

Abstract
Metabolic syndrome is a cluster of several clinical conditions characterized by insulin-resistance and high cardiovascular risk. Non-alcoholic fatty liver disease is the liver expression of the metabolic syndrome, and insulin resistance can be a frequent comorbidity in several chronic liver diseases, in particular hepatitis C virus infection and/or cirrhosis. Several studies have demonstrated that insulin action is not only relevant for glucose control, but also for vascular homeostasis. Insulin regulates nitric oxide production, which mediates to a large degree the vasodilating, anti-inflammatory and antithrombotic properties of a healthy endothelium, guaranteeing organ perfusion. The effects of insulin on the liver microvasculature and the effects of IR on sinusoidal endothelial cells have been studied in animal models of non-alcoholic fatty liver disease. The hypotheses derived from these studies and the potential translation of these results into humans are critically discussed in this review.
Keywords: Non-alcoholic fatty liver disease, Endothelial dysfunction, Insulin resistance, Metabolic syndrome
Core tip: Insulin-resistance participates in the development of endothelial dysfunction and interferes with vascular homeostasis in patients with metabolic syndrome. This has been demonstrated in large conductance vessels, promoting atherosclerosis, but also occurs at a microcirculation level, suggesting an important role for Insulin in controlling vascular resistance and, finally, organ perfusion. We offer an overview of those pre-clinical and clinical studies exploring the liver microcirculation, and discuss the importance of early vascular changes induced by insulin-resistance in non-alcoholic fatty liver disease and in the most common chronic hepatitis in which Insulin-Resistance is a comorbidity.
INTRODUCTION
Metabolic syndrome (MS) is a cluster of cardiovascular risk factors including glucose intolerance, hypertension, dyslipidemia, and visceral obesity, and has a prevalence of up to 25% in adults over 40 years old[1]. Non-alcoholic fatty liver disease (NAFLD) is the liver expression of MS, and constitutes a chronic disease associated with high cardio-vascular risk, with potential for progression to cirrhosis and hepatocellular carcinoma[2]. In other chronic liver conditions such as hepatitis C virus-related chronic hepatitis[3-5] and cirrhosis[6], MS can be an important comorbidity that potentially worsens liver histology[7] and increases the risk of decompensation[8]. Insulin-resistance (IR) is a common early finding in patients with MS and is the main pathogenic substrate for the development of NAFLD. IR is associated with endothelial dysfunction (ED)[9,10], a major pathogenic factor in arterial hypertension, coronary artery disease and atherosclerosis[11].
Several studies in animal models and humans have demonstrated that insulin action couples vascular and glucose homeostasis[12]. Indeed, in physiologic conditions, Insulin can influence the production of nitric oxide (NO) that mediates to a large part the vasodilating, anti-inflammatory and antithrombotic properties of a healthy endothelium. In patients with MS, the degree of IR parallels the severity of ED with important repercussion on structural and functional changes of the macro- and micro-circulation that may lead to impaired organ perfusion[11]. In the last few years, intrahepatic ED has been demonstrated in several models of liver disease including cirrhosis[13], ischemia-reperfusion[14], endotoxemia[15] and fatty liver[16,17]. As a consequence, vascular changes induced by IR, acting through the development of ED, are of potential therapeutic interest in the context of the most prevalent liver diseases. The present review offers an overview of the molecular mechanisms linking IR with ED in the control of vascular homeostasis, and reports the main biological and clinical findings on this topic in the context of the most common liver diseases.
INSULIN AND ENDOTHELIUM: FROM PHYSIOLOGY TO PATHOPHYSIOLOGY
Biological activity of NO is highly regulated in healthy conditions
NO is the main biochemical mediator of endothelium-dependent vasodilation in blood vessels. In physiologic conditions, it is constitutively synthesized by endothelial nitric oxide synthase (eNOS), whose activation results in a cascade of molecular events leading to smooth muscle relaxation[12]. The vasodilatory actions of NO play a key role in the renal control of extracellular fluid and is essential for the regulation of blood flow and blood pressure[18].
The endothelial NO production by eNOS is tightly regulated at the transcriptional and post-transcriptional level[19]. The expression of eNOS mRNA is largely restricted to the vascular endothelium[20]. Methylation is biologically associated with a marked impairment of promoter activity in mammalian cells and appears to play an important role in endothelial cell-specific expression of the human eNOS gene[21]. The eNOS promoter is heavily methylated in non-endothelial cells in comparison with endothelial cells. Kruppel-like factor 2 (KLF2) is a transcription factor that modulates the expression of multiple endothelial genes, including eNOS and thrombomodulin[22,23]. KLF2 is endothelial-specific and its expression, which is modulated by different flow patterns, confers endothelial protection against inflammation, thrombosis and excessive vasoconstriction[24,25]. Gracia-Sancho et al[26] have demonstrated that KLF2 can be activated by SIRT1 and that KLF2 overexpression activates vasoprotective genes in the vascular endothelium[27]. The same group demonstrated that simvastatin upregulates KLF2 expression in whole livers from cirrhotic rats[14] and in sinusoidal endothelial cells in culture[28]. This demonstrates that KLF2-mediated transcriptional regulation at the liver sinusoidal endothelial cells reproduces what occurs at peripheral endothelial cells.
