


Abstract
An in silico model for the 1:1 ferredoxin (Fd)/nitrate reductase (NR) complex, using the known structure of Synechocystis sp. PCC 6803 Fd and the in silico model of Synechococcus sp. PCC 7942 NR, is used to map the interaction sites that define the interface between Fd and NR. To test the electrostatic interactions predicted by the model complex, five positively charged NR amino acids (Arg43, Arg46, Arg197, Lys201, and Lys614) and a negatively charged amino acid (Glu219) were altered using site-directed mutagenesis and characterized by activity measurements, metal analysis, and electron paramagnetic resonance (EPR) studies. All of the charge replacement variants retained wild-type levels of activity with reduced methyl viologen (MV), but a significant decrease in activity was observed for the R43Q, R46Q, K201Q, and K614Q variants when reduced Fd served as the electron donor. EPR analysis as well as the Fe and Mo analyses showed that loss of activity observed with these variants was not the consequence of perturbation of the Mo center or [4Fe-4S] cluster. Therefore, the loss of the Fd-linked specific activity observed with these variants can be explained only by invoking a role for Arg43, Arg46, Lys201, and Lys614 in Fd binding. The R43Q, R46Q, K201Q, and K614Q NR variants also showed a decreased binding affinity for Fd, compared to that of wild-type NR, supporting a key role of these four positively charged residues in the productive binding of Fd.
Graphical Abstract
Nitrate serves as a readily available source of inorganic nitrogen for cyanobacteria and plants.1 In natural environments, fixed inorganic nitrogen (ammonium, nitrate, nitrite, and nitrous oxide) is available as a result of human agricultural and industrial activities and biological nitrogen fixation.1 Nitrifying bacteria convert most of the fixed inorganic nitrogen (ammonium and nitrite) into nitrate, which then serves as source of inorganic nitrogen for assimilation into amino acids or serves as an electron acceptor during respiration.2 The pathway by which nitrate is converted into amino acids in plants and cyanobacteria is collectively known as nitrate or nitrogen assimilation. In cyanobacteria, nitrate assimilation requires three redox reactions.3, 4 The first step is catalyzed by nitrate reductase (NR) and involves the initial two-electron reduction of nitrate to nitrite. This is followed by the six-electron reduction of nitrite to ammonia, which is catalyzed by nitrite reductase. The ammonia then reacts with glutamate to form glutamine in a reaction catalyzed by glutamine synthetase. Glutamine formation requires ATP but does not involve any redox chemistry. However, in the final step of the reductive portion of the nitrate assimilation pathway, glutamine reacts with one molecule of 2-oxoglutarate to form two molecules of glutamate in the two-electron reduction reaction catalyzed by glutamate synthase.3, 4 Cyanobacteria are unique among oxygenic photosynthetic organisms in using reduced Fd as the physiological electron donor for all three of the reductant-requiring steps of the nitrate assimilation pathway.3, 4 While all other oxygenic phototrophs use reduced Fd as the electron donor for both the reduction of nitrite to ammonia and the reaction of glutamine and 2-oxoglutarate to form glutamate, the two-electron reduction of nitrate to nitrite utilizes NADPH as the electron donor.4, 5
The Fd-dependent NR of the cyanobacterium Synechococcus sp. PCC 7942 is the product of the narB gene and is among the best characterized of the Fd-dependent nitrate reductases.6 The complete amino acid sequence, as deduced from the nucleotide sequence of the narB gene, indicates a 78 kDa protein, which contains one bis-molybdopterin guanine dinucleotide (MGD) Mo center and one [4Fe-4S]2+,+ cluster as prosthetic groups.2, 6 The redox and spectroscopic properties of these two prosthetic groups have been investigated by protein film voltammetry and electron paramagnetic resonance (EPR) spectroscopy.2 Titrations, using EPR spectroscopy to monitor the cofactor redox states, have shown that the oxidized form of the [4Fe-4S]2+ cluster is EPR silent, while one-electron reduction results in an EPR signal characteristic of an S = 1/2 [4Fe-4S]+ cluster with an Em value of –190 mV.2, 7 The Mo center of the enzyme was also characterized by EPR spectroscopy,2, 7 and an Em value of –150 mV was estimated for a Mo6+ to Mo5+ transition by EPR-monitored redox titrations.2 Protein film voltammetry suggested that during the catalytic cycle nitrate binds to a two-electron reduced form of the enzyme in which the cluster is in its [4Fe-4S]+ form and the molybdopterin is in the Mo5+ state.2 A [2Fe-2S] Fd serves as the electron donor for this catalytic reaction and forms a high-affinity 1:1 complex with NR at a low ionic strength.7–9 Jepson et al.2 proposed a pathway for electron transfer from reduced Fd to Synechococcus sp. PCC 7942 NR in which Fd first reduces the [4Fe-4S] cluster, which in turn reduces then Mo center, the site in NR where nitrate is bound and reduced. As Fd is a one-electron carrier and NR contains only one Fd-binding site, this process requires two separate, sequential electron transfers from Fd to the enzyme.9 The first reduced Fd transfers its electron to the [4Fe-4S] cluster, which subsequently reduces Mo, from Mo6+ to Mo5+ in the first step and from Mo5+ to Mo4+ in the second step.2, 5 A two-electron transfer from Mo4+ to bound nitrate completes the process, producing nitrite and returning the enzyme to its original state.2, 5 Flash photolysis experiments on Synechococcus sp. PCC 7942 NR showed that the enzyme completes one full catalytic cycle under the conditions favoring two sequential electron transfers from the reduced Fd to the NR.10
A combination of sequence alignments and site-directed mutagenesis identified four conserved cysteine residues in Synechococcus sp. PCC 7942 NR, i.e., Cys9, Cys12, Cys16, and Cys56, that most likely serve as ligands of the [4Fe-4S] cluster that is located at the N-terminus of the enzyme.6 Kinetic studies, substrate binding studies, computer modeling, and amino acid replacements have been used to investigate the molecular mechanism of the reaction catalyzed by this enzyme.7, 9 Site-directed mutagenesis study in our laboratory identified three highly conserved basic amino acids, i.e., Lys58, Arg70, and Lys130 in Synechococcus sp. PCC 7942 NR, that are essential for its full catalytic activity.7 This study also presented a computer-based in silico model of the three-dimensional structure of Synechococcus sp. PCC 7942 NR that has provided many details of the possible mechanism of action of this enzyme.7 As there is no three-dimensional structure available for any Fd-dependent NR, the in silico model of Synechococcus sp. PCC 7942 NR was calculated on the basis of the known structure of the dissimilatory NR from Desulfovibrio desulfuricans.11 On the basis of the in silico model7 and studies of the active sites in related enzymes,11–17 our recent characterization of site-directed variants of Synechococcus sp. PCC 7942 NR identified five amino acids, Cys148, Met149, Met306, Asp163, and Arg351, that are essential for activity and are involved in either nitrate binding, prosthetic group binding, or catalysis.9
In addition to characterizing amino acids likely to be present at or near the active site9 and highly conserved basic amino acids,7 we investigated the possible locations for the Fd-binding domain on Synechococcus sp. PCC 7942 NR. To map the amino acid residues forming the interface for Fd docking to the NR, an in silico model for the 1:1 Fd-NR complex was constructed, using the known structure of Synechocystis sp. PCC 6803 Fd [Protein Data Bank (PDB) entry 1OFF, ref 18] and the in silico model of Synechococcus sp. PCC 7942 NR.7, 9 This in silico model of the Fd–NR complex provided specific details about the interaction sites that are involved in the formation of the Fd–NR complex. In this work, site-directed mutagenesis was used to test this model of the complex and to identify specific amino acids of NR involved in the formation of the complex with Fd.
