GABA alone or (2) EC5 GABA plus a PAM (etomidate, propofol, pentobarbital, or diazepam). Concentrations of etomidate (3 μM), propofol (10 μM), and pentobarbital (120 μM) were chosen based on pilot experiments indicating that when combined with EC5 GABA, they activate the same fraction of GABAA receptors as EC50 GABA alone. We used a diazepam concentration of 1 μM to maximally – but selectively – potentiate GABAA receptors via the classical extracellular benzodiazepine binding site.49 Representative current traces recorded during these experiments are shown in figure 5. Figure 5A shows electrophysiological traces obtained upon perfusing an oocyte with either EC50 GABA alone or EC50 GABA plus 300 μM naphthalene-etomidate. It demonstrates that 300 μM naphthalene-etomidate minimally affected currents evoked by EC50 GABA. In contrast, figures 5B, 5C, 5D, and 5E respectively show that 300 μM naphthalene-etomidate reduced EC5 GABAA receptor currents that were potentiated by etomidate, propofol, or pentobarbital, but enhanced those potentiated by diazepam. The change in peak currents produced by naphthalene-etomidate upon activation with either EC50 GABA alone or EC5 GABA along with each of the four PAMs (n = 6 oocyte experiments per drug) is plotted in figure 5F with the mean values summarized in Table 1 under the simultaneous addition heading.
Figure 5.
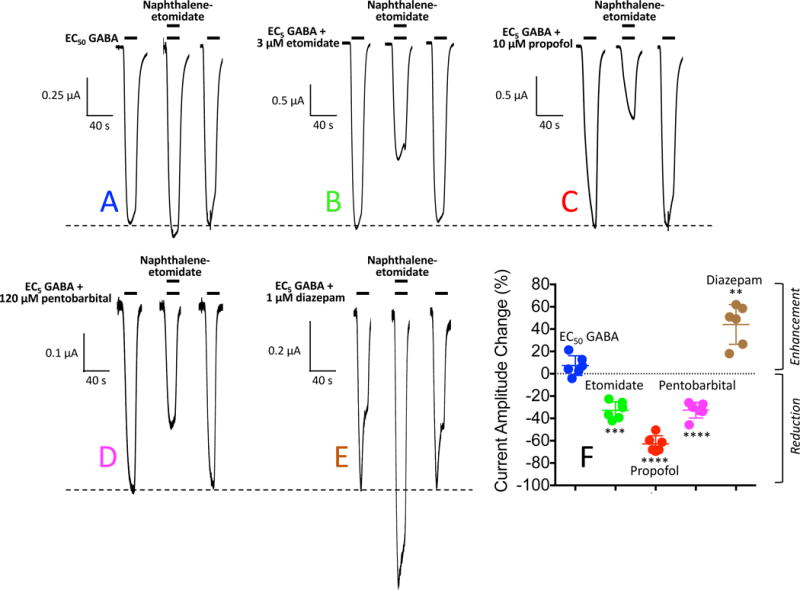
Naphthalene-etomidate modulation of α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor currents: Simultaneous Addition protocol. (A) Representative current traces obtained upon application of GABA at a concentration that evokes 50% of the current evoked by 1 mM GABA (EC50 GABA). The first and last traces were controls obtained in the absence of naphthalene-etomidate and the middle trace was obtained with simultaneous addition of 300 μM naphthalene-etomidate along with GABA. (B – E) Representative current traces obtained upon application of GABA at a concentration that evokes 5% of the current evoked by 1 mM GABA (EC5 GABA) along with the indicated positive allosteric modulator. In each panel, the first and last traces were controls obtained in the absence of naphthalene-etomidate and the middle trace was obtained with simultaneous addition of 300 μM naphthalene-etomidate along with GABA + modulator. In each panel, the dashed line shows the average control peak current produced in the absence of naphthalene-etomidate. (F) Percent change in peak current amplitude produced by 300 μM naphthalene-etomidate. Positive values indicate that naphthalene-etomidate enhanced peak currents whereas negative values indicate that it reduced peak currents. Each symbol represents data from a single oocyte experiment (n = 6 oocte experiments per drug). Mean ± SD are indicated for each dataset. Statistically significant change in current amplitude produced by naphthalene-etomidate: ** p<0.01; *** p<0.001 ; **** p<0.0001.
Table 1.
Percent Change in γ-Aminobutyric Acid Type A (GABAA) Receptor Peak Current Amplitude Produced by 300 μM Napthalene-etomidate
| Protocol | GABA EC50 | GABA EC5 + Etomidate |
GABA EC5 + Propofol |
GABA EC5 + Pentobarbital |
GABA EC5 + Diazepam |
|---|---|---|---|---|---|
| Simultaneous Addition | 7.3 ± 8.8 | −33 ± 7.8 | −63 ± 7.3 | −33 ± 7.1 | 44 ± 18 |
| Naphthalene-etomidate Pre-exposure | −8.8 ± 8.6 | −47 ± 4.1 | −68 ±6.0 | −45 ± 7.9 | 25 ± 14 |
| GABA Pre-exposure | 21 ± 9 | −24 ± 1.8 | −49 ± 4.69 | −21 ± 5.4 | 33 ± 6.9 |
All values are mean ± SD (n = 6 oocytes)
In a second set of studies, we pre-applied 300 μM naphthalene-etomidate for 10 s before activating with either EC50 GABA alone or EC5 GABA plus a PAM (figure 6). The results were similar to those described in the previous paragraph using the simultaneous addition protocol with naphthalene-etomidate having no significant effect on peak currents activated by EC50 GABA while significantly reducing EC5 GABA-evoked currents potentiated by etomidate, propofol, or pentobarbital, and enhancing EC5 GABA-evoked currents potentiated by diazepam. The mean values for these studies are summarized in Table 1 under the naphthalene-etomidate pre-exposure heading.