The activity of eNOS is also thoroughly regulated at the posttranslational level. These include protein-protein interactions, cofactors availability and protein phosphorylation[13]. The impact of these posttranslational modifications in liver endothelial eNOS has been thoroughly studied in animal models of liver disease[13]. Finally, the bioavailability of NO can be affected by the oxidative stress generated by several clinical conditions, and antioxidants have demonstrated the importance of redox environment to maintain microcirculation in conditions of compromised liver perfusion[29].
eNOS and iNOS: From physiology to pathophysiology
In addition to the constitutive form of eNOS, at least two other isoforms have been described: the inducible NOS (iNOS), and the neuronal NOS (nNOS). The potential pathogenic roles of eNOS and iNOS dysregulation have been assessed in several models of liver disease. Both isoforms produce NO, but their intracellular localization, activation, and concentration of NO produced are not the same, resulting in different biologic actions.
Physiological hepatic production of NO is derived from eNOS in response to stimuli such as shear stress and the presence of vasoconstrictors[30,31]. The classical activation of eNOS implies an increase of intracellular calcium (Ca2+) and binding of Ca2+/calmodulin to the enzyme. In addition, a Ca2+-independent pathway regulating eNOS has been recently described. This pathway can be stimulated by several factors, among them shear stress and insulin[32,33]. Both shear stress and insulin increase endothelial NO production via activation of PI-3-kinase and protein kinase B (PKB/Akt), which activate eNOS by Ser1179-phosphorylation[34]. In addition, insulin upregulates the transcription of eNOS in endothelial cells[35].
In the liver, eNOS-derived NO targets hepatic stellate cells (HSC), promoting the synthesis of cyclic GMP (cGMP)[36]. The most important target of cGMP is protein kinase cGMP-dependent (PKG), which phosphorylates numerous proteins involved in the regulation of Ca2+ homeostasis, among them inositol 1,4,5 triphosphate-receptor. This leads to a decrease in the concentration of intracellular Ca2+ in HSC and produces relaxation with enduing decreased intrahepatic vascular resistance[37]. Thus, a physiological production of NO in the healthy liver offsets vasoconstrictor stimuli[38]. Since increased intrahepatic vascular tone is a major factor leading to portal hypertension in cirrhosis, different pharmacological strategies have been explored to increase liver NO availability[39-42].
iNOS was initially identified for its vital role in the immune system. When activated, it produces continuously large amounts of NO since, in contrast to eNOS, the substrate and cofactors are not limiting. iNOS is upregulated in metabolic tissues under different conditions of stress[43]. Although it is important for the immune system, iNOS activity can be harmful for other cell types, including pancreatic β cells[44] and vascular cells[45]. Recent studies have shown that iNOS-derived NO may play a role in the pathophysiology of obesity-induced metabolic dysfunction[43]. Among other mechanisms, it has been shown that iNOS is a critical modulator of PPAR-γ activation (a target of insulin sensitizing drugs)[46] and can decrease insulin sensitivity through S-nytrosilation of the insulin receptor[47]. Indeed, the inhibition of iNOS reduces hyperglycemia, hyperinsulinemia and improves liver insulin sensitivity[48]. Moreover, several studies with animal models have demonstrated that the induction of iNOS can cause ED through increased nitro-oxidative stress[49-51] and downregulation of eNOS[52]. Finally, the inhibition of iNOS in animal models that overexpress this enzyme restores a normal endothelial function[53-55].
Under physiologic conditions, the only NOS expressed in the endothelium of the vessels is eNOS. During inflammation, blood vessels express iNOS and eNOS[56]. Overexpression of iNOS thus contributes to vascular dysfunction.
Insulin-resistance and eNOS activity
The binding of insulin to its receptors at the level of peripheral endothelial cells[57] activates the phosphorylation of the insulin-receptor substrate which initiates a phosphorylation of a series of down-stream substrates, among them the PI3K/Akt pathway[58,59], that finally activate eNOS[60,61]. The result of this set of reactions ultimately produces an increase in eNOS activity and increased production of nitric oxide (NO), leading to vasodilation (Figure 1).
Figure 1.
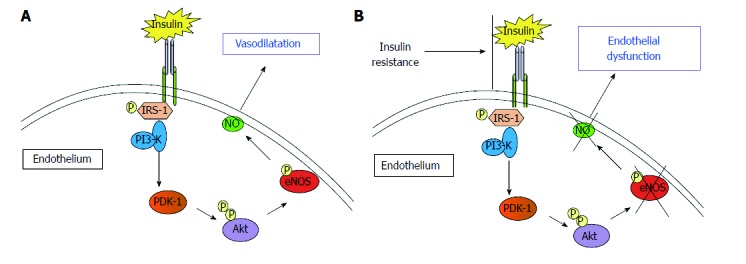
The binding of insulin to its receptor activates a series of phosphorylations of downstream receptors that finally activate nitric oxide-production by endothelial nitric oxide synthase. A: The release of nitric oxide (NO) causes endothelium dependent vasodilation; B: Insulin-resistance causes the reduction of Insulin-induced activation of endothelial nitric oxide synthase (eNOS). This is associated with reduction of NO bioavailability and, finally, endothelial dysfunction.