MATERIALS AND METHODS
Both recombinant wild-type Fd from the cyanobacterium Synechocystis sp. PCC 6803 and recombinant, C-terminally Histagged Synechococcus sp. PCC 7942 NR were expressed in Escherichia coli and purified as described previously.7, 9, 10 The QuikChange (QC) site-directed mutagenesis kit (Stratagene) was used to generate site-specific replacement variants of NR as described previously.7, 9 Polymerase chain reaction (PCR) was used to generate site-specific replacement variants of NR using mutagenic primers listed in Table 1. The same expression and purification procedure was followed for the variants of NR and for the wild-type enzyme.7, 9 The purity of all variants described in this study was judged to be >90% based upon a visual assessment analysis of Coomassie Brilliant Blue-stained SDS–PAGE.
Table 1.
Primers Used To Introduce Mutations into the pCSLM85 Nitrate Reductase Expression Plasmida
| primer name | sequence (5′–3′) |
|---|---|
| R43Q_F | CCGATCTGGCAGATTCAGGGCGATCGGCAACAT |
| R43Q_R | ATGTTGCCGATCGCCCTGAATCTGCCAGATCGG |
| R43K_F | CCGATCTGGCAGATTAAAGGCGATCGGCAACAT |
| R43K_R | ATGTTGCCGATCGCCTTTAATCTGCCAGATCGG |
| R46Q_F | CAGATTCGGGGCGATCAGCAACATCCCTCCAGT |
| R46Q_R | ACTGGAGGGATGTTGCTGATCGCCCCGAATCTG |
| R46K_F | CAGATTCGGGGCGATAAACAACATCCCTCCAGT |
| R46K_R | ACTGGAGGGATGTTGTTTATCGCCCCGAATCTG |
| R197Q_F | CTCTTCAATCGCTATCAGAAGCGCCATAAACAG |
| R197Q_R | CTGTTTATGGCGCTTCTGATAGCGATTGAAGAG |
| K201Q_F | TATCGCAAGCGCCATCAACAGGGTGGCACCAAC |
| K201Q_R | GTTGGTGCCACCCTGTTGATGGCGCTTGCGATA |
| K201R_F | TATCGCAAGCGCCATAGACAGGGTGGCACCAAC |
| K201R_R | GTTGGTGCCACCCTGTCTATGGCGCTTGCGATA |
| E219Q_F | TGCACACCAACGGCTCAGGTTGCCGATCTCCAC |
| E219Q_R | GTGGAGATCGGCAACCTGAGCCGTTGGTGTGCA |
| K614Q_F | ACCGGCCGCATCGACCAAATCAACAAGCTGCAT |
| K614Q_R | ATGCAGCTTGTTGATTTGGTCGATGCGGCCGGT |
| K614R_F | ACCGGCCGCATCGACAGAATCAACAAGCTGCAT |
| K614R_R | ATGCAGCTTGTTGATTCTGTCGATGCGGCCGGT |
F indicates the forward direction primer and R the reverse direction primer. The mutagenic base replacements are shown in bold.
Ultraviolet–visible (UV–vis) absorbance spectra and difference absorbance spectra for wild-type NR and its variant forms were recorded using a Shimadzu model UV-2401PC spectrophotometer with a spectral resolution of 0.5 nm. An OLIS model DSM-10 UV–vis CD spectrophotometer was used to record circular dichroism (CD) spectra for the wild-type enzyme and the variants at a spectral resolution of 1.0 nm. The Fd concentration was measured using an extinction coefficient of 9.7 mM−1 cm−1 at 420 nm.19 The Bradford method was used to measure NR protein concentrations, using bovine serum albumin as a standard.20 X-Band (~9.6 GHz) EPR spectra were recorded using a Bruker ESP-300E EPR spectrometer equipped with an ER-4116 dual-mode cavity and an Oxford Instruments ESR-9 liquid helium flow cryostat. Spin quantifications were performed under non-power saturation conditions using a 1 mM Cu2+ EDTA standard as described by Aasa and Vänngård.21 EPR spectra were recorded for Synechococcus sp. PCC 7942 NR samples as purified aerobically and reduced anaerobically with a 10-fold excess of dithionite. The iron (Fe) and molybdenum (Mo) contents were estimated using inductively coupled plasma (ICP) mass spectrometry in the School of Molecular Sciences at Arizona State University, as described previously.7, 9
NR activities with either reduced MV or reduced Fd as the electron donors were measured as previously described.7, 9 Fits to the Michaelis–Menten equation were obtained for all plots of NR activity as a function of nitrate concentration at a saturating Fd concentration and as a function of Fd concentration at a saturating nitrate concentration. Kinetic parameters were determined using GraphPad Prism 6 by fitting the data (i.e., initial velocity vs substrate concentration) to the Michaelis–Menten equation. Formation of a complex between Fd and NR was measured using the spectral perturbation method as described previously.7–9 Formation of a complex of wild-type NR with Fd was monitored at 420 nm minus 600 nm. However, variations were observed in the maxima of the difference spectra for different NR variants upon binding of Fd. Consequently, to maximize the magnitude of the absorbance changes used for Fd dissociation constant (Kd) determinations, the following wavelength pairs were used to monitor the binding of Fd to the NR variants: R43Q, 460 nm minus 520 nm; R43K, 450 nm minus 650 nm; R46Q, 420 nm minus 600 nm; R46K, 450 nm minus 600 nm; R197Q, 410 nm minus 470 nm; K201Q, 400 nm minus 650 nm; K201R, 410 nm minus 470 nm; E219Q, 500 nm minus 650 nm; K614R and K614Q, 500 nm minus 600 nm.
The docking of the Fd from Synechocystis sp. PCC 6803 (PDB entry 1OFF, ref 18) to the previously published in silico model7, 9 was performed using three programs, ZDOCK, ClusterPro, and Rosetta.22–26 These programs dock two structures by balancing a number of protein–protein interactions, including electrostatic interactions, van der Waals interactions, hydrogen bonds, solvation energies, and hydrophobic interactions. A fundamental assumption of these programs is that the backbone conformations of proteins typically do not change upon association; therefore, these programs treat the models as static and do not accommodate any flexibility of the proteins. Each program generates and scores thousands of different random positions. The most favorable models from each program were then evaluated on the basis of the relative positions and orientations of the two docked proteins.
