

Abstract
Many ERα-positive breast cancers develop resistance to endocrine therapy via mutation of estrogen receptors (ER) whose constitutive activation is associated with shorter patient survival. Because there is now a clinical need for new antiestrogens (AE) against these mutant ER, we describe here our development and characterization of three chemically novel AE that effectively suppress proliferation of breast cancer cells and tumors. Our AE are effective against wild type and Y537S and D538G ER, the two most commonly occurring constitutively active ER. The 3 new AE suppressed proliferation and estrogen target gene expression in WT and mutant ER-containing cells and were more effective in D538G than in Y537S cells and tumors. Compared to WT ER, mutants exhibited ~10 to 20-fold lower binding affinity for AE and a reduced ability to be blocked in coactivator interaction, likely contributing to their relative resistance to inhibition by AE. Comparisons between mutant ER-containing MCF7 and T47D cells revealed that AE responses were compound, cell-type and ERα-mutant dependent. These new ligands have favorable pharmacokinetic properties and effectively suppressed growth of WT and mutant ER-expressing tumor xenografts in NOD/SCID-gamma mice after oral or subcutaneous administration; D538G tumors were more potently inhibited by AE than Y537S tumors. These studies highlight the differential responsiveness of the mutant ER to different AE and make clear the value of having a toolkit of AE for treatment of endocrine therapy-resistant tumors driven by different constitutively active ER.
Keywords: estrogen receptor, antiestrogens, endocrine therapy resistance, breast cancer
Introduction
Estrogen receptor α (ER)-containing breast cancers account for about three-fourths of all breast cancers, and many of these primary breast cancers are effectively treated with antiestrogens (AEs) or aromatase inhibitors (1–4). Although these endocrine therapies have been mainstays in breast cancer treatment, breast cancers often become resistant to these initial treatments and recur. Therefore, second line endocrine treatments have been developed. These include most prominently, the selective estrogen receptor modulator (SERM) and downregulator (SERD), Fulvestrant (Fulv, ICI 182,780), which is not orally active and must be administered as a large volume intramuscular injection (5). Although two new orally active SERDs have been reported recently (GDC-0810 (6) and AZD9496 (7)), they have some troubling side effects, including causing diarrhea in one-third of patients (8), which could limit their clinical implementation and suggests the need for developing other new antiestrogens.
Of the ER-positive breast cancers that recur as metastatic cancers, often many years after the initial cancer diagnosis and treatment, approximately 40% have been found to possess mutations in the ER protein that confer constitutive activity and hormone-independent growth, and are associated with shorter patient overall survival (3,9–17). These ER mutations are all in the ligand binding domain of the receptor, and most frequently they result in changes at amino acid Y537 or D538 to generate Y537S (or N) or D538G constitutively active receptors (18,19). Because there is now a clinical need for new antiestrogens (AEs) that will effectively work against wild type (WT) and constitutively active mutant ERs, we describe herein studies in which we have developed and now characterize three chemically novel AEs that can suppress the proliferation of breast cancer cells and tumors driven by wild type ER and these mutant constitutively active ERs. These AE ligands might ultimately prove to be useful in treatment of breast cancer patients with endocrine therapy-resistant tumors driven by constitutively active ERs.
Materials and Methods
Cell cultures, reagents and ligands
17β-Estradiol (E2), 4-Hydroxytamoxifen (4-OHT) and Fulvestrant (ICI 182780, Fulv) were from Sigma-Aldrich. AZD9496 was from MedChemExpress. The preparations of the antiestrogens K-07, K-09 and K-62 have been reported (20). MCF7 and T47D cells from the ATCC were maintained and cultured as described (21,22). Cell lines were authenticated at the Genetics Core of the University of Arizona. T47D cells with both mutant ERα alleles were generated by CRISPR-Cas9 technology and mutation status of the clonal cell lines was verified by genotyping as described before (21). MCF7 cells or T47D cells containing 50% mutant Y537S- or D538G-ERα and 50% wild type ERα, determined by DNA sequencing and digital drop PCR analyses as detailed previously (17,23), were generated by adenovirus associated viral infection (MCF7) or CRISPR-Cas-9 methodology (T47D) (17,21,23,24). These cells were cultured as described(17,21,23,24). Serum used in all cell cultures was premium grade fetal bovine serum (VWR Life Science Seradigm, Cat# 97068-085). Cells were used in experiments within the first five passages after thawing. Cells were sometimes transferred to phenol-red free tissue culture media supplemented with 5% charcoal stripped FBS for 5 days so as to be in an estrogen-depleted condition before use in some experiments as noted. All cells were tested for mycoplasma using Real-Time PCR Mycoplasma Detection Kit (Akron Biotech, Boca Raton, FL).
Cell proliferation assay
WST-1 assay (Roche, Basel, Switzerland) was used to quantify cell viability as described (25). Absorbance was measured at 450 nm using a VICTOR X5 PerkinElmer 2030 Multilabel Plate Reader. All assays were performed in triplicate.
In-cell western and western blot assays
Cells were cultured in 96-well plates at 3000 cells/well, and treated with compound for 24 h. Cells were washed twice in PBS, fixed with 4% formaldehyde (Fisher Scientific) solution in PBS, permeabilized in 0.1% Triton X-100 in PBS, blocked with Odyssey Blocking Buffer (LI-COR), and incubated with rabbit HC-20 ERα antibody (Santa Cruz, Cat# SC-543) or mouse F10 ERα antibody (Santa Cruz, Cat# SC8002) at 4C overnight. Both IRDye 800 CW goat anti-rabbit secondary antibody (LI-COR, Cat# 926-32211) and Cell Tag 700 (LI-COR, Cat# 926-41090) were diluted (1:600) for incubation with cells. Plates were washed and ERα staining signals were quantified and normalized with Cell Tag signals (to control for any differences in cell number per well) using a LI-COR Odyssey infrared imaging system. Fold changes of ERα protein levels were calculated relative to the vehicle-treated WT samples. Results are the average ±SD from at least three independent experiments, each performed with 4 wells per treatment condition. Western immunoblots for GREB1 protein used rabbit polyclonal antibody (Sigma, HPA024616 at 1:1000 dilution) and for ERα used mouse monoclonal F10 antibody (Santa Cruz Cat# SC-8002 at 1:1000 dilution). β-actin was detected with mouse monoclonal antibody (Sigma-Aldrich at 1:10,000 dilution).