In the presence of IR, the PI3K/Akt pathway (involved in metabolic functions) is impaired, while other pathways of insulin signalling remain unaffected, including the Ras/MAPK pathway (involved in the control of cell proliferation), resulting in an imbalance between insulin functions performed by the PI3K pathways and MAPK[62,63]. This imbalance leads to decreased activation of eNOS and thus lowered production of NO, resulting in ED.
INTRAHEPATIC VASCULAR CHANGES IN NAFLD
Microvascular abnormalities in models of fatty liver: Structural and functional increase of resistance
The isolated liver perfusion technique has been instrumental in the assessment of liver microvascular changes in fatty liver[16] (Figure 2). In several studies, substantial changes in vascular function and liver blood flow have been demonstrated in fatty liver disease, as reviewed elsewhere[64]. Studies in rabbits with diet-induced steatosis of different severity confirmed that reduction in sinusoidal perfusion correlated with the severity of fat accumulation in parenchymal cells[65] and the severity of steatosis had a greater impact on microcirculation. These studies demonstrated that steatosis caused an increase in the mechanical component of intrahepatic vascular resistance to portal blood flow, independent of functional changes potentially induced by IR, a feature that could be observed also in patients with genetic susceptibility to NAFLD[66,67]. The potential impact of these hemodynamic changes on liver perfusion was subsequently explored in rats exposed to a high-cholesterol diet that developed steatosis. Compared to controls, steatotic rats had significantly reduced hepatic microcirculation and tissue oxygenation[68]. Interestingly, in this study, exposure to L-Arginine, the biochemical precursor of NO, improved tissue oxygenation, whereas L-NAME, a NOS inhibitor, further deteriorated hepatic microcirculation and hepatocyte oxygenation. These results allow for two major considerations. First, reduced oxygenation is an important issue if we consider the susceptibility of fatty livers to ischemic injury[69]. Second, all these results, from a biochemical point of view, suggest that NO, the marker of a healthy endothelium, is involved in the modulation of hepatic microcirculatory perfusion and oxygenation in rats with steatotic livers. Along these lines, we had previously demonstrated that intrahepatic ED in isolated and perfused livers from rats with several features of NAFLD and MS predated the development of fibrosis and inflammation[17], suggesting that liver ED contributes to increased intrahepatic resistance very early in the pathogenesis of NAFLD.
Figure 2.
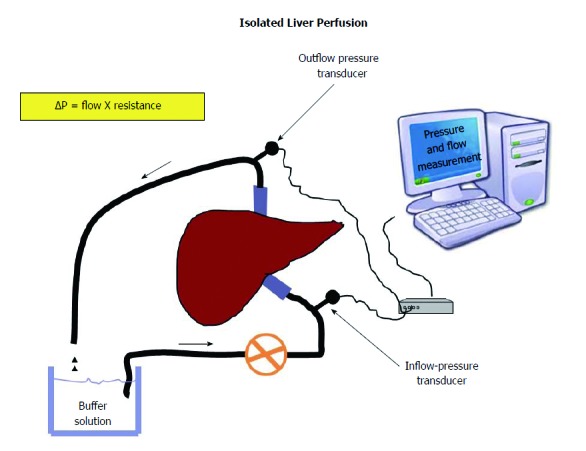
Livers are isolated and perfused at a constant velocity. Any change of pressure will be directly proportional to the resistance offered by sinusoids to the flow of a buffer solution according to Ohm’s low applied to fluid-dynamic (ΔP = flow X resistance). This allows measuring the intrahepatic resistance offered by sinusoids.
Sinusoidal endothelial dysfunction and fibrogenesis
Fibrogenesis is a complex biochemical process which represents the hallmark of any evolving chronic liver disease. Studies with animal models of fibrosis have demonstrated that fibrogenesis parallels neo-angiogenesis, confirming the importance of endothelial cells in this phenomenon[70]. In particular, the paracrine crosstalk of sinusoidal endothelial cells with hepatocytes, hepatic stellate cells and Kupffer cells is determinant in initiating and maintaining the fibrogenic reaction[71]. However, among all these cytotypes, there remains debate as to which cell initiates the process and when exactly these cells start changing their function towards a pathologic phenotype. In addition, the role of IR on this crosstalk and its impact on the pathogenesis and evolution of NAFLD has not been adequately addressed by in vitro and in vivo studies. Recently, Miyao et al[72], by using mice models of NAFLD/non-alcoholic steatohepatitis, confirmed that sinusoidal endothelial injury appeared in the steatotic phase, preceding the activation of Kupffer cells, hepatic stellate cells and, in turn, inflammation and fibrosis[72]. This suggests that sinusoidal endothelial cells may have a gatekeeper role in the progression from simple steatosis to steatohepatitis, but this would require confirmatory explorations. Interestingly, these functional changes in the endothelium parallel a replacement of regular sinusoidal anatomy into appearing disorganized and characterized by abnormal vascular interconnections[73]. Furthermore, the recent observation that fibrosis may be sustained by an abnormal activation of coagulation[74-76] suggests the theoretical scenario in which the loss of the anticoagulant properties of endothelium can play a mechanistic role in fibrogenesis even in NAFLD. Indeed, Kopec et al[77] demonstrated that steatosis due to 3 mo of high fat diet is associated with a pro-coagulant imbalance that has a cause-effect relation with the severity of liver damage. The real impact of all these microvascular abnormalities in the progression to steatohepatitis and cirrhosis is an intriguing question, and their role in the pathogenesis of NAFLD remains in need of further investigation.