RESULTS
Figure 1 shows the results of an in silico search for the energetically most favorable docking site(s) for Fd on Synechococcus sp. PCC 7942 NR. As there are no three-dimensional structures known for any Synechococcus Fd, the structure of the Fd from the cyanobacterium Synechocystis sp. 6803 was used in this model of the complex. Given the close similarity in tertiary structures of “plant-type” Fds,27, 28 the high degree of primary sequence homology of Synechocystis and Synechococcus Fds (Figure 2), and the essentially identical NR activities using the Synechocystis and Synechococcus Fds as electron donors,7 the use of Synechocystis Fd in this modeling exercise should not introduce any significant uncertainties (Figure 2). The two most favorable structures predicted by the modeling (one colored light red and one dark red), which used the known structure of Fd (PDB entry 1OFF, ref 18) and our previously published7, 9 in silico model for the enzyme, share a common binding site for Fd on the enzyme but predict somewhat different orientations for the bound Fd (Figure 1A). Of these two models, we have focused on the complex that has the Fd docked into a niche on the surface of NR, where a pocket is formed by a number of α-helices and β-strands of NR (Figure 1B). Fd slips into this site, assisted by interactions with several loops located near the [2Fe-2S] cluster of Fd. In the model, the [2Fe-2S] cluster of the Fd is 11.9 Å from the [4Fe-4S] cluster of NR. Also included are the locations of the [2Fe-2S] cluster of Fd and the two prosthetic groups of the enzyme. The model predicts specific amino acid residues to be present at the binding site, involving electrostatic interactions of ionizable side chains along with hydrophobic contributions from NR and Fd. As the surface of Synechocystis sp. PCC 6803 Fd would be negatively charged at physiological pH values27, 28 and because the predicted binding interface for NR on Fd is largely composed of negatively charged side aspartates and glutamates, the positively charged amino acids predicted to be present on the Fd-binding interface of NR are lysines and arginines.
Figure 1.

(A) In silico model for two possible Fd orientations in the NR–Fd complex. The structure of NR is shown as a surface model (blue) and two of the possible associations of Fd (light and dark red). (B) In silico model for the NR–Fd complex. Shown are the backbone models of Fd (purple) and nitrate reductase (wheat). Fd (purple) with its Fe–S cofactor (atom type) with the nitrate reductase cofactors (atom type).
Figure 2.

Amino acid sequence alignment of cyanobacterial ferredoxins. Both N-terminal and C-terminal regions of the protein are included. The following cyanobacterial ferredoxins are shown: PCC 6803, Synechocystis sp. PCC 6803; Anabaena, Anabaena cylindrica; P13C2, Limnothrix sp. P13C2; Lyngbya, Lyngbya aestuarii; NIES-3709, Geminocystis sp. NIES-3709; PCC 7942, Synechococcus sp. PCC 7942. Negatively charged amino acids at position 12 are highlighted in red, at position 31 in turquoise, at position 35 in green, at position 93 in magenta, and at position 94 in yellow. Hyphens indicate gaps introduced to optimize alignments. Asterisks denote fully conserved residues.
We assessed the involvement of individual amino acids on NR in binding Fd by mutagenic replacement of the positively charged side chains of Arg43, Arg46, Arg197, Lys201, and Lys614 and the negatively charged Glu219 that are modeled as being present at the NR–Fd complex interface. Activity was measured using both reduced Fd and the nonphysiological donor, reduced MV, as the electron donor.3 On the basis of the structural models, none of these selected residues are close to either of the enzyme’s prosthetic groups (Table 2). Site-specific variants at these positions were generated using mutagenic primers (see Table 1) by site-directed mutagenesis. PCR amplification with these primers was performed with the method as described previously.7, 9 The mutated plasmids were sequenced, using sequence-specific primers to confirm the mutations. The variants were expressed in E. coli, and their expression level was comparable to that of the wild-type enzyme. The yields of the variants of NR were approximately 8 mg of purified protein per liter of E. coli culture. Coomassie Brilliant Blue-stained SDS–PAGE confirmed the purity of these variants. SDS–PAGE analysis indicated that all of the enzyme samples used in this study were at least 90% pure and had an approximate molecular mass of 78 kDa. UV–vis absorption spectra for the wild-type enzyme and the variants were recorded over the spectral range from 250 to 700 nm. The absorbance spectra of the variants were similar to those of wild-type NR and showed a characteristic spectral feature (broad shoulder) at 320, 400, and 450 nm, consistent with the spectral features observed with the wild-type enzyme (data not shown). Circular dichroism (CD) spectra were recorded in the UV region (200–300 nm) for each of the variants. Comparison with the CD spectra of wild-type NR shows that none of the mutational replacements results in a significant conformational change. This is illustrated in Figure 3, which shows the UV– CD spectra of the wild-type enzyme and two of the variants used in this study, i.e. R46Q and K614Q.
Table 2.
Distances from Amino Acid Residues Altered by Mutagenesis to the FeS and Mo Centersa
| residue | distance to the Fe-S cluster (Å) | distance to Mo (Å) |
|---|---|---|
| R43 | 9.4 | 28.7 |
| R46 | 11.8 | 25.5 |
| R197 | 12.5 | 23.1 |
| K201 | 17.7 | 28.9 |
| E219 | 18.8 | 23.9 |
| K614 | 14.1 | 22.2 |
Distances are measured from the end of each side chain to the closest point of each cofactor.
Figure 3.

Circular dichroism spectra of wild-type nitrate reductase and its R46Q and K614Q variants. Spectra were measured at ambient temperature in a 1.0 cm cell using solutions of either wild-type nitrate reductase or the indicated variant. All samples contained 0.75 µM nitrate reductase in 10 mM potassium phosphate buffer (pH 7.5).
Table 3 summarizes the effects of mutagenic replacements at each of the selected NR docking site amino acids with the polar, but uncharged, amino acid glutamine on the kinetic properties of enzyme, with both reduced MV and Synechocystis sp. PCC 6803 Fd serving as the electron donors. Figure 4 shows representative plots of the enzyme activities of the wild-type enzyme and two variants (R46Q and R197Q) as a function of Fd concentration (at a saturating nitrate concentration) and nitrate concentration (at a saturating Fd concentration) with best fits to the Michaelis–Menten equation. All of the variants of Synechococcus sp. PCC 7942 NR retain wild-type levels of activity when reduced MV serves as the electron donor, but R43Q, R46Q, K201Q, and K614Q variants show activity that is substantially decreased compared to that of the wild type when reduced Fd serves as the electron donor. Specifically, the R43Q variant retains 91% of wild-type activity with reduced MV but only 16% of wild-type activity with reduced Fd. Similarly, R46Q retains 97% of wild-type activity with reduced MV and 24% of the Fd-dependent activity of the wild-type enzyme. With reduced MV as the electron donor, the K201Q and K614Q variants exhibit 99 and 98% of the wild-type enzyme activity, respectively, but show a dramatic loss of activity compared to that of the wild type with Fd as the electron donor, i.e., 85 and 88% loss of activity for K201Q and K614Q, respectively. In contrast, nonconservative replacement of Arg197 and Glu219 with glutamine has no effect on either MV- or Fd-dependent activity. Moreover, the R197Q and E219Q variants have KM values for both Fd and nitrate that are similar to that of the wild-type enzyme. The R46Q variant has inhibited Fd-dependent activity and exhibits a KM for Fd and nitrate that is 9-fold greater than the measured values for the wild-type enzyme. The rates observed with the R43Q, K201Q, and K614Q variants were too low to allow an accurate determination of the KM value for its substrates. These observations are consistent with those expected if Arg43, Arg46, Lys201, and Lys614 all play important roles in binding and orienting Fd for productive electron transfer.
Table 3.