Immunohistochemistry
Immunohistochemistry was performed on paraffin-embedded tissue sections as before (26). A mouse monoclonal anti-human ERα antibody (Novacastra, Cat# NCL-L-ER-6F11) was used at 1:300 dilution.
RNA isolation and real-time PCR
Total RNA was isolated using TRIzol (Invitrogen) and reverse transcribed using MMTV reverse transcriptase (New England BioLabs). Real-time PCR was performed using SYBRgreen PCR Master Mix (Roche) as described (27). Relative mRNA levels of genes were normalized to the housekeeping gene 36B4, and fold change calculated relative to the vehicle-treated samples. Results are the average ±SD from at least two independent experiments carried out in triplicate.
Receptor ligand binding and coregulator interaction assays
Binding of E2 to WT and mutant ERs was determined by direct Scatchard binding assay using tritiated E2 (28). Binding of other ligands was determined by a competitive radiometric assay with tritiated E2 and increasing concentrations of ligand (28).
A time-resolved Fluorescence Resonance Energy Transfer (FRET) assay was used to determine the ability of AE compounds to reduce the interaction of WT or mutant ERs with the coactivator SCR3/AIB1, which is often amplified and expressed at high levels in ER-positive breast cancers. The assay used site-specific labeled biotin-streptavidin/terbium WT, Y537S or D538G ER ligand binding domain, fluorescein-labeled SRC3 nuclear receptor interaction domain, and E2 and increasing concentrations of K-07, K-09, K-62 or trans-hydroxytamoxifen, and FRET was monitored as described (11,29).
Pharmacokinetic studies
All experiments involving animals were conducted in accordance with National Institutes of Health (NIH) standards for the care and use of animals, with protocols approved by the University of Illinois IACUC. The pharmacokinetics of compounds K-09, K-07 and K-62 were monitored after single dose administration into female CD1 mice (7–9 weeks of age) via sc injection or oral gavage. For sc injection, each compound was dissolved in DMSO and then mixed with corn oil for a total injection volume of 100 μl (10% DSMO + 90% corn oil) per mouse. For oral gavage, compounds were administered in a 200μl formulation of 9/0.5/0.5/90 parts of PEG400/Tween80/Povidone/0.5% Carboxymethylcellulose. Multiple plasma samples were collected from each mouse (n=4 for each experiment) over the course of 48 h after compound administration. Compounds were quantified by LC-MS/MS at the University of Illinois Metabolomics Core Facility. The data were fitted to a non-compartment model.
In vivo breast cancer xenograft studies
For examination of the growth of tumors containing Wild Type ER, female NOD/SCID-gamma (NSG) mice were ovariectomized at 7–8 weeks of age, and 2 weeks after ovariectomy, animals were supplemented with 0.36 mg E2-pellets (60-day release, Innovative Research of America) to support ER+ tumor growth. Cell suspensions of wild type MCF7 cells (2 x 106 cells/mouse) were injected orthotopically into the right axial mammary gland. When tumors reached 100–150 mm3 in size, mice were randomized and received compound or control vehicle (corn oil) by daily sc injection or oral gavage (PEG400/PVP/Tween/CMC as Veh) per day, for approximately 26 days, until Veh-treated tumors grew to ca. 1000 mm3. Tumor volume (length × width2/2) and animal body weights were monitored over time.
For examination of the growth of tumors containing mutant ERs, female NSG mice were ovariectomized but received no E2 pellets, to mimic the low estrogen environment of postmenopausal women. At 3 weeks after ovariectomy, cells were injected orthotopically into the axial mammary gland. Animals received daily sc injection or oral gavage of vehicle or compound, and tumor growth and animal body weights were monitored over time.
Results
Structurally Novel Antiestrogens Suppress Growth of Breast Cancer Cells with Constitutively Active Mutant ERα
We first examined the effects of three new antiestrogens developed in our laboratory (Fig. 1A) in T47D cells containing all wild type ER or all mutant ER (Y537S or D538G). The mutant ERs were introduced by homology directed repair using CRISPR-Cas9 technology (21). The mutants examined, Y537S and D538G, are the two most commonly occurring mutant ERα’s in humans with therapy-resistant breast cancers. The three antiestrogens have an adamantyl core and were selected through structure-activity relationship studies on breast cancer cells with wild type ER(20). Hence, the current studies are the first to examine their effects on cells and tumors with growth driven by constitutively active ERs.
Fig. 1. Proliferation of T47D cells harboring homozygous mutant ERα (introduced by CRISPR-Cas9) is suppressed by antiestrogens (AEs).