Pathogenic link between intrahepatic microvascular abnormalities and IR
Many mechanisms observed in patients with IR, among them, lipotoxicity[78], oxidative stress[79,80], changes in local renin-angiotensin system[81], increased sensitivity to adrenergic stimuli of vascular smooth muscle cells[82], glucotoxicity (via oxidative stress, increased flow, activation of diacylglycerol, among others[83,84]) and inflammation[85,86], could explain the development of ED.
In a rat model of simple steatosis, we demonstrated the presence of insulin resistance in the liver sinusoidal endothelium that was mediated, at least in part, by the upregulation of iNOS[16]. As occurs in the peripheral circulation, in the normal liver, insulin results in liver vasodilation. In rats with fatty liver, insulin-dependent vasodilation in the liver vasculature was significantly impaired as compared to livers from control rats. This was partially restored in rats treated with the iNOS specific inhibitor 1400 W. Moreover, the insulin sensitizer metformin also restored hepatic vascular sensitivity to insulin while diminishing hepatic iNOS expression. Recently, the interaction between sinusoidal ED and iNOS has been explored in a rat model of endotoxemia (characterized by an overexpression of iNOS). These data further demonstrated that iNOS upregulation can induce, per se, sinusoidal ED[87]. All these findings support that in the hepatic vasculature, IR can be detected early in the course of the disease and may contribute to disease progression. A recent work by Gonzalez-Paredes et al[88]. confirmed the occurrence of intra-hepatic ED in rats with several features of MS and disclosed an important role of oxidative stress and cyclooxygenase end products in determining these functional abnormalities of the vasculature after 6 weeks of exposure to a high fat diet[88].
FROM BENCH TO BEDSIDE: POTENTIAL CLINICAL CONSEQUENCES
The effects of IR on hepatic vasculature could be of relevance in the pathogenesis and progression of NAFLD. The development of ED induced by IR may promote a pro-fibrogenic, pro-inflammatory and a pro-thrombotic environment, and impair regeneration after liver injury; aspects all related with the transition from steatosis to steatohepatitis and cirrhosis[72]. Unfortunately, in humans, the vascular abnormalities described in the liver of animal models with IR have been poorly investigated. Several authors have shown a correlation between IR and the severity of NAFLD[89], and have tested pharmacologic and non-pharmacologic strategies to improve IR in patients with NAFLD[90]. However, the association of these results with intra-hepatic vascular abnormalities, although theoretically possible, has not been demonstrated so far.
The study of intrahepatic ED in clinical practice is demanding and probably represents the Achilles’ heel to translating experimental observations of vascular abnormalities from animal models to humans. Catheterization of the suprahepatic vein and measurement of the hepatic venous pressure gradient (HVPG) and hepatic blood flow with indicator-dilution techniques is the most reliable tool to measure hepatic resistance to portal blood flow[91]. These measurements can be more informative if performed dynamically, after stimulating the hepatic circulation by increased splanchnic blood flow, for example with a test meal. The rationale for this is that if ED occurs in the liver, the liver circulation is less efficient in accommodating an increase in blood flow, with a consequent abrupt increase in portal pressure[92]. Unfortunately, the use of invasive methods in patients with early disease is challenging. Notwithstanding, Francque et al[93] published a series of 50 patients with biopsy-proven NAFLD who underwent liver hemodynamic studies. They found that up to 28% of patients had a HVPG over 5 mmHg (the threshold indicating sinusoidal portal hypertension), even though only one patient had histological documentation of cirrhosis. In that series, HVPG significantly correlated with the degree of steatosis, suggesting that the ballooning of hepatocytes causes narrowing of sinusoidal spaces and consequently increases intrahepatic resistance to portal blood flow, in keeping with the experimental observations in animal models of steatosis. The investigation of intra-hepatic hemodynamic changes induced by IR has been addressed by studies including patients with cirrhosis and comorbidities related to MS. Cirrhosis is frequently associated with IR[6,94]. Berzigotti et al[8] have demonstrated that obesity is an independent risk factor for higher portal pressure and disease progression in patients with cirrhosis (independent of the etiology) and might suggest that clinical features of IR could worsen the degree of intrahepatic ED. Interestingly, the same authors demonstrated that a program of weight loss through diet and physical exercise reduced portal pressure in overweight/obese patients with cirrhosis with positive results[95].