Kinetic Properties of Wild-Type Nitrate Reductase and Its Docking Site Variants
| enzyme | MV-linked specific activitya |
Fd-linked specific activitya |
KM with nitrate (µM) |
KM with Fd (µM) |
|---|---|---|---|---|
| wild type | 790 (100%) | 30.5 (100%) | 6.0 ± 1.0 | 38.0 ± 3.7 |
| R43Q | 721 (91.2%) | 5.0 (16.4%) | NDb | NDb |
| R43K | 734 (92.9%) | 27.8 (91%) | 8.1 ± 0.7 | 48.6 ± 3.9 |
| R46Q | 768 (97.2%) | 7.4 (24.3%) | 55 ± 20 | 364 ± 94 |
| R46K | 783 (99.1%) | 29.8 (97.7%) | 6.6 ± 1.0 | 49.6 ± 16.7 |
| R197Q | 738 (93.4%) | 27.9 (91.8%) | 7.0 ± 0.7 | 41.9 ± 3.8 |
| K201Q | 779 (98.6%) | 4.5 (14.8%) | NDb | NDb |
| K201R | 775 (98.11%) | 29.4 (91.8%) | 9.6 ± 1.0 | 45.6 ± 3.1 |
| E219Q | 749 (94.8%) | 29.3 (96.1%) | 4.5 ± 0.4 | 42.2 ± 5.1 |
| K614Q | 771 (97.6%) | 3.5 (11.5%) | NDb | NDb |
| K614R | 761 (96.3%) | 19.3 (63.3%) | 4.2 ± 0.5 | 44.5 ± 16.2 |
In units of micromoles of nitrite produced per minute per milligram of enzyme.
Not determined (because of the very low activity).
Figure 4.

Michaelis-Menten plots of the activity of wild-type, R46Q, and R197Q nitrate reductase as a function of Fd concentration (at a saturating nitrate concentration) and nitrate concentration (at a saturating Fd concentration). The solid lines are best fits to the Michaelis–Menten equation (data presented here are from a single measurement). Values of KM for nitrate and Fd are listed in Table 3.
To investigate if arginine at positions 43 and 46 and lysine at positions 201 and 614 are specifically required for effective electron donation by Fd, or whether it is simply a positively charged side chain that is required at these positions, the R43K, R46K, K201R, and K614R charge-conserving variants were prepared and characterized. The charge-conserving variants of Arg43, Arg46, Lys201, and Lys614 retain essentially full wild-type activity with MV as the electron donor (Table 3). With Fd as the electron donor, these variants also exhibit levels of activity that are generally close to the wild-type level and much higher than those measured for the glutamine-substituted variants (i.e., 91% of wild-type activity for the R43K variant, 98% of wild-type activity for the R46K variant, 92% of wild-type activity for the K201R variant, and 63% of wild-type activity for the K614R variant). The activity with Fd as the electron donor was sufficiently high to allow determinations of the KM values for both nitrate and Fd for the reaction catalyzed by these charge-conserving variants (Table 3). The KM values for both Fd and nitrate obtained with R43K, R46K, K210R, and K614R variants are quite similar to those observed for the wild-type enzyme [i.e., 38 µM for Fd and 6.0 µM for nitrate (see Table 3)]. These results suggest that the presence of a positive charge at these positions is the most important factor for productive Fd binding and that there is not a strict requirement for arginine or lysine per se.
In principle, the significant loss of Fd-dependent activity observed for the R43Q, R46Q, K201Q, and K614Q variants could also arise from a decrease in Fd binding affinities. This possibility was addressed by measuring the Fd dissociation constant (Kd) of the wild type and variants using a spectral perturbation method developed previously for the wild-type enzyme.8 Table 4 summarizes the Fd Kd values obtained for the wild-type and variant enzymes. Figure 5 shows representative plots of difference absorption versus Fd concentration and the fits that were used to calculate Fd Kd values for the wild-type, R43Q, R46Q, R46K, K201Q, and K614Q enzymes. Spectral perturbation measurements showed decreased Fd binding affinities for the R43Q (5-fold), R46Q (2.5-fold), and K201Q (3-fold) variants. No spectral perturbations were observed for K614Q, which prevented assessment of the Kd value. The R197Q and E219Q variants that retained wild-type level activity with both electron donors showed almost the same binding affinities for Fd as the wild-type enzyme. The conservative replacement variants generally exhibited enhanced Fd binding affinities compared to those of the equivalent nonconservative replacement variants. Wild-type Fd binding affinities were observed for R46K and K201R, and the Fd binding affinity for R43K was enhanced 2.5-fold compared to that of R43Q. In contrast to the K614Q variant in which no spectral perturbations were detected anywhere in the visible region when Fd was mixed with the enzyme, the K614R variant showed spectral changes after mixing with Fd that made it possible to estimate the binding affinity of the variant for Fd. In fact, this NR variant showed an affinity for Fd (>5-fold) significantly lower than that of the wild-type enzyme. These observations are consistent with the hypothesis that the side chains of Arg43, Arg46, Lys201, and Lys614 play an important role in Fd binding.
Table 4.
Prosthetic Group Content and Ferredoxin Binding Affinities of Wild-Type Nitrate Reductase and Its Docking Site Variants
| enzyme | irona | molybdenuma | Kd with Fd (µM) |
|---|---|---|---|
| wild type | 3.6 ± 0.45 | 0.93 ± 0.04 | 7.8 ± 2.1 |
| R43Q | 3.6 ± 0.20 | 0.95 ± 0.10 | 38.6 ± 14 |
| R43K | 3.7 ± 0.20 | 0.95 ± 0.10 | 20.2 ± 1.1 |
| R46Q | 2.8 ± 0.3 | 0.70 ± 0.10 | 19.8 ± 5.1 |
| R46K | 4.0 ± 0.1 | 0.80 ± 0.30 | 3.7 ± 0.3 |
| R197Q | 3.0 ± 0.20 | 0.70 ± 0.10 | 7.5 ± 1.9 |
| K201Q | 4.0 ± 0.20 | 1.0 ± 0.10 | 26.3 ± 12 |
| K201R | 3.9 ± 0.20 | 0.95 ± 0.10 | 4.2 ± 1.4 |
| E219Q | 4.0 ± 0.20 | 1.0 ± 0.10 | 8.3 ± 3.0 |
| K614Q | 3.8 ± 0.34 | 1.04 ± 0.18 | b |
| K614R | 3.8 ± 0.39 | 1.11 ± 0.14 | 39.8 ± 17.6 |
In units of moles per mole of enzyme.
Cannot be detected by spectral perturbations.
Figure 5.

Measurement of ferredoxin dissociation constants (Kd) by difference UV–vis absorption measurements for wild-type, R43Q, R46Q, R46K, K201Q, and K614R nitrate reductases. Solid lines show best fits using the Kd values listed in Table 4. Titrations were conducted in samples containing 10 µM nitrate reductase in 25 mM HEPES buffer (pH 8.0).