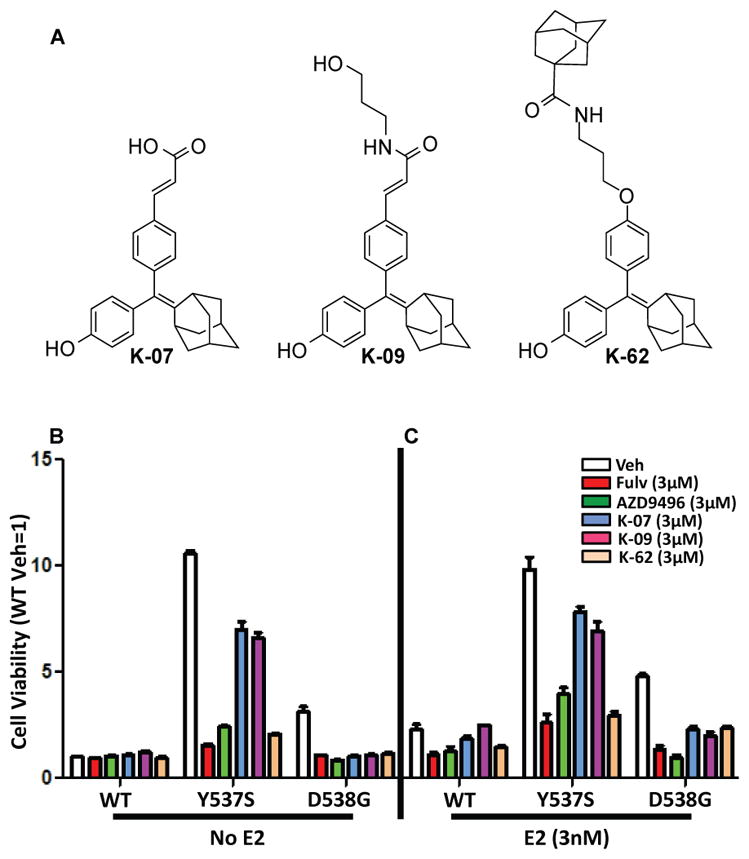
(A) Structures of the 3 AEs studied. T47D cells with WT-ERα, Y537S-ERα or D538G-ERα were cultured in (B) E2-deprived conditions or (C) with E2 (3×10−9 M) and compounds at 3×10−6 M for 6 days to evaluate the impact of compounds on cell viability. Values are the mean ± SD of 3 determinations from 3 separate experiments.
As seen in Fig. 1B, cells containing the Y537S and D538G ER mutants showed high constitutive activity in the absence of estrogen, having 11-times and four-times higher proliferative activity, respectively, than that of cells containing wild type (WT) ER. Treatments with the known antiestrogens fulvestrant (Fulv) or AZD9496 resulted in marked suppression of this constitutive activity in both mutants. All three of our new compounds fully suppressed proliferative activity of the D538G cells, whereas in Y537S cells, good suppression was effected by K-62, but to only a more limited extent by the K-07 and K-09 compounds. In the presence of E2 (Fig. 1C), proliferation of wild type and D538G cells was increased, whereas cells with Y537S ER proliferated rapidly in the absence of added E2 and showed no further increase with E2. Again, all five compounds suppressed proliferation of the mutant ER-containing cell lines, with D538G cell proliferation being more effectively suppressed by the antiestrogens than Y537S cell proliferation. In cells with wild type and Y537S ER, K-62 was as inhibitory as Fulv and AZD, with K-07 and K-09 being less effective anti-proliferative agents.
Because the studies in Fig. 1 were conducted at only one high concentration of each compound (3 μM), we carried out proliferation dose-response studies with each of these compounds in the three cell types (i.e., containing WT or the two mutant ERs). As seen in Fig. 2 (panels A–E), the mutants again showed strong constitutive proliferative ER activity, whereas E2 increased WT ER cell proliferation to that of the constitutively active mutant ER cells. With all five compounds, dose-dependent inhibition of cell proliferation revealed that mutant Y537S-ER and D538G-ER cells showed more resistance to suppression by all of the antiestrogens, with Y537S cells requiring the highest concentrations of antiestrogens to bring about growth inhibition. Approximately 10–100 times higher concentrations of compounds were needed for equal suppression of growth in Y537S cells compared to D538G cells. Comparisons of cell proliferation conducted in WT and mutant ERα-expressing T47D cells in estrogen-replete conditions (i.e., full medium with 5% fetal bovine serum) again showed high resistance of Y537S ER-containing cells to growth suppression by all antiestrogens (Fig. S1, panels A–E).
Fig. 2. Dose-dependent inhibition of T47D cell proliferation and gene expression by AEs: D538G-ERα is more effectively suppressed by AEs than Y537S-ERα.

(A–E) T47D cells with WT- ERα, Y537S-ERα or D538G-ERα were cultured under estrogen-deprived conditions (in phenol red-free medium with 5% charcoal dextran-treated serum). They were treated with ligands at the concentrations indicated (3×10−11 to 3×10−6 M), and cell proliferation was monitored after 6 days. Values are mean ± SD of 3 determinations from 3 separate experiments. (F, G) Effects of E2 and ligands on expression of the ER target genes GREB1 and PGR in T47D cells were monitored after treatments with Veh, E2 and ligands for 24 h, followed by RNA extraction and qPCR analysis. Black horizontal line indicates mRNA expression in Veh-treated WT T47D cells (set as 1), and dashed horizontal line indicates mRNA expression level in E2-treated cells. Values are the mean ± SD of 3 determinations from 2 separate experiments.
Reduced Binding Affinities and Lower Potencies in Reversing Coactivator Binding by Antiestrogen Ligands for Mutant-ERs vs. WT ER
Table 1A summarizes the binding affinity of these ligands to WT, Y537S and D538G ERs. Of note, antiestrogens bound about 10–40x less well to these mutant ERs vs. WT ER. E2 was also found to bind 7x less well to the mutants. These differences in ligand binding to the mutant ERs could in part explain their relative resistance to inhibition of proliferation by the AE compounds, but not the greater resistance of the Y537S mutant compared to the D538G mutant. In assays monitoring the reversal of coactivator SRC3/AIB1 binding to WT and mutant ERs, we again found that 10–50x higher concentrations of AE ligands were required with mutant ERs than with WT ER, but in this assay the interaction of SRC3 with the Y537S mutant was more difficult to reverse than with the D538G mutant (Table 1B and Fig. S2), better reflecting the results from the proliferation inhibition assays.