Beyond HVPG measurement, some authors have separately explored less invasive methodologies or biomarkers of inflammation, coagulation, platelet activation that may be linked to ED in NAFLD. In human studies, imaging techniques such as Doppler flowmetry[96], positron emission tomography[97] and magnetic resonance[98] have demonstrated the potential for investigating intravascular changes and hepatic perfusion within the steatotic liver. It has been much harder to identify biomarkers specific for intrahepatic ED. Selectins (such as P-Selectin, E-Selectin), von Willebrand Factor, Isoprostans, asymmetric dimethyl-arginine, endothelins and a series of molecules involved in the inflammatory and hemostatic activity of the endothelium have been used by cardiologists to describe and monitor ED[99]. However, unless these biomarkers are measured at the level of the hepatic veins or, alternatively, be discriminable by their organ-origin, the impairment of all these tests can only be interpreted as consequence of IR/MS but is not specifically liver-related. This underlines once again that NAFLD is the liver expression of a systemic disease. As matter of fact, several authors have demonstrated that the severity of NAFLD correlates with some key features of the systemic cardiovascular risk described in patients with MS/IR, among them, peripheral ED measured by vasodilatory response of the brachial artery to ischemia[10], mild chronic inflammation and low fibrinolytic activity[100,101]. Presently, it would be hard to recommend a pharmacologic strategy specifically targeting intrahepatic ED. However, any strategy of treatment recommended for NAFLD[102] which can modulate MS/IR has the potential to benefit sinusoidal endothelial cells in this clinical setting (Figure 3). Furthermore, the ongoing research on new drugs targeted against apoptosis, inflammation, fibrogenesis could offer in the next future an alternative/adjuvant therapy to contrast the downstream effects of the vascular changes induced by IR.
Figure 3.
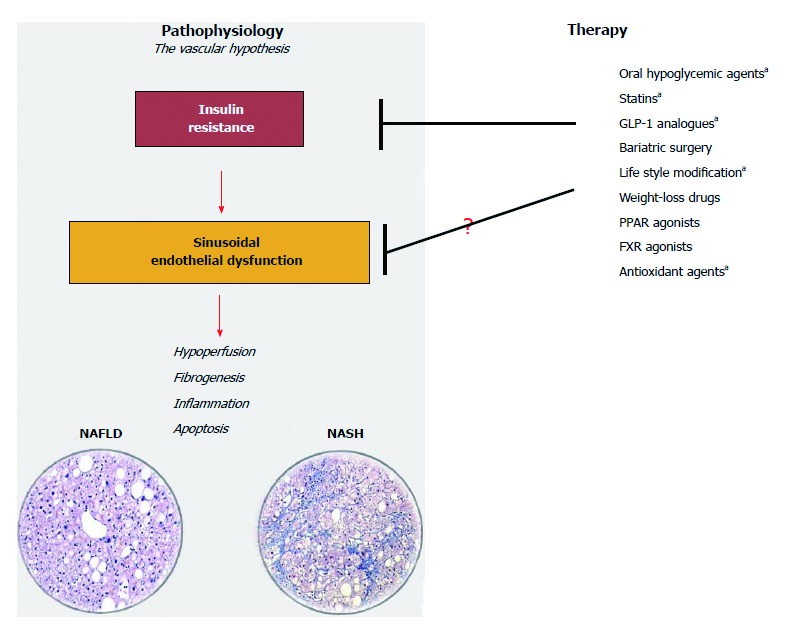
The vascular-hypothesis of liver damage in non-alcoholic fatty liver disease considers sinusoidal endothelial dysfunction due to insulin-resistance a key factor for the initiation and perpetuation of liver damage from simple steatosis to steatohepatitis. Any strategy of treatment ameliorating insulin-resistance may be efficacious in ameliorating sinusoidal endothelial dysfunction. Drugs marked with a are those with a proven efficacy on liver microcirculation (Ref. [16,42,95,103,104]). (Histological images are courtesy of Dr. Marco Maggioni, IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy). NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis.
CONCLUSION
Several studies with animal models have demonstrated that IR is associated with narrow sinusoids and sinusoidal endothelial dysfunction, which cause both a mechanical and functional increase of hepatic vascular resistance to portal blood flow, even in the absence of cirrhosis. Due to the high prevalence of metabolic syndrome in patients with chronic liver diseases, the influence of these vascular changes on the natural history of NAFLD and of cirrhosis of other etiologies is highly plausible and should be explored by specifically designed human studies. This could certainly result in new strategies for the treatment of patients with chronic liver disease.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C, C, C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: No potential conflicts of interest.