To assess if any changes in Fd-linked activity for the variants compared to that of wild-type Synechococcus NR were a consequence of loss or perturbation of the Mo or [4Fe-4S] centers, EPR studies as well as Fe and Mo analyses were performed for each variant. The analytical data in Table 4 show that the wild type and essentially all variants investigated contained approximately one [4Fe-4S] cluster and one Mo center per nitrate reductase monomer. The only exceptions were the R46Q and R197Q variants, which were approximately 25% deficient in both the [4Fe-4S] cluster and the Mo center. The effects of the K614Q, K614R, R46Q, R46K, R43Q, R43K, R197Q, K201Q, K201R, and E219Q mutations on the EPR properties of the Mo and [4Fe-4S] centers are shown in Figure 6. At 70 K, air-oxidized samples of all nine mutants exhibit the same high-g proton-split S = 1/2 Mo5+ resonance (g1 = 2.023, g2 = 1.998, and g3 = 1.993; A1 = 6.8 G, A2 = 7.5 G, and A3 = 6.8 G) accounting for 20–30% of the Mo content, as previously observed for the wild-type enzyme2, 7, 9 (see Figure 6A). On the basis of crystallographic and density functional theory studies,16, 29, 30 the structure of this Mo5+ species is proposed to be a bis-MGD Mo center ligated by cysteinate and a terminal sulfido, with the nonexchangeable proton hyperfine splitting originating from the cysteine β-CH2 protons.30, 31 After dithionite reduction, the spectra of the wild-type enzyme and all nine variants recorded at 10 K exhibit the characteristic fast-relaxing rhombic resonance (g1 = 2.06, g2 = 1.95, and g3 = 1.91) associated with the S = 1/2 [4Fe-4S]+ cluster and accounting for 90–100% of the Fe content2, 7, 9 (see Figure 6B).
Figure 6.

EPR spectra of wild-type Synechococcus sp. PCC 7942 nitrate reductase and the R43Q, R43K, K614Q, K614R, R46Q, R46K, R197Q, K201Q, K201R, and E219Q variants. (A) Air-oxidized samples recorded at 70 K and a microwave power of 2 mW. (B) Dithionite-reduced samples recorded at 20 K and 1 mW. (C) Dithionite-reduced samples recorded at 70 K and 2 mW. All spectra were recorded at 9.60 GHz with a modulation amplitude of 6.3 G. The intensities of all spectra have been normalized with respect to the wild-type enzyme, to correct for differences in the spectrometer gain and the sample protein concentration.
EPR studies of dithionite-reduced samples at 70 K facilitated characterization of the slower-relaxing Mo5+ species in isolation (see Figure 6C). Two distinct types of Mo5+ species have been observed for the dithionite-reduced wild-type enzyme, and we have yet to define the specific conditions required to elicit each type of resonance. One is termed the high-g proton-split Mo5+ signal (g1 = 1.997, g2 = 1.990, and g3 = 1.982; A1 = 6.5 G, A2 = 6.0 G, and A3 = 5.0 G) that accounts for 0–20% of the Mo in wild-type samples.2, 9 The other is termed the low-potential Mo5+ signal (g1 = 2.017, g2 = 1.988, and g3 = 1.962 with negligible proton splitting) that accounts for 0–10% of the Mo content in wild-type samples.7, 9 The observation of two distinct Mo5+ species in dithionite-reduced wild-type samples illustrates the plasticity of the Mo active site and suggests facile interconversion between these two Mo5+ species. The high-g proton-split Mo5+ signal is proposed to comprise a bis-MGD Mo center ligated by a side-on η2-cysteine persulfide with a partial disulfide bond. The nonexchangeable proton hyperfine splitting originates from the cysteine β-CH2 protons.9, 16, 29–31 Because the low-potential Mo5+ signal lacks observable proton splitting, this suggests dissociation of the cysteine or conversion of the η2-cysteine persulfide ligand into a terminal cysteine persulfide ligand, which requires cleavage of only one cysteine S–Mo bond.9 Figure 6C shows that the dithionite-reduced K614Q and K614R samples exhibit weak low-potential Mo5+ resonances accounting for 4% of the Mo. All the other variants investigated, i.e., R46Q, R46K, R43Q, R43K, R197Q, K201Q, K201R, and E219Q, exhibit high-g proton-split Mo5+ resonances accounting for 3–20% of the Mo. While the proton hyperfine splitting in the high-g proton-split Mo5+ resonances shown in Figure 6C cannot be observed because of the spectra being recorded with a 6.3 G modulation amplitude, they are observed in spectra recorded with a 2.9 G modulation amplitude. In each case, the spectra recorded with a 2.9 G modulation amplitude exhibited proton splittings identical to those reported for the wild type (see ref 9).
On the basis of the analytical data and the EPR properties of air-oxidized and dithionite-reduced samples, we conclude that the Mo and [4Fe-4S] prosthetic groups remain intact and essentially unperturbed compared to the wild type in all the Synechococcus NR mutants investigated in this work. While there are significant differences in the magnitude of the high-g proton-split Mo5+ resonances (3–20% of Mo), these differences do not correlate with either the reduced MV-dependent or the reduced Fd-dependent specific activities listed in Table 3. Hence, the dramatic decrease in the Fd-linked specific activity that is observed for the R46Q, K614Q, R43Q, and K201Q variants can be interpreted only in terms of changes in the Fd-binding region.
DISCUSSION
In addition to providing a basis for designing mutagenic replacements that can provide information about the interaction domains involved in the formation of the complex between Fd and NR, NR–Fd complex models also provide information about the distances between and orientations of the electron transfer prosthetic groups involved in the reaction catalyzed by this Fd-dependent NR. In the model shown in Figure 1, the distance from the Fd [2Fe-2S] cluster to the NR [4Fe-4S] cluster is 11.9 Å. In the lowest-energy, most favorable models, this distance ranged from 10 to 17 Å, suggesting that a reasonable estimate for this distance predicted by the modeling is 13.5 ± 3.5 Å. It is this distance that is likely to play an important role in determining the rate of electron transfer from reduced Fd to NR. The most probable mechanism for electron transfer from Fd to NR involves an initial electron transfer from the Fd [2Fe-2S]+ cluster to the NR [4Fe-4S]2+ cluster, followed by intramolecular electron transfer from the [4Fe-4S]+ cluster to the Mo center of NR.2
The structural model for the Fd–NR complex presented above not only provides estimates of distances between electron-carrying prosthetic groups but also identifies specific amino acids at the protein–protein interaction interface. Five positively charged amino acids (Arg43, Arg46, Arg197, Lys201, and Lys614) and a negatively charged amino acid (Glu219) were chosen to test the in silico NR–Fd complex model. Charge replacement variants (R43Q, R46Q, K201Q, and K614Q) retained wild-type levels of activity with MV, but substantial losses of activity were found when reduced Fd served as the electron donor. These observations, along with the higher KM observed for Fd with the R46Q variant when compared to that of wild-type NR, indicate that Arg43, Arg46, Lys201, and Lys614 play an important role in binding Fd to facilitate the electron transfer to the Mo active site via the [4Fe-4S]2+,+ cluster. R43Q, R46Q, and K201Q variants also showed decreased binding affinity for Fd, when compared to that of wild-type NR, which further supports the role of Arg43, Arg46, and Lys201 in Fd binding. As the K614Q variant did not show any spectral perturbations upon being titrated with Fd, no conclusions about its ability to bind Fd could be drawn. Charge-conserving replacement mutations at Arg43, Arg46, Lys201, and Lys614 (i.e., R43K, R46K, K201R, and K614R, respectively) yielded Fd-dependent activities much higher than those of the charge-eliminating replacements. In addition to the increase in Fd-dependent activity, these four charge-conserving replacement variants showed increased binding affinities for Fd and KM values for both of substrates similar to those observed for the wild-type enzyme, indicating that the presence of a positive charge at these positions is the most important factor for productive Fd binding and electron transfer.