Table 1.
| Table 1A. Ligand Binding to Wild Type (WT) and Mutant ERs a | |||
|---|---|---|---|
| Ligand | Ki (nM) [Fold Increase Over WT] |
||
| WT-ER | Y537S-ER | D538G-ER | |
| E2 (Kd) (nM) | 0.22 ± 0.11 | 1.40 ± 0.54 [6.4x] | 1.77 ± 0.66 [8.0x] |
| Trans-hydroxytamoxifen | 0.12 ± 0.01 | 2.64 ± 0.4 [22.0x] | 3.28 ± 0.7 [27.3x] |
| Fulvestrant | 0.13 ± 0.03 | 3.68 ± 0.77 [28.3x] | 5.06 ± 1.16 [38.9x] |
| AZD 9496 | 0.19 ± 0.05 | 5.20 ± 1.16 [26.9x] | 7.52 ± 1.9 [39.0x] |
| K-07 | 0.30 ± 0.01 | 3.94 ± 1.10 [13.1x] | 4.60 ± 1.28 [15.3x] |
| K-09 | 0.45 ± 0.07 | 2.88 ± 0.37 [6.4x] | 7.00 ± 1.36 [15.6x] |
| K-62 | 0.47 ± 0.02 | 5.35 ± 1.37 [11.4x] | 10.31±2.93 [21.9x] |
| Table 1B. Reversal of Coactivator SRC3/AIB1 Binding to Wild Type (WT) and Mutant ERs | ||||
|---|---|---|---|---|
|
| ||||
| WT-ER | Y537S-ER | D538G-ER | ||
|
| ||||
| E2 Priming Conc. (nM) b | 9 | 0.3 | 2.5 | |
|
| ||||
| E2 (Kd) (nM) | 0.22 ± 0.11 | 1.40 ± 0.54 | 1.77 ± 0.66 | |
|
| ||||
| Cheng-Prusoff Correction: 1 + ([E2]/Kd) c | 42 | 1.2 | 2.4 | |
|
| ||||
| Ligand |
Ki (nM) [Fold Increase Over WT] |
|||
|
| ||||
| Trans-hydroxytamoxifen | IC50 nM d | 11 | 3.4 | 6.0 |
| Ki nM e | 0.26 | 2.8 | 1.3 | |
| [fold change from WT] f | [1x] | [11x] | [5x] | |
|
| ||||
| K-07 | IC50 nM d | 32 | 49 | 38 |
| Ki nM e | 0.76 | 41 | 16 | |
| [fold change from WT] f | [1x] | [54x] | [21x] | |
|
| ||||
| K-09 | IC50 nM d | 50 | 29 | 26 |
| Ki nM e | 1.2 | 24 | 11 | |
| [fold change from WT] f | [1x] | [20x] | [9.2x] | |
|
| ||||
| K-62 | IC50 nM d | 54 | 53 | 36 |
| Ki nM e | 1.3 | 44 | 16 | |
| [fold change from WT] f | [1x] | [34x] | [12x] | |
Binding of E2 was determined by direct Scatchard binding assay using tritiated E2 (28). Binding of other ligands was determined by a competitive radiometric assay using tritiated E2 and increasing concentrations of ligand (28).
The concentration of E2 needed to recruit SRC3 binding to about 50% of maximum to each receptor. It differs from WT vs. mutant ERs because of different levels of constitutive activity and E2 binding affinity of these ERs.
This factor is required to convert IC50 values to Ki values.
IC50 values are read from the inhibition curves shown in Fig. S2.
Ki values are determined by dividing the IC50 values by the Cheng-Prusoff correction factor.
Fold change from WT is determined for each AE by dividing the Ki value for the mutant-ER by the Ki value for WT-ER.
The Constitutive Gene Expression Activity of Mutant ERs Is Suppressed by Novel Antiestrogen Compounds
We observed constitutive expression of the ER target genes GREB1 and PGR in cells with mutant ERs, and consistent with our findings of anti-proliferative activities of Fulv and our three new compounds, we found that Fulv, K-07, K-09 and K-62 effectively suppressed the expression of these genes in a dose-dependent manner (Fig. 2F, G). Stimulation was most effectively turned off in the WT cells, with both GREB1 and PGR gene expression also being suppressed in both mutants by all compounds; however, higher concentrations of AEs were needed for suppression of gene expression in mutant ER- versus WT ER-containing cells. The lower potency of the AEs in suppressing gene expression in the mutant ERs is again consonant with their lower binding affinity and reduced potency in reversing coactivator binding to the mutants than WT ER (Table 1).
Antiestrogenic Compounds Differentially Downregulate WT and Mutant ERα Proteins in Cells
We next compared the abilities of the compounds to elicit downregulation of WT ERα and mutant ERα proteins in the T47D cells. Levels of WT- and D538G-ERα were markedly reduced by Fulv and by our three antiestrogens, whereas the Y537S-ERα protein was very resistant to downregulation by all ligands (Fig. 3A–E). Of note, 4-OHT did not downregulate mutant or WT-ERα (Fig. 3B), although it reduced proliferation of WT and mutant ER-expressing cells (Fig. 2B). In fact, 4-OHT reproducibly and dose-dependently upregulated Y537S ERα levels, implying a possible change in the dynamics of turnover of this particular mutant ER protein.
Fig. 3. Differences in the ability of compounds to induce the down-regulation of WT or mutant ERα protein in cells.

T47D cells were treated with Vehicle Control, E2 or compounds (A, Fulv; B, 4-OHT; C, K-07; D, K-09; E, K-62) alone for 24 h at the concentrations indicated, and cells were subjected to in cell Western (ICW) blot analysis for evaluation of ERα protein levels.
Comparison of Ligand Effects in T47D and MCF7 Cells Containing Both WT and Mutant ERs
We also carried out comparison studies using MCF7 cells and T47D cells containing 50% mutant ER and 50% WT ER (Fig. S3). In these cells, we found that high concentrations of Fulv, 4-OHT, and K-07, K-09, and K-62 were again needed to obtain suppression of cell proliferation, similar to that observed in the T47D cells containing only mutant ERs (Fig. S1). This suggests that the mutant ERα is dominant in determining cell phenotypic behavior when both receptors are present, as also seems to be the case in human metastatic breast tumors with these mutant ERs. Of interest, some differences were seen in ligand potency and efficacy when monitored in MCF7 or T47D cells. Hence, K-07 and K-09 worked more effectively as growth suppressors in MCF7 cells with mutant ERs, whereas Fulv was somewhat more potent in T47D cells with mutant ERs (Figs. S3A,B). Likewise, our compounds and Fulv could reduce the ER protein level in these MCF7 and T47D cells, with Fulv generally eliciting a somewhat greater magnitude of down-regulation (Figs. S4A,B).