Figure 6.
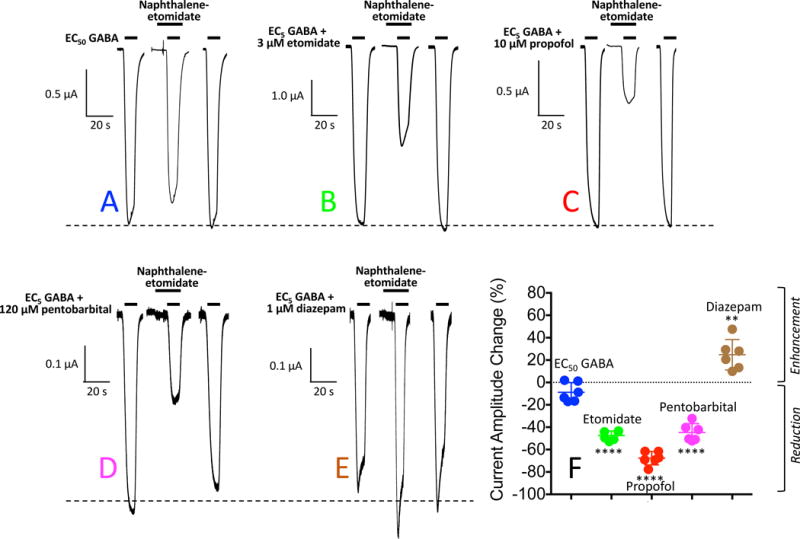
Naphthalene-etomidate modulation of α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor currents: Naphthalene-etomidate Pre-exposure protocol. (A) Representative current traces obtained upon application of GABA at a concentration that evokes 50% of the current evoked by 1 mM GABA (EC50 GABA). The first and last traces were controls obtained in the absence of naphthalene-etomidate and the middle trace was obtained with a 10 s pre-exposure of 300 μM naphthalene-etomidate along with GABA. (B – E) Representative current traces obtained upon application of GABA at a concentration that evokes 5% of the current evoked by 1 mM GABA (EC5 GABA) along with the indicated positive allosteric modulator. In each panel, the first and last traces were controls obtained in the absence of naphthalene-etomidate and the middle trace was obtained with a 10 s pre-exposure of 300 μM naphthalene-etomidate. In each panel, the dashed line shows the average control peak current produced in the absence of naphthalene-etomidate. (F) Percent change in peak current amplitude produced by 300 μM naphthalene-etomidate. Positive values indicate that naphthalene-etomidate enhanced peak currents whereas negative values indicate that it reduced peak currents. Each symbol represents data from a single oocyte experiment (n = 6 oocte experiments per drug). Mean ± SD are indicated for each dataset. Statistically significant change in current amplitude produced by naphthalene-etomidate: ** p<0.01; **** p<0.0001.
In a third set of studies, we first activated GABAA receptors using either EC50 GABA alone or EC5 GABA plus a PAM before adding 300 μM naphthalene-etomidate (figure 7). With this protocol, we observed significant potentiation during naphthalene-etomidate administration when receptors where activated by either EC50 GABA alone or EC5 GABA plus diazepam. However, naphthalene-etomidate again significantly reduced EC5 GABA-evoked currents potentiated by etomidate, propofol, or pentobarbital. The mean values for these studies are summarized in Table 1 under the GABA pre-exposure heading.
Figure 7.
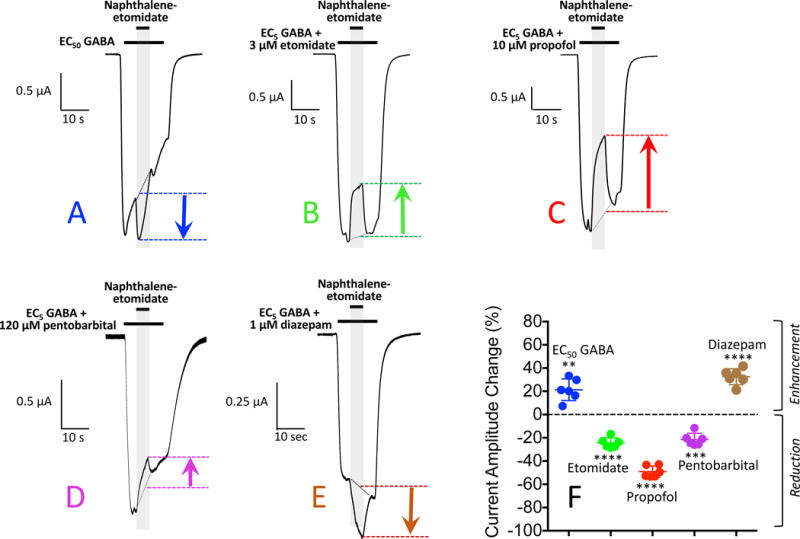
Naphthalene-etomidate modulation of α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor currents: GABA Pre-exposure protocol. (A) Representative current trace obtained upon receptor activation for 30 s with GABA at a concentration that evokes 50% of the current evoked by 1 mM GABA (EC50 GABA). Ten seconds into this activation period, 300 μM naphthalene-etomidate was added for 10 s. (B – E) Representative current trace obtained upon receptor activation for 30 s with GABA at a concentration that evokes 5% of the current evoked by 1 mM GABA (EC5 GABA) along with the indicated positive allosteric modulator. Ten seconds into this activation period, 300 μM naphthalene-etomidate was added for 10 s. (F) Percent change in peak current amplitude produced by 300 μM naphthalene-etomidate. Positive values indicate that naphthalene-etomidate enhanced peak currents whereas negative values indicate that it reduced peak currents. Each symbol represents data from a single oocyte experiment (n = 6 oocte experiments per drug). Mean ± SD are indicated for each dataset. Statistically significant change in current amplitude produced by naphthalene-etomidate: ** p<0.01; *** p<0.001; **** p<0.0001.