The involvement of Arg43, Arg46, Lys201, and Lys614 in ferredoxin binding is also consistent with amino acid sequence alignments of Fd-dependent cyanobacterial and non-Fd-dependent photosynthetic nitrate reductases (Figure 7). The sequence alignments show that all four of the positively charged amino acids at positions 43, 46, 201, and 614 are conserved in ferredoxin-dependent cyanobacterial NRs, but only one (Arg43) is conserved in non-Fd-dependent photosynthetic NRs. The sequence alignments also show that the amino acids at positions 197 and 219 in Synechococcus sp. PCC 7942 NR are not conserved in either cyanobacterial or other photosynthetic NRs. This is in agreement with the observation that variants at positions 197 and 219 showed wild-type levels of activity with ferredoxin as the electron donor.
Figure 7.

Amino acid sequence alignment of ferredoxin-dependent cyanobacterial and other photosynthetic nitrate reductases. The N-terminal and C-terminal regions of the enzymes are not shown. The following ferredoxin-dependent cyanobacterial nitrate reducatases are shown: PCC 7942, Synechococcus sp. PCC 7942; PCC 7421, Gloeobacter violaceus PCC 7421; PCC 6803, Synechocystis sp. PCC 6803; Tricho, Trichodesmium erytraeum; PCC 7108, Anabaena sp. PCC 7108; Nostoc, Nostoc punctiforme. The following non-ferredoxin-dependent photosynthetic nitrate reductases are shown: Chlorella, Chlorella vulgari; Solanum, Solanum tuberosum; Cucurbita, Cucurbita maxima. Amino acids at position 43 are highlighted in yellow, at position 46 in green, at position 197 in turquoise, at position 201 in magenta, at position 219 in gray, and at position 614 in red.
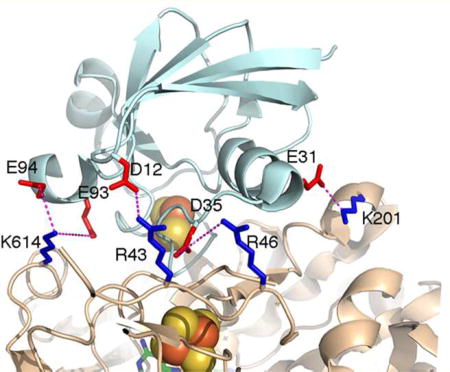
The mutagenesis results presented in this work provide convincing support for the prediction based on the NR–Fd complex model that the four positively charged amino acids (Arg43, Arg46, Lys201, and Lys614) of NR are involved in binding Fd. While there are a number of other interactions that would stabilize the complex, for example, polar interactions involving Fd residues Tyr38, Ser39, and Ser63 and a hydrophobic interaction involving hydrophobic residue Phe64 of Fd, the results presented herein demonstrate that electrostatic interactions are the crucial element. The in silico NR–Fd complex model shown in Figure 8 allowed identification of the specific negatively charged Fd amino acids (Asp12, Asp35, Glu31, Glu93, and Glu94) that are putatively involved in electrostatic interactions with the four positively charged amino acid residues of NR. The specific interactions are R43–D12, R46–D35, K201–E31, K614–E93, and K614–E94, where the pairs are residues of NR and Fd, respectively. All five of these negatively charged Fd amino acid residues are highly conserved (Figure 2). The exception of D12, which is equivalent to A12 in Synechococcus sp. PCC 7942, suggests that R43 may interact with a different amino acid residue, probably D35 after small rotations of the side chains upon binding. The largest impact on Kd for binding of Fd to NR was the alteration of K614, which may be due to the loss of two electrostatic interactions involving K614.
Figure 8.

Fd-NR complex in silico model showing specific Fd amino acids that are putatively involved in electrostatic interactions with Arg43, Arg46, Lys201, and Lys614 of Synechococcus sp. PCC 7942 nitrate reductase.
The results presented herein are consistent with earlier reports that the negatively charged residues of Fd play an important role in efficient electron transfer from Fd to the Fd-dependent enzymes.8, 32, 33 Data for the ionic strength dependence of the stability constants for these complexes led to the proposal that they were stabilized predominantly by electrostatic forces, with Fd supplying the negatively charged groups and the enzymes supplying the positively charged groups involved in complex formation.3, 34 Chemical modification studies have been used to explore the effects of eliminating these negative charges on the ability of Fd to interact productively with target enzymes, and the results of these studies support the hypothesis that Fd does indeed contribute negative charges to the stabilization of these complexes.35 Additionally, site-directed mutagenesis has also provided evidence that negatively charged Fd amino acids of Anabaena sp. PCC 712036 and spinach33 are crucial for electron transfer from Fd to ferredoxin:NADP+ reductase (FNR) and involved in the formation of an electrostatic complex with positively charged amino acids of FNR. Chemical modification studies have yielded results supporting the proposal that positive charges on the target enzymes, e.g., FNR,37 glutamate synthase,38 and nitrite reductase,39 play an important role in the formation of the complex with Fd.
Electrostatic interactions have also been found to be important for interactions of [2Fe-2S] Fd with mammalian and bacterial enzymes. For example, mitochondrial cytochrome P450 (P450) and Fd reductase have also been shown to form an electrostatic complex with Fd, in which Fd provides negative charges and P450s and Fd reductases provide the positive charges.40 Site-directed mutagenesis identified the specific negatively charged amino acids of human Fd that are important for forming a complex with Fd reductase and mitochondrial cytochrome P450scc (CYP11A1).41 In addition to the Fd amino acids, site-directed mutagenesis has also shown that positively charged amino acids of mammalian mitochondrial P450s [P450scc (CYP11A1) and P450c27 (CYP27A1)] are important for the formation of a complex with adrenodoxin, a [2Fe-2S] Fd in adrenal glands.42 In bacterial systems, electrostatic interactions were also found to be crucial for the formation of the complex between Fd and cytochrome P450s. Positively charged amino acid residues in cytochrome P450 (CYP199A2) from Rhodopseudomonas palustris were found to be important for the formation of a complex with the [2Fe-2S] Fd palustrisredoxin.43
On the basis of the studies described above and the fact that plant-type Fds typically have isoelectric points well below 7.0 and would thus exhibit net negative surface charges at physiological pH values, it seemed logical to assume that Fd would supply the negative charges and NR would provide the positive charges. Indeed, this was shown to be the case with Synechococcus sp. PCC 7942 NR in which positively charged amino acids were shown to be essential for the formation of the complex with Fd.8 Chemical modification of Synechococcus sp. PCC 7942 NR, in which positive charges were eliminated from NR with either phenylglyoxal or N-acetylsuccinimide, was found to interfere with the formation of the complex between NR and Fd.8 The data presented in this study are consistent with the observations made by Hirasawa et al.8 that the positively charged residues of NR are involved in the formation of the complex with Fd and provide specific details about the positively charged amino acids of Synechococcus sp. PCC 7942 NR that are involved in the formation of the complex with Fd.