When expression of estrogen target genes GREB1 and PGR was assessed in MCF7 and T47D cells with 50% mutant and 50% WT ER, gene expression was constitutively high (Fig. S5) and increased only little with added E2, similar to the response in cells with only mutant ER. Gene expression was generally more fully suppressed by compounds in MCF7 cells versus T47D cells. Of note, the D538G ER was more fully inhibited than Y537S ER, regardless of the cell background. These findings suggest that beyond the status of ER itself (WT or mutant), other genomic and cellular alterations in the different cell backgrounds may well contribute to differences in responsiveness to treatment with different antiestrogens.
Pharmacokinetic Properties of Antiestrogens K-07, K-09 and K-62
Prior to in vivo xenograft tumor studies, we monitored the pharmacokinetic properties of antiestrogens K-07, K-09, and K-62 after sc and oral administration in mice (Fig. 4A–F). Overall, K-07 showed the most optimal PK properties by both sc and oral routes, having the highest blood levels and a very long terminal half-life in blood (t½= 14.0 h after sc injection or t½= 97.2 h after oral administration). Although K-09 and K-62 had long half-lives after sc injection, their blood levels were much lower compared with those of compound K-07 (note changes in y-axis). Because of its especially high blood levels and long half-life after oral administration, we also examined the oral efficacy of K-07 in subsequent tumor growth experiments.
Fig. 4. Pharmacokinetics and half-lives of new AEs in mice after sc or oral administration.

(A) Pharmacokinetics (PK) of K-07 after single dose administration via sc injection (20mg/kg) or (B) oral gavage (20mg/kg). (C) PK of K-09 after single dose sc injection at 20 mg/kg or (D) oral gavage at 40 mg/kg. (E) PK of K-62 after single dose sc injection at 40 mg/kg and (F) oral gavage at 20 mg/kg. After compounds were administered, multiple plasma samples were collected from each mouse (n=4 for each experiment) over the course of 48 h. Compounds were quantified using LC-MS/MS. The data were fitted to a non-compartment PK model.
Structurally Novel Compounds Inhibit Tumor Growth of Wild Type and Mutant ER-containing Breast Cancer Cells In Vivo
Because our new compounds suppressed cell proliferation and ER target gene expression, we investigated the ability of K-07, K-09 and K-62 to suppress growth of ER-positive breast cancer xenografts in NSG mice. Studies were first conducted in MCF7 xenografts containing wild type ERα. We did not study T47D xenografts containing wild type ERα because we found that wild type T47D cells with E2 supplementation and mutant ER T47D cells without E2 pellets formed only very small tumors, whereas MCF7 cells formed much larger and faster-growing tumors. Mice receiving E2 pellet supplementation were injected with wild type MCF7 cells orthotopically into the mammary fat pad, and when tumors reached 100–150 mm3 in size, animals were randomized to groups and treated with compounds or Fulv administered daily sc at 80 mg/kg per day (Fig. 5). All compounds reduced tumor growth, with K-09 and K-62 being as effective as Fulv, and K-07 being the most effective in growth suppression (Fig. 5A). The favorable PK profile of K-07 likely underlies the observation that K-07 was most efficacious in inhibiting tumor growth. All compounds greatly reduced expression of the GREB1 and PGR genes monitored in tumors harvested at day 26 (Fig. 5B).
Fig. 5. New AEs show good growth suppression of MCF7 xenograft tumors and inhibition of estrogen target gene expression.

NSG mice were supplemented with E2 (0.36 mg, 60-day release) pellets, injected with WT MCF7 cells to generate xenograft tumors, and dosed with 80 mg/kg of Fulv, K-07, K09 or K-62 by daily sc injection. (A) Tumor volume was monitored over time (2-way ANOVA, Bonferroni post-test, ****, P<0.0001, n=8–9 per group). (B) Harvested tumors were analyzed by qPCR for GREB1 and PGR RNA levels (1-way ANOVA, Tukey post-test, n=8–9/group). (C) E2-supplemented mice bearing MCF7 tumors were dosed daily with vehicle or 80 mg/kg of K-07 by oral gavage. Tumor volumes of Veh and K-07 treated tumors were monitored (2-way ANOVA, Bonferroni post-test, ****, P<0.0001, n=8 per group). (D) After 28 days, tumors were harvested at 24 h after the last control Veh or K-07 oral treatment for gene expression analysis (T-test, ****, P<0.0001, n=8 per group) and (E) Western blot examination of GREB1 and ERα protein in individual tumors. β-actin serves as the loading control. (F) Quantitation of ERα and GREB1 protein in Veh and K-07 treated tumors. (T-test, ****, P<0.0005 n=5 Veh and n=6 K-07 tumors).
K-07 is a Potent Oral Inhibitor of Tumor Progression In Vivo
Orally administered K-07 very effectively reduced tumor growth within a few days of the start of treatment (Fig. 5C). ER target gene (GREB1, PgR) expression was also fully suppressed in tumors harvested from these animals (Fig. 5D). Analysis of GREB1 and ERα protein showed almost complete loss of GREB1 protein expression and a 51% reduction of ERα in the tumors of K-07 treated animals (Fig. 5E, F). Although K-07, K-09 and K-62 by sc and K-07 given by oral routes suppressed tumor growth substantially in these animals supplemented with E2 pellets, there was no effect on animal body weight compared to Veh control animals over the time course of the treatments (Fig. S6A,B).