Peer-review started: May 19, 2017
First decision: June 22, 2017
Article in press: September 19, 2017
P- Reviewer: Bubnov RV, Kai K, Lee HC, Tacke F S- Editor: Ma YJ L- Editor: A E- Editor: Huang Y
Contributor Information
Marcos Pasarín, Hepatic Hemodynamic Laboratory, Liver Unit, Hospital Clinic, IDIBAPS (Institut d’Investigacions Biomèdiques August Pi i Sunyer), University of Barcelona, 08036 Barcelona, Spain. vincenzo.lamura@unimi.it.
Juan G Abraldes, Cirrhosis Care Clinic, Division of Gastroenterology (Liver Unit), CEGIIR, University of Alberta, AB T6G 2R3 Edmonton, Canada.
Eleonora Liguori, Internal Medicine, IRCCS San Donato, Department of Biomedical Sciences for Health, University of Milan, 20097 San Donato Milanese, Italy.
Beverley Kok, Cirrhosis Care Clinic, Division of Gastroenterology (Liver Unit), CEGIIR, University of Alberta, AB T6G 2R3 Edmonton, Canada.
Vincenzo La Mura, Internal Medicine, IRCCS San Donato, Department of Biomedical Sciences for Health, University of Milan, 20097 San Donato Milanese, Italy.
References
- 1.Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, Tuomilehto J, Salonen JT. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA. 2002;288:2709–2716. doi: 10.1001/jama.288.21.2709. [DOI] [PubMed] [Google Scholar]
- 2.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 3.Petta S, Macaluso FS, Craxì A. Cardiovascular diseases and HCV infection: a simple association or more? Gut. 2014;63:369–375. doi: 10.1136/gutjnl-2013-306102. [DOI] [PubMed] [Google Scholar]
- 4.Persico M, Masarone M, La Mura V, Persico E, Moschella F, Svelto M, Bruno S, Torella R. Clinical expression of insulin resistance in hepatitis C and B virus-related chronic hepatitis: differences and similarities. World J Gastroenterol. 2009;15:462–466. doi: 10.3748/wjg.15.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soverini V, Persico M, Bugianesi E, Forlani G, Salamone F, Massarone M, La Mura V, Mazzotti A, Bruno A, Marchesini G. HBV and HCV infection in type 2 diabetes mellitus: a survey in three diabetes units in different Italian areas. Acta Diabetol. 2011;48:337–343. doi: 10.1007/s00592-011-0293-x. [DOI] [PubMed] [Google Scholar]
- 6.Cammà C, Petta S, Di Marco V, Bronte F, Ciminnisi S, Licata G, Peralta S, Simone F, Marchesini G, Craxì A. Insulin resistance is a risk factor for esophageal varices in hepatitis C virus cirrhosis. Hepatology. 2009;49:195–203. doi: 10.1002/hep.22655. [DOI] [PubMed] [Google Scholar]
- 7.Petta S, Cammà C, Di Marco V, Alessi N, Cabibi D, Caldarella R, Licata A, Massenti F, Tarantino G, Marchesini G, et al. Insulin resistance and diabetes increase fibrosis in the liver of patients with genotype 1 HCV infection. Am J Gastroenterol. 2008;103:1136–1144. doi: 10.1111/j.1572-0241.2008.01813.x. [DOI] [PubMed] [Google Scholar]
- 8.Berzigotti A, Garcia-Tsao G, Bosch J, Grace ND, Burroughs AK, Morillas R, Escorsell A, Garcia-Pagan JC, Patch D, Matloff DS, et al. Obesity is an independent risk factor for clinical decompensation in patients with cirrhosis. Hepatology. 2011;54:555–561. doi: 10.1002/hep.24418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 10.Villanova N, Moscatiello S, Ramilli S, Bugianesi E, Magalotti D, Vanni E, Zoli M, Marchesini G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology. 2005;42:473–480. doi: 10.1002/hep.20781. [DOI] [PubMed] [Google Scholar]
- 11.Reaven GM. Role of insulin resistance in human disease (syndrome X): an expanded definition. Annu Rev Med. 1993;44:121–131. doi: 10.1146/annurev.me.44.020193.001005. [DOI] [PubMed] [Google Scholar]
- 12.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 13.Bosch J, Abraldes JG, Fernández M, García-Pagán JC. Hepatic endothelial dysfunction and abnormal angiogenesis: new targets in the treatment of portal hypertension. J Hepatol. 2010;53:558–567. doi: 10.1016/j.jhep.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 14.Russo L, Gracia-Sancho J, García-Calderó H, Marrone G, García-Pagán JC, García-Cardeña G, Bosch J. Addition of simvastatin to cold storage solution prevents endothelial dysfunction in explanted rat livers. Hepatology. 2012;55:921–930. doi: 10.1002/hep.24755. [DOI] [PubMed] [Google Scholar]
- 15.La Mura V, Pasarín M, Meireles CZ, Miquel R, Rodríguez-Vilarrupla A, Hide D, Gracia-Sancho J, García-Pagán JC, Bosch J, Abraldes JG. Effects of simvastatin administration on rodents with lipopolysaccharide-induced liver microvascular dysfunction. Hepatology. 2013;57:1172–1181. doi: 10.1002/hep.26127. [DOI] [PubMed] [Google Scholar]