Acknowledgments
The authors thank Prof. Enrique Flores (University of Sevilla, Sevilla, Spain) for providing the initial clone used to express wild-type Synechococcus sp. PCC 7942 NR The authors thank Dr. Masakazu Hirasawa for his help with gel filtration chromatography and for many helpful suggestions. The authors also thank Dr. Pierre Sétif (iBiTec-S, CNRS UMR 8221, CEA Saclay, Saclay, France) for helpful discussions.
Funding
The mutagenesis, protein expression and purification, kinetic measurements, and substrate binding determinations performed at Texas Tech University were funded by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy, through Grant DE-FG03-99ER20346 (to D.B.K.). The in silico structural modeling and the Fe and Mo determinations performed at Arizona State University were supported by National Science Foundation Grant CHE 1505874 (to J.P.A.). Funding for the EPR spectrometry and spin quantitation measurements was provided by the National Institutes of Health, through Grant GM62524 (to M.K.J.).
DEDICATION
This work is dedicated to the memory of David B. Knaff (1941–2016).
ABBREVIATIONS
- CD
circular dichroism
- EPR
electron paramagnetic resonance
- Fd
ferredoxin
- HEPES
4-(2-hydroxyethyl)-1-piperazineetha-nesulfonic acid
- ICP
inductively coupled plasma
- IPTG
isopropyl β-D-thiogalactopyranoside
- LB
Luria-Bertani
- MGD
molybdopterin guanine dinucleotide
- MV
methyl viologen
- PCR
polymerase chain reaction
- PMSF
phenylmethanesul-fonyl fluoride
- SDS–PAGE
sodium dodecyl sulfate–poly-acrylamide gel electrophoresis
- UV
ultraviolet
Footnotes
AUTHOR INFORMATION
Notes
The authors declare no competing financial interest.
References
- 1.Falkowski PG. Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean. Nature. 1997;387:272–275. [Google Scholar]
- 2.Jepson BJN, Anderson LJ, Rubio LM, Taylor CJ, Butler CS, Flores E, Herrero A, Butt JN, Richardson DJ. Tuning a nitrate reductase for function. The first spectropotentiometric characterization of a bacterial assimilatory nitrate reductase reveals novel redox properties. J. Biol. Chem. 2004;279:32212–32218. doi: 10.1074/jbc.M402669200. [DOI] [PubMed] [Google Scholar]
- 3.Hase T, Schürmann P, Knaff DB. The interaction of ferredoxin with ferredoxin-dependent enzymes. In: Golbeck JH, editor. Photosystem. Vol. 1. Springer; Dordrecht, The Netherlands: 2006. pp. 477–498. [Google Scholar]
- 4.Suzuki A, Knaff DB. Nitrogen metabolism and roles of glutamate synthase in higher plants. Photosynth. Res. 2005;83:191–217. doi: 10.1007/s11120-004-3478-0. [DOI] [PubMed] [Google Scholar]
- 5.Flores E, Frías JE, Rubio LM, Herrero A. Photosynthetic nitrate assimilation in cyanobacteria. Photosynth. Res. 2005;83:117–133. doi: 10.1007/s11120-004-5830-9. [DOI] [PubMed] [Google Scholar]
- 6.Rubio LM, Flores E, Herrero A. Purification, cofactor analysis, and site-directed mutagenesis of Synechococcus ferredoxin-nitrate reductase. Photosynth. Res. 2002;72:13–26. doi: 10.1023/A:1016078700839. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava AP, Hirasawa M, Bhalla M, Chung J-S, Allen JP, Johnson MK, Tripathy JN, Rubio LM, Vaccaro B, Subramanian S, Flores E, Zabet-Moghaddam M, Stitle K, Knaff DB. The roles of four conserved basic amino acids in a ferredoxin-dependent cyanobacterial nitrate reductase. Biochemistry. 2013;52:4343–4353. doi: 10.1021/bi400354n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirasawa M, Rubio LM, Griffin JL, Flores E, Herrero A, Li J, Kim S-K, Hurley JK, Tollin G, Knaff DB. Complex formation between ferredoxin and Synechococcus ferredoxin: nitrate oxidoreductase. Biochim. Biophys. Acta, Bioenerg. 2004;1608:155–162. doi: 10.1016/j.bbabio.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Srivastava AP, Allen JP, Vaccaro BJ, Hirasawa M, Alkul S, Johnson MK, Knaff DB. Identification of amino acids at the catalytic site of a ferredoxin-dependent cyanobacterial nitrate reductase. Biochemistry. 2015;54:5557–5568. doi: 10.1021/acs.biochem.5b00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava AP, Knaff DB, Sétif P. Kinetic Studies of a ferredoxin-dependent cyanobacterial nitrate reductase. Biochemistry. 2014;53:5092–5101. doi: 10.1021/bi500386n. [DOI] [PubMed] [Google Scholar]
- 11.Dias JM, Than ME, Humm A, Huber R, Bourenkov GP, Bartunik HD, Bursakov S, Calvete J, Caldeira J, Carneiro C, Moura JJG, Moura I, Romao MJ. Crystal structure of the first dissimilatory nitrate reductase at 1.9 Å solved by MAD methods. Structure. 1999;7:65–79. doi: 10.1016/s0969-2126(99)80010-0. [DOI] [PubMed] [Google Scholar]
- 12.Boyington JC, Gladyshev VN, Khangulov SV, Stadtman TC, Sun PD. Crystal structure of formate dehydrogenase H: Catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science. 1997;275:1305–1308. doi: 10.1126/science.275.5304.1305. [DOI] [PubMed] [Google Scholar]
- 13.Raaijmakers H, Macieira S, Dias JM, Teixeira S, Bursakov S, Huber R, Moura JJG, Moura I, Romão MJ. Gene sequence and the 1.8Å crystal structure of the tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Structure. 2002;10:1261–1272. doi: 10.1016/s0969-2126(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 14.Arnoux P, Sabaty M, Alric J, Frangioni B, Guigliarelli B, Adriano J-M, Pignol D. Structural and redox plasticity in the heterodimeric periplasmic nitrate reductase. Nat. Struct. Biol. 2003;10:928–934. doi: 10.1038/nsb994. [DOI] [PubMed] [Google Scholar]
- 15.Dementin S, Arnoux P, Frangioni B, Grosse S, Leger C, Burlat B, Guigliarelli B, Sabaty M, Pignol D. Access to the active site of periplasmic nitrate reductase: Insights from site-directed mutagenesis and zinc inhibition studies. Biochemistry. 2007;46:9713–9721. doi: 10.1021/bi700928m. [DOI] [PubMed] [Google Scholar]
- 16.Najmudin S, González PJ, Trincão J, Coelho C, Mukhopadhyay A, Cerqueira NM, Romao FSA, Moura I, Moura JJG, Brondino CD, Romão MJ. Periplasmic nitrate reductase revisited: a sulfur atom completes the sixth coordination of the catalytic molybdenum. JBIC, J. Biol. Inorg. Chem. 2008;13:737–753. doi: 10.1007/s00775-008-0359-6. [DOI] [PubMed] [Google Scholar]