Naphthalene-etomidate Rightward Shifts the Propofol Concentration-Response Curve for EC5 GABA Potentiation
We then assessed the impact of naphthalene-etomidate on the anesthetic concentration-response curve for EC5 potentiation using the simultaneous addition protocol. We chose to study propofol as a representative anesthetic not only because it is the most widely used anesthetic, but also because our data showed that that naphthalene-etomidate produced an approximately 2-fold greater reduction in currents potentiated by propofol as compared to currents potentiated by etomidate or pentobarbital (Table 1). Thus, we expected that any change in the anesthetic concentration-response curve produced by naphthalene-etomidate would be greater if we used propofol to potentiate currents rather than etomidate or pentobarbital. Figure 8A shows the propofol concentration-response relationship for EC5 potentiation in the absence and presence of 300 μM naphthalene-etomidate. The curves are fits of the two datasets to equation 1. In the absence and presence of naphthalene-etomidate, the respective minima were constrained to 5% (by definition for EC5 GABA evoked currents) and 11% (the peak current amplitude produced by 300 μM naphthalene-etomidate in the absence of propofol). The EC50 for propofol potentiation of EC5 GABA-evoked currents was 6.0 μM (95% CI, 4.4 to 8.0 μM) in the absence of naphthalene-etomidate and increased 6-fold to 36 μM (95% CI, 17 to 78 μM) in its presence. The maximal peak current response at high propofol concentrations was virtually unchanged by 300 μM naphthalene-etomidate with values of 88% (95% CI, 81 to 96%) in the absence of naphthalene-etomidate and 87% (95% CI, 66 to 107%) in its presence.
Figure 8.
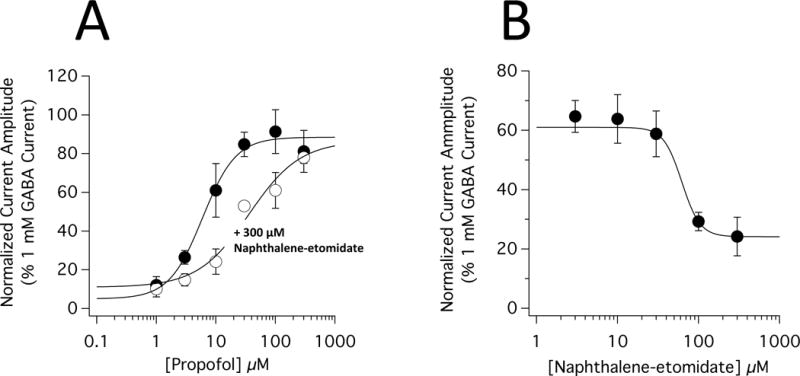
Inhibition of propofol-mediated potentiation of α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor currents by naphthalene-etomidate. (A) Propofol concentration-response curves for potentiation of GABA-evoked currents in the absence and presence of 300 μM naphthalene-etomidate. The curves are fits of the datasets to equation 1. The propofol concentration that half-maximally potentiated GABA-evoked currents (EC50) was 6.0 μM (95% CI, 4.4 to 8.0 μM) in the absence of naphthalene-etomidate and 36 μM (95% CI, 17 to 78 μM) in the presence of 300 μM etomidate. The respective slopes were 1.5 (95% CI, 0.97 to 2.1) and 1.0 (95% CI, 0.54 to 1.5 μM). In the absence and presence of 300 μM naphthalene-etomidate, the maximal responses at high propofol concentrations were essentially identical with values of 88% (95% CI, 81 to 96%) and 87% (95% CI, 66 to 107%), respectively. (B) Naphthalene-etomidate concentration-response curves for inhibition of GABA-evoked currents potentiated by 10 μM propofol. The curves are fits of the datasets to equation 2. The naphthalene-etomidate concentration that half-maximally inhibited potentiated currents (IC50) was 62 μM (95% CI, 38 to 103 μM) with a minimum value of 24% (95% CI, 17 to 31%) and a slope of −3.9 (95% CI, −9.7 to −0.4). In both panels, each data point is the mean ± SD of 4 oocyte experiments.
Naphthalene-etomidate Antagonizes EC5 GABA Potentiation by Propofol in a Concentration-Dependent Manner
Again using the simultaneous addition protocol, we defined the naphthalene-etomidate concentration-dependence for inhibiting the peak amplitude of EC5 GABA-evoked currents potentiated by 10 μM propofol. We found that the peak amplitude of propofol-potentiated currents decreased steeply with naphthalene-etomidate concentration (figure 8B). A fit of this relationship to equation 2 with the maximum constrained to 61% (the peak current amplitude produced by 10 μM propofol in the absence of naphthalene-etomidate) yielded an IC50 of 62 μM (95% CI, 38 to 103 μM), a minimum value of 24% (95% CI, 17 to 31%), and a slope of −3.9 (95% CI, −9.7 to −0.4).