- 16.Pasarín M, Abraldes JG, Rodríguez-Vilarrupla A, La Mura V, García-Pagán JC, Bosch J. Insulin resistance and liver microcirculation in a rat model of early NAFLD. J Hepatol. 2011;55:1095–1102. doi: 10.1016/j.jhep.2011.01.053. [DOI] [PubMed] [Google Scholar]
- 17.Pasarín M, La Mura V, Gracia-Sancho J, García-Calderó H, Rodríguez-Vilarrupla A, García-Pagán JC, Bosch J, Abraldes JG. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One. 2012;7:e32785. doi: 10.1371/journal.pone.0032785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon Y, Song J, Hong SH, Kim JQ. Plasma nitric oxide concentrations and nitric oxide synthase gene polymorphisms in coronary artery disease. Clin Chem. 2000;46:1626–1630. [PubMed] [Google Scholar]
- 19.Searles CD. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol Cell Physiol. 2006;291:C803–C816. doi: 10.1152/ajpcell.00457.2005. [DOI] [PubMed] [Google Scholar]
- 20.Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, Harrison DG, Marsden PA. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 1997;17:2479–2488. doi: 10.1161/01.atv.17.11.2479. [DOI] [PubMed] [Google Scholar]
- 21.Chan Y, Fish JE, D’Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, Marsden PA. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem. 2004;279:35087–35100. doi: 10.1074/jbc.M405063200. [DOI] [PubMed] [Google Scholar]
- 22.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA Jr, Balasubramanian V, García-Cardeña G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–e57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- 24.Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJ, Biessen EA, van Berkel TJ, Pannekoek H, et al. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167:609–618. doi: 10.1016/S0002-9440(10)63002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, García-Cardeña G, Gimbrone MA Jr. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci USA. 2004;101:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gracia-Sancho J, Villarreal G Jr, Zhang Y, García-Cardeña G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Res. 2010;85:514–519. doi: 10.1093/cvr/cvp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gracia-Sancho J, Russo L, García-Calderó H, García-Pagán JC, García-Cardeña G, Bosch J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011;60:517–524. doi: 10.1136/gut.2010.220913. [DOI] [PubMed] [Google Scholar]
- 28.Marrone G, Russo L, Rosado E, Hide D, García-Cardeña G, García-Pagán JC, Bosch J, Gracia-Sancho J. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J Hepatol. 2013;58:98–103. doi: 10.1016/j.jhep.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 29.Gracia-Sancho J, Laviña B, Rodríguez-Vilarrupla A, García-Calderó H, Fernández M, Bosch J, García-Pagán JC. Increased oxidative stress in cirrhotic rat livers: A potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology. 2008;47:1248–1256. doi: 10.1002/hep.22166. [DOI] [PubMed] [Google Scholar]
- 30.Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ, Sessa WC. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest. 1997;100:2923–2930. doi: 10.1172/JCI119842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastor CM, Hadengue A. Shear stress modulates the vascular tone in perfused livers isolated from normal rats. Hepatology. 2000;32:786–791. doi: 10.1053/jhep.2000.17739. [DOI] [PubMed] [Google Scholar]
- 32.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartell NA, Archer HE, Bailey CJ. Insulin-stimulated endothelial nitric oxide release is calcium independent and mediated via protein kinase B. Biochem Pharmacol. 2005;69:781–790. doi: 10.1016/j.bcp.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 34.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179) J Biol Chem. 2001;276:30392–30398. doi: 10.1074/jbc.M103702200. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Wallerath T, Förstermann U. Physiological mechanisms regulating the expression of endothelial-type NO synthase. Nitric Oxide. 2002;7:132–147. doi: 10.1016/s1089-8603(02)00127-1. [DOI] [PubMed] [Google Scholar]
- 36.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 37.Wiest R, Groszmann RJ. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology. 2002;35:478–491. doi: 10.1053/jhep.2002.31432. [DOI] [PubMed] [Google Scholar]
- 38.Mittal MK, Gupta TK, Lee FY, Sieber CC, Groszmann RJ. Nitric oxide modulates hepatic vascular tone in normal rat liver. Am J Physiol. 1994;267:G416–G422. doi: 10.1152/ajpgi.1994.267.3.G416. [DOI] [PubMed] [Google Scholar]