Y537S-ER and D538G-ER-containing Cells Form Tumors in the Absence of Estrogen and are Arrested by K-07 Treatment
To mirror the low estrogen environment in postmenopausal women, tumor xenograft studies with Y537S and D538G-containing MCF7 cells were conducted in ovariectomized NSG mice in the absence of any added E2. Y537S and D538G tumors grew well under these conditions (Fig. 6), and growth of these constitutively active tumors was acutely arrested by sc treatment with K-07, as effectively as by Fulv (Fig. 6A, B). Notably, oral K-07 was also very effective in arresting growth of the mutant ER tumors (Fig. 6C), and D538G tumors were more fully suppressed by K-07 treatment compared to Y537S tumors, consistent with the greater resistance of Y537S cells to antiproliferative effects of antiestrogens that we found in cell cultures in vitro. Expression of estrogen-regulated genes, monitored in the small mutant-ER-containing tumors at the end of the study, was almost fully suppressed by K-07 in both Y537S and D538G tumors (Fig. 6D,E). Analysis of ERα protein by Western immunoblot in the small tumors harvested at the end of the study revealed a 60% and 75% decrease in ERα in Y537S and D538G tumors, respectively (Fig. 6F). This substantial decrease in ERα in Y537S and D538G tumors receiving K-07 vs. vehicle treatment was also seen by immunohistochemistry of tumors as a marked decrease in nuclear ERα staining (Fig. 6G).
Figure 6. Y537S ERα- and D538G ERα-containing tumors grow in the absence of estrogen and are arrested by K-07 treatment.

NSG mice were ovariectomized and 3 weeks later received MCF7 cells containing half Y537S ER or D538G ER and half wild type ER. (A, B) Mice then received daily sc injection with Vehicle or 80 mg/kg of K-07 or Fulv, and tumor volumes were monitored over time (2-way ANOVA, Bonferroni post-test, ****, P<0.0001, n=8 per group). (C) Mice received daily treatment with oral vehicle or oral K-07 at 80 mg/kg, and tumor volumes were monitored over time (2-way ANOVA, Bonferroni post-test, P<0.0001, n=8 per group). (D, E) Tumors harvested at day 26 were analyzed for expression of the estrogen target genes GREB1 and PGR. In D, 1-way ANOVA, Tukey post-test, n = 8 per group. In E, T-test, ****, P<0.0001, n=8 per group. (F) Harvested tumors were also analyzed for ERα protein by Western blot of tumor lysates [some D538G tumors were too small for Western analyses so only 3 tumors are shown here]; and by (G) immunohistochemistry for ERα in tumor tissue sections.
Discussion
Our studies show that three antiestrogens with novel chemical structures have efficacy in suppressing growth of breast cancer cells and tumors containing WT ERs and that, at higher concentrations, these compounds can also inhibit ER-regulated gene expression and proliferation of breast cancer cells containing constitutively active mutant ERs. K-07 had the most optimal pharmacokinetic profile, and it was the most effective in suppression of WT and Y537S and D538G mutant tumor growth in vivo.
The studies provide new insights about mutant ERs that drive hormone-independent constitutive activity. Of note, we observed that the nature of the mutant ER (D538G or Y537S), the cell background (T47D or MCF7), and the chemical structure of the antiestrogens (K-07 vs. K-09 vs. K-62 vs. Fulv) all affected response to AE ligands. Thus, these mutant ERs showed differential responsiveness to chemically distinct AEs, with cells containing the mutant Y537S-ER being more resistant to the AEs compared to the mutant D538G receptor and requiring higher compound concentrations for growth suppression. Other key regulators in breast cancer, such as the histone methyl transferase EZH2, also have various activating mutations that differ in their sensitivities to different EZH2 inhibitors (30). In our studies, the cell background was also very crucial. Thus, in T47D cells, Y537S elicited a more endocrine-resistant phenotype, whereas in the MCF7 cell background the Y537S and D538G mutant ERs were more similar to each other in their response to antiestrogens. This suggests that cell context, including alterations in genomic and cell signaling pathways, may work with ERs to confer different cell behaviors and responsiveness or resistance to treatment with different endocrine agents (23,31,32). This is not surprising, since it is known, for example, that among other differences, MCF7 and T47D cells carry different mutant forms of PI3K. Also, GATA3, an important factor for ER activity, is often mutated in breast cancers (16) and could influence responsiveness of WT and mutant ERs to antiestrogens.
Our studies also document that the mutant ER allele fraction is crucial in the extent of endocrine treatment resistance observed. In fact, T47D cells with both alleles homozygous for the mutant ER showed greater resistance to antiestrogens than did cells with 50% mutant and 50% wild type ER. Notably however, even 50% mutant ER conferred a dominant antiestrogen-resistant phenotype. This is of importance because metastatic breast cancers usually contain a mixture of mutant and wild type ERs (1,3,14–19,32).
The pharmacokinetic properties of the antiestrogens proved to be very important in their tumor suppressive efficacies in vivo. K-07 displayed the best pharmacokinetic properties by either sc or oral routes. Despite the fact that all three antiestrogens showed good anti-proliferative and target gene inhibitory activities in cells in culture, K-07, which displayed the best pharmacokinetic properties, was the most effective growth inhibitor in breast tumor xenografts in vivo.
It is now recognized that approximately 40% of ER-positive breast cancers that become resistant to endocrine treatment and recur, contain ER mutations (1,3,17–19). Most of these mutations are found in the C-terminal transactivation domain in the ligand-binding domain of the ER and result in changes at amino acids Y537, D538, L536, P535, or V534 and also at E380. Several large analyses have shown that the two most common mutations are at Y537 (changing Y to S, but also less commonly to N or C) and at D538 (always changed to G). These arise from single nucleotide changes in the codons for these amino acids, resulting in the alteration of one amino acid. X-ray crystallography and molecular modeling have shown that Y537 and D538 are present at key locations in the activation function-2 region of the ligand binding domain that determine the three-dimensional structure of helix 12 of the ER that is key in interactions with coregulators. These changes in ER structure (11,33) result in ligand-independent interaction with coactivators that is normally seen in wild type receptor only in the presence of estrogenic hormones (34–37). Notably, we report here that these changes in ER also reduce the receptor binding affinity for antiestrogen ligands. This reduced binding affinity, shown in Table 1A, likely contributes to the relative resistance of these ER mutant cells to antiestrogens. However, since Y537S-ER was always more resistant to the suppressive effects of ligands compared to D538G-ER even though both mutant ERs showed similar reductions in their binding affinities for antiestrogens, other factors are clearly involved, such as the more greatly reduced potency of the AEs in suppressing coregulator interaction to Y537S-ER than to D538G-ER (Table 1B). As we and others (38) have observed, tumors with mutant Y537S ERs are more antiestrogen resistant than those with D538G ERs.