Discussion
This report describes a novel etomidate analog that exhibits the pharmacology of an anesthetic-selective competitive antagonist. Specifically, our studies show that naphthalene-etomidate (1) inhibits photoaffinity labeling of the two classes of GABAA receptor transmembrane anesthetic binding sites with similar affinities, but possesses low intrinsic efficacy for positively modulating GABAA receptor function; (2) reduces the positive modulatory actions of drugs that bind to these receptor sites (propofol > etomidate ~ pentobarbital) but not those of drugs that bind elsewhere on the receptor (GABA and diazepam); and (3) shifts the anesthetic (propofol) concentration-response curve for potentiation rightward without affecting the maximal response obtained at high anesthetic concentrations.
Within the context of Monod-Wyman-Changeux allosteric models of receptor function, the intrinsic efficacy of a ligand is defined by its relative affinity for the open versus closed receptor states (figure 9A).43,50,51 Ligands with high efficacies bind with much higher affinity to the open state than to the closed state. Such state-selective binding shifts the preexisting receptor equilibrium between closed and open states towards the open state. For propofol and etomidate, the relative binding affinities for the open versus closed states of the GABAA receptor have been estimated to be on the order of 100:1, quantitatively accounting for both their agonist potentiating and direct activating actions.41,44 In contrast, ligands with very low intrinsic efficacies (i.e. competitive antagonists) bind with similar affinities to both receptor states.52 Consequently, they minimally perturb the closed:open state equilibrium and have little functional effect on their own. However, they can competitively inhibit the binding – and thus the actions – of more efficacious ligands.
Figure 9.
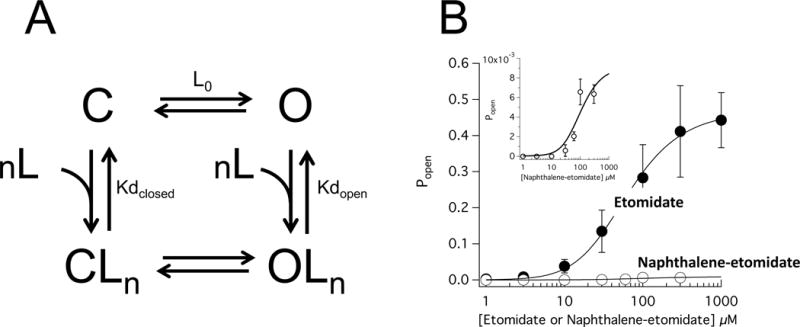
Allosteric analysis of α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor direct activation by etomidate and naphthalene-etomidate. (A) Allosteric model for receptor activation. C and O are the closed and open states, respectively. CLn and OLn are the liganded closed and open states, respectively, and n is the number of ligand (etomidate or naphthalene-etomidate) binding sites. Lo is the open state:closed state ratio in the absence of any modulatory ligands. Kdclosed and Kdopen are the ligand microscopic dissociation constants in the closed and open state, respectively. (B) GABAA receptor open state probability (Popen) as a function of etomidate or naphthalene-etomidate concentration. The inset shows the naphthalene-etomidate data on an expanded verticle axis. The curves are fits of the datasets to equation 3 yielding respective Kdclosed and Kdopen values of 0.23 μM (95% CI, 0.15 to 0.31 μM) and 44 μM (95% CI, 26 to 62 μM) for etomidate and 6.2 μM and 27 μM for naphthalene-etomidate. For etomidate and naphthalene-etomidate, the number of binding sites (n) was assumed to be 2 and 4, respectively. L0 was constrained at 40,000 for both fits.
The affinities of a ligand for the open and closed states can be quantified from the relationship between the ligand concentration and the fraction of receptors that it opens (Popen). For the allosteric model shown in figure 9A, this relationship is defined by equation 3. Figure 9B plots that relationship using the direct activation data for etomidate and naphthalene-etomidate where Popen was determined at each drug concentration from the peak amplitude of the directly activated current normalized to that evoked by a maximally activating GABA concentration (i.e. 1 mM) and assuming a maximum Popen value for GABA of 0.85 in this receptor subtype.42 The curved lines in this figure are fits of this relationship to equation 3 with the number of anesthetic binding sites n constrained to 2 for etomidate (at the two β+–α− subunit interfacial sites) and 4 for naphthalene-etomidate (because our photoaffinity labeling studies suggest that it binds to the two β+–α− subunit interfacial sites and to the α+−β− and γ+−β− interfacial sites with similar affinities).31,45,51 Based on this analysis, we determined that etomidate binds to the open state with an affinity that is 190-fold higher than to the closed state with microscopic dissociation constants of 0.23 μM (95% CI, 0.15 to 0.31 μM) and 44 μM (95% CI, 26 to 62 μM), respectively. In contrast, naphthalene-etomidate binds to the open state with an affinity that is only 4.4-fold higher than to the closed state with microscopic dissociation constants of 6.2 μM and 27 μM, respectively. Comparison of the dissociation constants for the two drugs reveals that the much lower intrinsic efficacy of naphthalene-etomidate can be almost entirely attributed to a 27-fold lower affinity of for the open state of the receptor.