- 17.Wang TH, Chen YH, Huang JY, Liu KC, Ke SC, Chu HA. Enzyme kinetics, inhibitors, mutagenesis and electron paramagnetic resonance analysis of dual-affinity nitrate reductase in unicellular N2-fixing cyanobacterium Cyanothece sp. PCC 8801. Plant Physiol. Biochem. 2011;49:1369–1376. doi: 10.1016/j.plaphy.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 18.van den Heuvel RHH, Svergun DI, Petoukhov MV, Coda B, Curti S, Ravasio MV, Vanoni MA, Mattevi A. The active conformation of glutamate synthase and its binding to ferredoxin. J. Mol. Biol. 2003;330:113–128. doi: 10.1016/s0022-2836(03)00522-9. [DOI] [PubMed] [Google Scholar]
- 19.Tagawa K, Arnon DI. Ferredoxins as electron carriers in photosynthesis and in the biochemical production and consumption of hydrogen gas. Nature. 1962;195:537–543. doi: 10.1038/195537a0. [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. A rapid and sensitive for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Aasa R, Vänngård T. EPR signal intensity and powder shapes: A reexamination. J. Magn. Reson. 1975;19:308–315. [Google Scholar]
- 22.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 23.Mintseris J, Pierce B, Wiehe K, Anderson R, Chen R, Weng Z. Integrating statistical pair potentials into protein complex prediction. Proteins: Struct., Funct., Genet. 2007;69:511–520. doi: 10.1002/prot.21502. [DOI] [PubMed] [Google Scholar]
- 24.Lyskov S, Gray JJ. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008;36:W233–W238. doi: 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozakov D, Hall DR, Beglov D, Brenke R, Comeau SR, Shen Y, Li K, Zheng J, Vakili P, Paschalidis IC, Vajda S. Achieving reliability and high accuracy in automated protein docking: Cluspro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins: Struct., Funct., Genet. 2010;78:3124–3130. doi: 10.1002/prot.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce BG, Hourai Y, Weng Z. Accelerating Protein Docking in ZDOCK Using an Advanced 3D Convolution Library. PLoS One. 2011;6:e24657. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bottin H, Lagoutte B. Ferredoxin and flavodoxin from the cyanobacterium Synechocystis sp. PCC 6803. Biochim. Biophys. Acta, Bioenerg. 1992;1101:48–56. doi: 10.1016/0167-4838(92)90465-p. [DOI] [PubMed] [Google Scholar]
- 28.Hanke G, Mulo P. Plant-type ferredoxins and ferredoxin-dependent enzymes. Plant, Cell Environ. 2013;36:1071–1084. doi: 10.1111/pce.12046. [DOI] [PubMed] [Google Scholar]
- 29.Coelho C, González PJ, Moura JJG, Moura I, Trincão J, João Romão M. The crystal structure of Cupriavidus necator nitrate reductase in oxidized and partially reduced states. J. Mol. Biol. 2011;408:932–948. doi: 10.1016/j.jmb.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Biaso F, Burlat B, Guigliarelli B. DFT investigation of the molybdenum cofactor in periplasmic nitrate reductases: Structure of the Mo(V) EPR-active species. Inorg. Chem. 2012;51:3409–3419. doi: 10.1021/ic201533p. [DOI] [PubMed] [Google Scholar]
- 31.Butler CS, Fairhurst SA, Ferguson SJ, Thomson AJ, Berks BS, Richardson DJ, Lowe DJ. Mo(V) coordination in the periplasmic nitrate reductase from Paracoccus pantotrophus probed by electron nuclear double resonance (ENDOR) spectroscopy. Biochem. J. 2002;363:817–823. doi: 10.1042/0264-6021:3630817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurley JK, Hazzard JT, Martínez-Júlvez M, Medina M, Gomez-Moreno C, Tollin G. Electrostatic forces involved in orienting Anabaena ferredoxin during binding to Anabaena ferredoxin:NADP+ reductase: Site-specific mutagenesis, transient kinetic measurements, and electrostatic surface potentials. Protein Sci. 1999;8:1614–1622. doi: 10.1110/ps.8.8.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aliverti A, Livraghi A, Piubelli L, Zanetti G. On the role of the acidic cluster Glu 92–94 of spinach ferredoxin I. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1997;1342:45–50. doi: 10.1016/s0167-4838(97)00079-4. [DOI] [PubMed] [Google Scholar]
- 34.Knaff DB. In: Oxygenic Photosynthesis: The Light Reactions. Ort DR, Yocum CF, editors. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1996. pp. 333–361. [Google Scholar]
- 35.Vieira BJ, Davis B. Interaction of ferredoxin with ferredoxin:NADP+ reductase: effects of chemical modification of ferredoxin. Arch. Biochem. Biophys. 1986;247:140–146. doi: 10.1016/0003-9861(86)90542-4. [DOI] [PubMed] [Google Scholar]
- 36.Hurley JK, Salamon Z, Meyer TE, Fitch JC, Cusanovich MA, Markley JL, Cheng H, Xia B, Chae YK, Medina M, Gomez-Moreno C, Tollin G. Amino-acid residues in Anabaena ferredoxin crucial to interaction with ferredoxin NADP+ reductase: Site-directed mutagenesis and laser flash photolysis. Biochemistry. 1993;32:9346–9354. doi: 10.1021/bi00087a013. [DOI] [PubMed] [Google Scholar]
- 37.Jelesarov I, De Pascalis AR, Koppenol WH, Hirasawa M, Knaff DB, Bosshard HR. Ferredoxin binding site on ferredoxin:NADP+ reductase. Differential chemical modification of free and ferredoxin-bound enzyme. Eur. J. Biochem. 1993;216:57–66. doi: 10.1111/j.1432-1033.1993.tb18116.x. [DOI] [PubMed] [Google Scholar]
- 38.Hirasawa M, Knaff DB. The role of lysine and arginine residues at the ferredoxin-binding site of spinach glutamate synthase. Biochim. Biophys. Acta, Bioenerg. 1993;1144:85–91. [Google Scholar]
- 39.Hirasawa M, de Best JH, Knaff DB. The effect of lysine- and arginine- modifying reagents on spinach ferredoxin:nitrite oxidoreductase. Biochim. Biophys. Acta, Bioenerg. 1993;1140:304–312. [Google Scholar]
- 40.Lewis DFV, Hlavica P. Interactions between redox partners in various cytochrome P450 systems: functional and structural aspects. Biochim. Biophys. Acta, Bioenerg. 2000;1460:353–374. doi: 10.1016/s0005-2728(00)00202-4. [DOI] [PubMed] [Google Scholar]
- 41.Coghlan VM, Vickery LE. Site-specific mutations in human ferredoxin that affect binding to ferredoxin reductase and cytochrome P450scc. J. Biol. Chem. 1991;266:18606–18612. [PubMed] [Google Scholar]
- 42.Pikuleva IA, Tesh K, Waterman MR, Kim Y. An additional electrostatic interaction between adrenodoxin and P450c27 (CYP27A1) results in tighter binding than between adrenodoxin and P450scc (CYP11A1)*. Arch. Biochem. Biophys. 2000;373:44–55. doi: 10.1074/jbc.274.4.2045. [DOI] [PubMed] [Google Scholar]
- 43.Xu F, Bell SG, Peng Y, Johnson EO, Bartlam M, Rao Z, Wong LL. Crystal structure of a ferredoxin reductase for the CYP199A2 system from Rhodopseudomonas palustris. Proteins: Struct., Funct., Genet. 2009;77:867–880. doi: 10.1002/prot.22510. [DOI] [PubMed] [Google Scholar]