When breast cancer patients no longer respond to treatment with tamoxifen or aromatase inhibitors, second line treatment with Fulvestrant is often used (39). However, Fulvestrant is not orally available, so large volumes of Fulvestrant in an oil-containing excipient are administered intramuscularly, which can be painful and is generally not liked by patients (4). Therefore, there is considerable interest in the development of new orally active antiestrogens for treatment of these recurrent, usually metastatic, breast cancers. Recently, two orally available antiestrogens have been reported, AZD9496 from AstraZeneca and GDC-0810 from Genentech/Seragon (6,7). While effective, each is not fully optimal, and some troubling side effects that may limit their clinical utility have been noted (8). RAD-0910 (40,41) and boronic acid derivatives of Fulvestrant and GW7604 (42,43) show promise, but clinical evaluations are at very early stages. Thus, there is an unmet need for orally active antiestrogens with an improved clinical profile and reduced side effects. Compound K-07 appears to be a potential alternative orally effective antiestrogen. Although in our studies no impact on overall animal health was observed with this compound, or with K-09 or K-62, further investigations of ultimate safety and clinical effectiveness will be needed with K-07 or related antiestrogens.
Because clinical studies have shown that patients with recurrent ER-positive breast cancer can respond to second and often third-line endocrine treatments, having a toolkit of new antiestrogens of different chemical classes should increase the beneficial options for such patients. It could bring therapeutic advances and result in inhibitors better matched for effectiveness in subsets of patients with breast tumors carrying differing ER mutations. We found differences in antiestrogen response of MCF7 and T47D cells containing mutant ERs, implying that different AEs may work better in different cell contexts. In this regard, it is noteworthy that in the BOLERO-2 Trial, only patients with breast tumors carrying D538G ER mutations, but not Y537S mutations, showed increased progression free survival when treated with the mTOR inhibitor everolimus plus the aromatase inhibitor exemestane, indicating that patients with breast cancers driven by different mutant ERs may benefit differentially from certain cancer treatments (9,12). Patients with ER-positive tumors containing breast cancer cells with mutant ERs present in different tumor microenvironments may also differentially benefit, since it is now appreciated that breast tumors are quite heterogeneous in their cellular compositions (44–46), which can impact clinical outcomes of patients to endocrine treatments with antiestrogens.
Supplementary Material
Acknowledgments
Financial Support This study was funded by Grants from the Breast Cancer Research Foundation (BCRF-16-083 to B.S. Katzenellenbogen and BCRF 082854 to J.A. Katzenellenbogen), the National Institutes of Health (DK015556 to J.A. Katzenellenbogen; DK071909 to D.J. Shapiro; CA132022 and DK077085 to K.W. Nettles), the Avon Foundation (to B.H. Park), the Meloche Memorial Funds (to S. Oesterreich), and the Department of Defense (BCRP W81XWH-13 to D.J. Shapiro).
We thank Kathryn Carlson for excellent assistance in some of the assays.
Abbreviations
- AE
antiestrogen
- AF2
activation function 2
- ER
estrogen receptor α
- E2
17β-estradiol
- ICW
in cell western
- KD
knockdown
- KI
knock in
- PK
pharmacokinetics
- SERM
selective estrogen receptor modulator
- SERD
selective estrogen receptor down-regulator
- SRC3/AIB1
steroid receptor coactivator 3/amplified in breast cancer 1
Footnotes
Ethical Approval All animal experiments were performed in accordance with institutional protocols approval by the University of Illinois IACUC and the National Institutes of Health Guide for the Care and Use of Laboratory Animals guidelines. This article does not contain any studies with human participants performed by any of the authors.
References
- 1.Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12:573–83. doi: 10.1038/nrclinonc.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jeselsohn R, De Angelis C, Brown M, Schiff R. The Evolving Role of the Estrogen Receptor Mutations in Endocrine Therapy-Resistant Breast Cancer. Curr Oncol Rep. 2017;19:35. doi: 10.1007/s11912-017-0591-8. [DOI] [PubMed] [Google Scholar]
- 3.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clinical Cancer Research. 2014;20:1757–67. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tryfonidis K, Zardavas D, Katzenellenbogen BS, Piccart M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat Rev. 2016;50:68–81. doi: 10.1016/j.ctrv.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Robertson JF. Fulvestrant (Faslodex) -- how to make a good drug better. The oncologist. 2007;12:774–84. doi: 10.1634/theoncologist.12-7-774. [DOI] [PubMed] [Google Scholar]
- 6.Joseph JD, Darimont B, Zhou W, Arrazate A, Young A, Ingalla E, et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. Elife. 2016 Jul 13;5:5. doi: 10.7554/eLife. pii: e15828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, et al. AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models. Cancer research. 2016;76:3307–18. doi: 10.1158/0008-5472.CAN-15-2357. [DOI] [PubMed] [Google Scholar]
- 8.Dickler M, Bardia A, Mayer I, Winer E, Rix P, Hager J, et al. A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader GDC-0810 (ARN-810) in postmenopausal women with estrogen receptor+HER2-, advanced/metastatic breast cancer. In Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015. Abstract nr CT231. [Google Scholar]