Our studies also showed that naphthalene-etomidate inhibited photoaffinity labeling by 3[H]azi-etomidate and R-3[H]mTFD-MPAB with similar IC50s. This suggests that in addition to markedly reducing binding selectivity for the open versus closed receptor, adding the phenyl substituent group also abolished the 100-fold binding selectivity that etomidate has to the receptor’s two β+−α− sites versus its α+−β−/γ+−β− sites. Thus, the selectivity of naphthalene-etomidate for these two classes of transmembrane anesthetic binding sites more closely resembles that of propofol (which has virtually no binding site selectivity) and other phenyl-substituted etomidate analogs than etomidate or pentobarbital (which respectively bind selectively to the receptor’s two β+−α− sites and to its α+−β−/γ+−β− sites).32
To examine the impact of naphthalene-etomidate on currents activated by EC50 GABA alone or by EC5 GABA potentiated by various PAMs, we utilized three protocols that added naphthalene-etomidate at different times relative to receptor activation (before, after, or during activation). In general, the effect was similar regardless of when naphthalene-etomidate was added (Figures 5F, 6F, 7F, and Table 1). In all three cases, naphthalene-etomidate significantly reduced EC5 GABA-evoked currents potentiated by etomidate, propofol, or pentobarbital but enhanced those potentiated by diazepam. Additionally, the inhibitory effects of naphthalene-etomidate on propofol-potentiated currents were approximately twice as large as those on etomidate-potentiated and pentobarbital-potentiated currents. The effects of naphthalene-etomidate on EC50 GABA-evoked currents were somewhat more protocol-dependent as it had little or no effect when added before or simultaneously with GABA application, but significantly potentiated currents when added after GABA application.
The hallmark of a competitive antagonist is that it produces a rightward shift in the concentration-response curve of the drug with which it competes (i.e. it increases the drug’s EC50) without reducing the maximal response produced by the drug at high concentrations. This may be contrasted with the effects of non-competitive antagonists that classically reduce the maximal response without shifting the concentration-response curve. Our data show that naphthalene-etomidate exhibits the pharmacology of an anesthetic competitive antagonist as it increased the EC50 for propofol potentiation of EC5 GABA-evoked currents by 6-fold without reducing the maximal response recorded at high propofol concentrations. Although not tested, we expect that naphthalene-etomidate would have similarly shifted the concentration-response curves for etomidate and pentobarbital as they also bind to the transmembrane anesthetic binding sites on the GABAA receptor. However, the magnitude of those shifts would almost certainly have been smaller because the inhibitory actions of naphthalene-etomidate on etomidate-potentiated and pentobarbital-potentiated currents are less than those on propofol-potentiated ones.
Based on our experimental observations, we propose the following conceptual model to explain key results of our studies (figure 10). In the absence of any other positive modulatory ligands, naphthalene-etomidate binds to the two classes of transmembrane anesthetic binding sites and very modestly (because it has low intrinsic efficacy) positively modulates receptors (figure 10A). In the presence of positive modulatory ligands that act at sites other than these anesthetic binding sites (e.g. GABA and diazepam), naphthalene-etomidate similarly binds and modestly positively modulates receptors (figure 10B and 10C). However, when a general anesthetic is bound to a transmembrane anesthetic binding site and positively modulating the GABAA receptor, the net effect of naphthalene-etomidate binding to that site is inhibitory because it displaces the higher efficacy anesthetic. In the case of propofol (which positively modulates by binding to both classes of anesthetic binding sites), naphthalene-etomidate has the greatest inhibitory effect on potentiated currents because it displaces anesthetic binding from both classes of anesthetic binding sites (figure 10D). Thus, the net effect of naphthalene-etomidate binding to each of the two classes of anesthetic binding sites is inhibitory. In the case of etomidate (which positively modulates by binding to the β+−α− anesthetic binding sites), naphthalene-etomidate has two opposing effects on receptor function (figure 10E). It has an inhibitory effect by displacing etomidate from the β+−α− anesthetic binding sites. However, it also has a modest positive modulatory effect by binding to the unoccupied α+−β−/γ+−β− anesthetic binding sites. The result of these opposing actions at the two classes of sites is that naphthalene-etomidate is only about half as effective at reducing etomidate-potentiated currents as propofol-potentiated currents. The case of pentobarbital (which positively modulates by binding to the α+−β−/γ+−β− anesthetic binding sites) is identical to that of etomidate except that actions at the two classes of anesthetic binding sites are exactly reversed (figure 10F). Although our model dose not explicitly include the possibility of other anesthetic binding sites besides those identified by 3[H]azi-etomidate and R-3[H]mTFD-MPAB photolabeling, it does not provide evidence against their existence as naphthalene-etomidate may bind to these sites as well.
Figure 10.
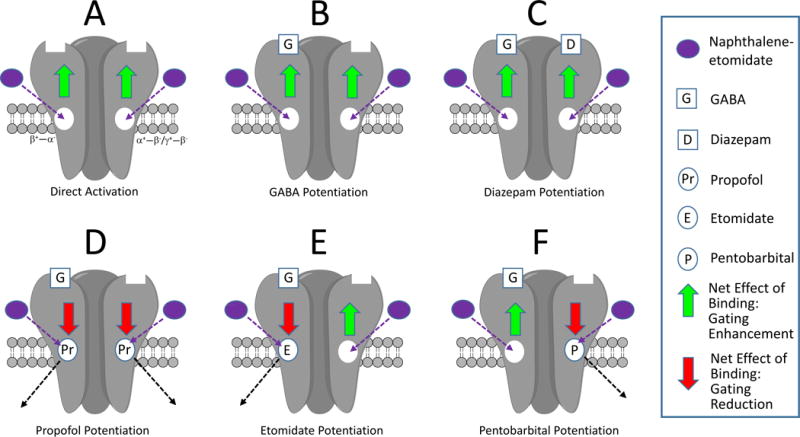
Conceptual model of the actions of naphthalene-etomidate on γ-aminobutyric acid type A (GABAA) receptor pharmacology. They key features are that naphthalene-etomidate (1) binds to both classes of transmembrane anesthetic binding sites; (2) has lower intrinsic positive modulatory efficacy than propofol, etomidate, and pentobarbital; and (3) competitively antagonizes the binding of these three anesthetics, but not GABA or diazepam because they bind elsewhere. Thus when the transmembrane anesthetic binding sites are unoccupied (A – C), naphthalene-etomidate weakly enhances channel gating (green arrows). However when such sites are occupied by an anesthetic possessing higher efficacy (D – F), the net effect of naphthalene-etomidate binding to that site (and competitively displacing the anesthetic) is to reduce gating efficacy. Inhibitory effect of naphthalene-etomidate on currents potentiated by propofol (D) is greater than currents potentiated by either etomidate (E) or pentobarbital (F) because the latter two anesthetics bind selectively to only one class of sites. This allows naphthalene-etomidate to bind to the other (unoccupied) class of sites where its effect is to enhance channel gating efficacy.
There are several potential clinical and experimental uses for anesthetic competitive antagonists. Currently, recovery from anesthesia must occur as a passive process whose time course is dictated by the rate of anesthetic drug clearance rather than the actual clinical need. The development of competitive antagonists for general anesthetics that act via the GABAA receptor could change this paradigm if they allow anesthesia to be reversed immediately and on demand. This direct competitive approach may be contrasted with ones that utilize stimulants that target other proteins and achieve emergence from anesthesia presumably by producing central nervous system arousal/stimulation.5,53–55 Beyond their potential clinical utility as anesthetic reversal agents, members of this new class of drugs would also be extremely valuable research tools. They would help scientists locate functionally important anesthetic binding sites on GABAA receptors, rationally design new exogenous ligands for these sites, and define the role that GABAA receptors play in producing particular anesthetic pharmacological, physiological, toxicological, and behavioral actions. Naphthalene-etomidate provides proof-of-concept for the design of anesthetic analogues with low intrinsic efficacies that may act as anesthetic-selective competitive antagonists at receptor targets. Such compounds would be valuable drugs for patient care and pharmacological tools to define the mechanisms of anesthetic action.
Acknowledgments
Funded by grants GM087316, GM58448, and GM122806 from the National Institutes of Health, Bethesda, MD, and the Department of Anesthesia, Critical Care, and Pain Medicine, Massachusetts General Hospital, Boston, Massachusetts.
Footnotes
Conflicts of Interest: None.
References
- 1.Sivilotti ML. Flumazenil, naloxone and the ‘coma cocktail’. Br J Clin Pharmacol. 2016;81:428–36. doi: 10.1111/bcp.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brull SJ, Kopman AF. Current Status of Neuromuscular Reversal and Monitoring: Challenges and Opportunities. Anesthesiology. 2017;126:173–190. doi: 10.1097/ALN.0000000000001409. [DOI] [PubMed] [Google Scholar]
- 3.Kakisis JD, Antonopoulos CN, Moulakakis KG, Schneider F, Geroulakos G, Ricco JB. Protamine Reduces Bleeding Complications without Increasing the Risk of Stroke after Carotid Endarterectomy: A Meta-analysis. Eur J Vasc Endovasc Surg. 2016;52:296–307. doi: 10.1016/j.ejvs.2016.05.033. [DOI] [PubMed] [Google Scholar]
- 4.Pani N, Dongare PA, Mishra RK. Reversal agents in anaesthesia and critical care. Indian J Anaesth. 2015;59:664–9. doi: 10.4103/0019-5049.167484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chemali JJ, Van Dort CJ, Brown EN, Solt K. Active emergence from propofol general anesthesia is induced by methylphenidate. Anesthesiology. 2012;116:998–1005. doi: 10.1097/ALN.0b013e3182518bfc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava A, Hunter JM. Reversal of neuromuscular block. Br J Anaesth. 2009;103:115–29. doi: 10.1093/bja/aep093. [DOI] [PubMed] [Google Scholar]
- 7.Pert CB, Snyder SH. Properties of opiate-receptor binding in rat brain. Proc Natl Acad Sci U S A. 1973;70:2243–7. doi: 10.1073/pnas.70.8.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon EJ, Hiller JM, Edelman I. Stereospecific binding of the potent narcotic analgesic (3H) Etorphine to rat-brain homogenate. Proc Natl Acad Sci U S A. 1973;70:1947–9. doi: 10.1073/pnas.70.7.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohler H, Richards JG. Agonist and antagonist benzodiazepine receptor interaction in vitro. Nature. 1981;294:763–5. doi: 10.1038/294763a0. [DOI] [PubMed] [Google Scholar]
- 10.Ahn KH, Sewell A, Elander J, Pittman B, Ranganathan M, Gunduz-Bruce H, Krystal J, D’Souza DC. Role of GABA Deficit in Sensitivity to the Psychotomimetic Effects of Amphetamine. Neuropsychopharmacology. 2015;40:2822–31. doi: 10.1038/npp.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller KW, Paton WD, Smith EB. Site of action of general anaesthetics. Nature. 1965;206:574–7. doi: 10.1038/206574a0. [DOI] [PubMed] [Google Scholar]
- 12.Miller KW, Pang KY. General anaesthetics can selectively perturb lipid bilayer membranes. Nature. 1976;263:253–5. doi: 10.1038/263253a0. [DOI] [PubMed] [Google Scholar]
- 13.Kaufman RD. Biophysical mechanisms of anesthetic action: historical perspective and review of current concepts. Anesthesiology. 1977;46:49–62. doi: 10.1097/00000542-197701000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Matubayasi N, Ueda I. Is membrane expansion relevant to anesthesia? Mean excess volume. Anesthesiology. 1983;59:541–6. doi: 10.1097/00000542-198312000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Raines DE, Cafiso DS. The enhancement of proton/hydroxyl flow across lipid vesicles by inhalation anesthetics. Anesthesiology. 1989;70:57–63. doi: 10.1097/00000542-198901000-00013. [DOI] [PubMed] [Google Scholar]