- 9.Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016;2:1310–5. doi: 10.1001/jamaoncol.2016.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu D, Paoletti C, Gersch C, VanDenBerg DA, Zabransky DJ, Cochran RL, et al. ESR1 Mutations in Circulating Plasma Tumor DNA from Metastatic Breast Cancer Patients. Clinical Cancer Research. 2016;22:993–9. doi: 10.1158/1078-0432.CCR-15-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife. 2016 Feb 2;5:5. doi: 10.7554/eLife. pii: e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuqua SA, Rechoum Y, Gu G. ESR1 Mutations in Cell-Free DNA of Breast Cancer: Predictive “Tip of the Iceberg”. JAMA Oncol. 2016;2:1315–6. doi: 10.1001/jamaoncol.2016.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu G, Fuqua SA. ESR1 Mutations in Breast Cancer: Proof-of-Concept Challenges Clinical Action. Clin Cancer Res. 2016;22:1034–6. doi: 10.1158/1078-0432.CCR-15-2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell reports. 2013;4:1116–30. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nature genetics. 2013;45:1446–51. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, et al. Sensitive Detection of Mono- and Polyclonal ESR1 Mutations in Primary Tumors, Metastatic Lesions, and Cell-Free DNA of Breast Cancer Patients. Clin Cancer Res. 2016;22:1130–7. doi: 10.1158/1078-0432.CCR-15-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J Clin Oncol. 2016;34:2961–8. doi: 10.1200/JCO.2016.67.3061. [DOI] [PubMed] [Google Scholar]
- 19.Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 2016;7:11579. doi: 10.1038/ncomms11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Min J, Guillen VS, Sharma A, Zhao Y, Ziegler Y, Gong P, et al. Adamantyl Antiestrogens with Novel Side Chains Reveal a Spectrum of Activities in Suppressing Estrogen Receptor Mediated Activities in Breast Cancer Cells. Journal of medicinal chemistry. 2017;60:6321–36. doi: 10.1021/acs.jmedchem.7b00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao C, Livezey M, Kim JE, Shapiro DJ. Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor alpha Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Scientific reports. 2016;6:34753. doi: 10.1038/srep34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sengupta S, Schiff R, Katzenellenbogen BS. Post-transcriptional regulation of chemokine receptor CXCR4 by estrogen in HER2 overexpressing, estrogen receptor-positive breast cancer cells. Breast Cancer Res Treat. 2009;117:243–51. doi: 10.1007/s10549-008-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017;19:60. doi: 10.1186/s13058-017-0851-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott GK, Chu D, Kaur R, Malato J, Rothschild DE, Frazier K, et al. ERpS294 is a biomarker of ligand or mutational ERalpha activation and a breast cancer target for CDK2 inhibition. Oncotarget. 2016 Oct 18; doi: 10.18632/oncotarget.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong P, Madak-Erdogan Z, Flaws JA, Shapiro DJ, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen receptor-alpha and aryl hydrocarbon receptor involvement in the actions of botanical estrogens in target cells. Molecular and cellular endocrinology. 2016;437:190–200. doi: 10.1016/j.mce.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y, Gong P, Chen Y, Nwachukwu JC, Srinivasan S, Ko C, et al. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Science translational medicine. 2015;7:271ra9. doi: 10.1126/scitranslmed.3010626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y, Chen Y, Kuang Y, Bagchi MK, Taylor RN, Katzenellenbogen JA, et al. Multiple Beneficial Roles of Repressor of Estrogen Receptor Activity (REA) in Suppressing the Progression of Endometriosis. Endocrinology. 2016;157:900–12. doi: 10.1210/en.2015-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 29.Jeyakumar M, Carlson KE, Gunther JR, Katzenellenbogen JA. Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J Biol Chem. 2011;286:12971–82. doi: 10.1074/jbc.M110.205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014;25:1729–35. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Science translational medicine. 2015;7:313ra182. doi: 10.1126/scitranslmed.aac7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nature chemical biology. 2008;4:241–7. doi: 10.1038/nchembio.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazennec G, Ediger TR, Petz LN, Nardulli AM, Katzenellenbogen BS. Mechanistic aspects of estrogen receptor activation probed with constitutively active estrogen receptors: correlations with DNA and coregulator interactions and receptor conformational changes. Mol Endocrinol. 1997;11:1375–86. doi: 10.1210/mend.11.9.9983. [DOI] [PubMed] [Google Scholar]
- 35.Pakdel F, Reese JC, Katzenellenbogen BS. Identification of charged residues in an N-terminal portion of the hormone-binding domain of the human estrogen receptor important in transcriptional activity of the receptor. Mol Endocrinol. 1993;7:1408–17. doi: 10.1210/mend.7.11.8114756. [DOI] [PubMed] [Google Scholar]
- 36.Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. 1996;10:1388–98. doi: 10.1210/mend.10.11.8923465. [DOI] [PubMed] [Google Scholar]
- 37.Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer research. 1997;57:1244–9. [PubMed] [Google Scholar]
- 38.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discovery. 2017;7:277–87. doi: 10.1158/2159-8290.CD-15-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nardone A, De Angelis C, Trivedi MV, Osborne CK, Schiff R. The changing role of ER in endocrine resistance. Breast. 2015;24(Suppl 2):S60–6. doi: 10.1016/j.breast.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs. 2015;26:948–56. doi: 10.1097/CAD.0000000000000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer. 2015;22:713–24. doi: 10.1530/ERC-15-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, Zheng S, Akerstrom VL, Yuan C, Ma Y, Zhong Q, et al. Fulvestrant-3 Boronic Acid (ZB716): An Orally Bioavailable Selective Estrogen Receptor Downregulator (SERD) Journal of medicinal chemistry. 2016;59:8134–40. doi: 10.1021/acs.jmedchem.6b00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Zheng S, Guo S, Zhang C, Zhong Q, Zhang Q, et al. Rational Design of a Boron-Modified Triphenylethylene (GLL398) as an Oral Selective Estrogen Receptor Downregulator. ACS Med Chem Lett. 2017;8:102–6. doi: 10.1021/acsmedchemlett.6b00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–9. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holton SE, Bergamaschi A, Katzenellenbogen BS, Bhargava R. Integration of molecular profiling and chemical imaging to elucidate fibroblast-microenvironment impact on cancer cell phenotype and endocrine resistance in breast cancer. PLoS One. 2014;9:e96878. doi: 10.1371/journal.pone.0096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, et al. A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell. 2016;29:407–22. doi: 10.1016/j.ccell.2016.02.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.