- 16.North C, Cafiso DS. Contrasting membrane localization and behavior of halogenated cyclobutanes that follow or violate the Meyer-Overton hypothesis of general anesthetic potency. Biophys J. 1997;72:1754–61. doi: 10.1016/S0006-3495(97)78821-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perouansky M. Coagulation, flocculation, and denaturation: a century of research into protoplasmic theories of anesthesia. Anesth Analg. 2014;119:311–20. doi: 10.1213/ANE.0000000000000287. [DOI] [PubMed] [Google Scholar]
- 18.Franks NP, Lieb WR. Which molecular targets are most relevant to general anaesthesia? Toxicol Lett. 1998;100–101:1–8. doi: 10.1016/s0378-4274(98)00158-1. [DOI] [PubMed] [Google Scholar]
- 19.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–20. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- 20.Franks NP. Molecular targets underlying general anaesthesia. Br J Pharmacol. 2006;147(Suppl 1):S72–81. doi: 10.1038/sj.bjp.0706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belelli D, Muntoni AL, Merrywest SD, Gentet LJ, Casula A, Callachan H, Madau P, Gemmell DK, Hamilton NM, Lambert JJ, Sillar KT, Peters JA. The in vitro and in vivo enantioselectivity of etomidate implicates the GABAA receptor in general anaesthesia. Neuropharmacology. 2003;45:57–71. doi: 10.1016/s0028-3908(03)00144-8. [DOI] [PubMed] [Google Scholar]
- 22.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. Faseb J. 2003;17:250–2. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- 23.Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HT, Taverna FA, Roder JC, Macdonald JF, Bhambri A, Collinson N, Wafford KA, Orser BA. Alpha5GABAA receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate. J Neurosci. 2006;26:3713–20. doi: 10.1523/JNEUROSCI.5024-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solt K, Forman SA. Correlating the clinical actions and molecular mechanisms of general anesthetics. Curr Opin Anaesthesiol. 2007;20:300–6. doi: 10.1097/ACO.0b013e32816678a5. [DOI] [PubMed] [Google Scholar]
- 25.Zarnowska ED, Rodgers FC, Oh I, Rau V, Lor C, Laha KT, Jurd R, Rudolph U, Eger EI, 2nd, Pearce RA. Etomidate blocks LTP and impairs learning but does not enhance tonic inhibition in mice carrying the N265M point mutation in the beta3 subunit of the GABA(A) receptor. Neuropharmacology. 2015;93:171–8. doi: 10.1016/j.neuropharm.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belelli D, Peden DR, Rosahl TW, Wafford KA, Lambert JJ. Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J Neurosci. 2005;25:11513–20. doi: 10.1523/JNEUROSCI.2679-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hill-Venning C, Belelli D, Peters JA, Lambert JJ. Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br J Pharmacol. 1997;120:749–56. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krasowski MD, Koltchine VV, Rick CE, Ye Q, Finn SE, Harrison NL. Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyric acid type A receptor distinct from that for isoflurane. Mol Pharmacol. 1998;53:530–8. doi: 10.1124/mol.53.3.530. [DOI] [PubMed] [Google Scholar]
- 29.Ge R, Pejo E, Gallin H, Jeffrey S, Cotten JF, Raines DE. The pharmacology of cyclopropyl-methoxycarbonyl metomidate: a comparison with propofol. Anesth Analg. 2014;118:563–7. doi: 10.1213/ANE.0000000000000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pejo E, Santer P, Wang L, Dershwitz P, Husain SS, Raines DE. gamma-Aminobutyric Acid Type A Receptor Modulation by Etomidate Analogs. Anesthesiology. 2015 doi: 10.1097/ALN.0000000000000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB. Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human alpha1beta3gamma2 gamma-aminobutyric acid type A (GABAA) receptor. J Biol Chem. 2013;288:19343–57. doi: 10.1074/jbc.M113.479725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jayakar SS, Zhou X, Savechenkov PY, Chiara DC, Desai R, Bruzik KS, Miller KW, Cohen JB. Positive and Negative Allosteric Modulation of an alpha1beta3gamma2 gamma-Aminobutyric Acid Type A (GABAA) Receptor by Binding to a Site in the Transmembrane Domain at the gamma+-beta- Interface. J Biol Chem. 2015;290:23432–46. doi: 10.1074/jbc.M115.672006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith GB, Olsen RW. Functional domains of GABAA receptors. Trends Pharmacol Sci. 1995;16:162–8. doi: 10.1016/s0165-6147(00)89009-4. [DOI] [PubMed] [Google Scholar]
- 35.Sigel E, Buhr A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci. 1997;18:425–9. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- 36.Husain SS, Ziebell MR, Ruesch D, Hong F, Arevalo E, Kosterlitz JA, Olsen RW, Forman SA, Cohen JB, Miller KW. 2-(3-Methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate: a derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels. J Med Chem. 2003;46:1257–65. doi: 10.1021/jm020465v. [DOI] [PubMed] [Google Scholar]
- 37.Savechenkov PY, Zhang X, Chiara DC, Stewart DS, Ge R, Zhou X, Raines DE, Cohen JB, Forman SA, Miller KW, Bruzik KS. Allyl m-trifluoromethyldiazirine mephobarbital: an unusually potent enantioselective and photoreactive barbiturate general anesthetic. J Med Chem. 2012;55:6554–65. doi: 10.1021/jm300631e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pejo E, Cotten JF, Kelly EW, Le Ge R, Cuny GD, Laha JK, Liu J, Lin XJ, Raines DE. In vivo and in vitro pharmacological studies of methoxycarbonyl-carboetomidate. Anesth Analg. 2012;115:297–304. doi: 10.1213/ANE.0b013e3182320559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dostalova Z, Liu A, Zhou X, Farmer SL, Krenzel ES, Arevalo E, Desai R, Feinberg-Zadek PL, Davies PA, Yamodo IH, Forman SA, Miller KW. High-level expression and purification of Cys-loop ligand-gated ion channels in a tetracycline-inducible stable mammalian cell line: GABAA and serotonin receptors. Protein Sci. 2010;19:1728–38. doi: 10.1002/pro.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiara DC, Dostalova Z, Jayakar SS, Zhou X, Miller KW, Cohen JB. Mapping general anesthetic binding site(s) in human alpha1beta3 gamma-aminobutyric acid type A receptors with [(3)H]TDBzl-etomidate, a photoreactive etomidate analogue. Biochemistry. 2012;51:836–47. doi: 10.1021/bi201772m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rusch D, Zhong H, Forman SA. Gating allosterism at a single class of etomidate sites on alpha1beta2gamma2L GABA A receptors accounts for both direct activation and agonist modulation. J Biol Chem. 2004;279:20982–92. doi: 10.1074/jbc.M400472200. [DOI] [PubMed] [Google Scholar]
- 42.Ziemba AM, Forman SA. Correction for Inhibition Leads to an Allosteric Co-Agonist Model for Pentobarbital Modulation and Activation of alpha1beta3gamma2L GABAA Receptors. PLoS One. 2016;11:e0154031. doi: 10.1371/journal.pone.0154031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang Y, Weiss DS. Allosteric activation mechanism of the alpha 1 beta 2 gamma 2 gamma-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J. 1999;77:2542–51. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruesch D, Neumann E, Wulf H, Forman SA. An allosteric coagonist model for propofol effects on alpha1beta2gamma2L gamma-aminobutyric acid type A receptors. Anesthesiology. 2012;116:47–55. doi: 10.1097/ALN.0b013e31823d0c36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guitchounts G, Stewart DS, Forman SA. Two etomidate sites in alpha1beta2gamma2 gamma-aminobutyric acid type A receptors contribute equally and noncooperatively to modulation of channel gating. Anesthesiology. 2012;116:1235–44. doi: 10.1097/ALN.0b013e3182567df3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stewart DS, Pierce DW, Hotta M, Stern AT, Forman SA. Mutations at beta N265 in gamma-aminobutyric acid type A receptors alter both binding affinity and efficacy of potent anesthetics. PLoS One. 2014;9:e111470. doi: 10.1371/journal.pone.0111470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ge RL, Pejo E, Haburcak M, Husain SS, Forman SA, Raines DE. Pharmacological studies of methoxycarbonyl etomidate’s carboxylic acid metabolite. Anesth Analg. 2012;115:305–8. doi: 10.1213/ANE.0b013e318239c6ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pejo E, Santer P, Jeffrey S, Gallin H, Husain SS, Raines DE. Analogues of etomidate: modifications around etomidate’s chiral carbon and the impact on in vitro and in vivo pharmacology. Anesthesiology. 2014;121:290–301. doi: 10.1097/ALN.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walters RJ, Hadley SH, Morris KD, Amin J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci. 2000;3:1274–81. doi: 10.1038/81800. [DOI] [PubMed] [Google Scholar]
- 50.Changeux JP. 50 years of allosteric interactions: the twists and turns of the models. Nat Rev Mol Cell Biol. 2013;14:819–29. doi: 10.1038/nrm3695. [DOI] [PubMed] [Google Scholar]
- 51.Forman SA. Monod-Wyman-Changeux allosteric mechanisms of action and the pharmacology of etomidate. Curr Opin Anaesthesiol. 2012;25:411–8. doi: 10.1097/ACO.0b013e328354feea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rusch D, Forman SA. Classic benzodiazepines modulate the open-close equilibrium in alpha1beta2gamma2L gamma-aminobutyric acid type A receptors. Anesthesiology. 2005;102:783–92. doi: 10.1097/00000542-200504000-00014. [DOI] [PubMed] [Google Scholar]
- 53.Waine TE, Dinmore P. Thiopentone anaesthesia terminated by bemegride. Anaesthesia. 1958;13:324–8. doi: 10.1111/j.1365-2044.1958.tb08101.x. [DOI] [PubMed] [Google Scholar]
- 54.Plourde G, Chartrand D, Fiset P, Font S, Backman SB. Antagonism of sevoflurane anaesthesia by physostigmine: effects on the auditory steady-state response and bispectral index. Br J Anaesth. 2003;91:583–6. doi: 10.1093/bja/aeg209. [DOI] [PubMed] [Google Scholar]
- 55.Gale AS. The effect of methylphenidate (ritalin) on thiopental recovery. Anesthesiology. 1958;19:521–31. doi: 10.1097/00000542-195807000-00009. [DOI] [PubMed] [Google Scholar]