- 39.Yu Q, Shao R, Qian HS, George SE, Rockey DC. Gene transfer of the neuronal NO synthase isoform to cirrhotic rat liver ameliorates portal hypertension. J Clin Invest. 2000;105:741–748. doi: 10.1172/JCI7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah V, Chen AF, Cao S, Hendrickson H, Weiler D, Smith L, Yao J, Katusic ZS. Gene transfer of recombinant endothelial nitric oxide synthase to liver in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol. 2000;279:G1023–G1030. doi: 10.1152/ajpgi.2000.279.5.G1023. [DOI] [PubMed] [Google Scholar]
- 41.Fiorucci S, Antonelli E, Morelli O, Mencarelli A, Casini A, Mello T, Palazzetti B, Tallet D, del Soldato P, Morelli A. NCX-1000, a NO-releasing derivative of ursodeoxycholic acid, selectively delivers NO to the liver and protects against development of portal hypertension. Proc Natl Acad Sci USA. 2001;98:8897–8902. doi: 10.1073/pnas.151136298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abraldes JG, Rodríguez-Vilarrupla A, Graupera M, Zafra C, García-Calderó H, García-Pagán JC, Bosch J. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J Hepatol. 2007;46:1040–1046. doi: 10.1016/j.jhep.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. 2001;7:1138–1143. doi: 10.1038/nm1001-1138. [DOI] [PubMed] [Google Scholar]
- 44.Shimabukuro M, Ohneda M, Lee Y, Unger RH. Role of nitric oxide in obesity-induced beta cell disease. J Clin Invest. 1997;100:290–295. doi: 10.1172/JCI119534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwashina M, Shichiri M, Marumo F, Hirata Y. Transfection of inducible nitric oxide synthase gene causes apoptosis in vascular smooth muscle cells. Circulation. 1998;98:1212–1218. doi: 10.1161/01.cir.98.12.1212. [DOI] [PubMed] [Google Scholar]
- 46.Dallaire P, Bellmann K, Laplante M, Gélinas S, Centeno-Baez C, Penfornis P, Peyot ML, Latour MG, Lamontagne J, Trujillo ME, et al. Obese mice lacking inducible nitric oxide synthase are sensitized to the metabolic actions of peroxisome proliferator-activated receptor-gamma agonism. Diabetes. 2008;57:1999–2011. doi: 10.2337/db08-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carvalho-Filho MA, Ueno M, Hirabara SM, Seabra AB, Carvalheira JB, de Oliveira MG, Velloso LA, Curi R, Saad MJ. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes. 2005;54:959–967. doi: 10.2337/diabetes.54.4.959. [DOI] [PubMed] [Google Scholar]
- 48.Fujimoto M, Shimizu N, Kunii K, Martyn JA, Ueki K, Kaneki M. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes. 2005;54:1340–1348. doi: 10.2337/diabetes.54.5.1340. [DOI] [PubMed] [Google Scholar]
- 49.Gunnett CA, Lund DD, Chu Y, Brooks RM 2nd, Faraci FM, Heistad DD. NO-dependent vasorelaxation is impaired after gene transfer of inducible NO-synthase. Arterioscler Thromb Vasc Biol. 2001;21:1281–1287. doi: 10.1161/hq0801.093509. [DOI] [PubMed] [Google Scholar]
- 50.Zanetti M, d’Uscio LV, Kovesdi I, Katusic ZS, O’Brien T. In vivo gene transfer of inducible nitric oxide synthase to carotid arteries from hypercholesterolemic rabbits. Stroke. 2003;34:1293–1298. doi: 10.1161/01.STR.0000068366.00173.E7. [DOI] [PubMed] [Google Scholar]
- 51.Gunnett CA, Lund DD, Howard MA 3rd, Chu Y, Faraci FM, Heistad DD. Gene transfer of inducible nitric oxide synthase impairs relaxation in human and rabbit cerebral arteries. Stroke. 2002;33:2292–2296. doi: 10.1161/01.str.0000027427.86177.d4. [DOI] [PubMed] [Google Scholar]
- 52.Chauhan SD, Seggara G, Vo PA, Macallister RJ, Hobbs AJ, Ahluwalia A. Protection against lipopolysaccharide-induced endothelial dysfunction in resistance and conduit vasculature of iNOS knockout mice. FASEB J. 2003;17:773–775. doi: 10.1096/fj.02-0668fje. [DOI] [PubMed] [Google Scholar]
- 53.Gunnett CA, Chu Y, Heistad DD, Loihl A, Faraci FM. Vascular effects of LPS in mice deficient in expression of the gene for inducible nitric oxide synthase. Am J Physiol. 1998;275:H416–H421. doi: 10.1152/ajpheart.1998.275.2.H416. [DOI] [PubMed] [Google Scholar]
- 54.Gunnett CA, Heistad DD, Faraci FM. Gene-targeted mice reveal a critical role for inducible nitric oxide synthase in vascular dysfunction during diabetes. Stroke. 2003;34:2970–2974. doi: 10.1161/01.STR.0000099123.55171.3F. [DOI] [PubMed] [Google Scholar]
- 55.Kessler P, Bauersachs J, Busse R, Schini-Kerth VB. Inhibition of inducible nitric oxide synthase restores endothelium-dependent relaxations in proinflammatory mediator-induced blood vessels. Arterioscler Thromb Vasc Biol. 1997;17:1746–1755. doi: 10.1161/01.atv.17.9.1746. [DOI] [PubMed] [Google Scholar]
- 56.Gunnett CA, Heistad DD, Loihl A, Faraci FM. Tumor necrosis factor-alpha impairs contraction but not relaxation in carotid arteries from iNOS-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1558–R1564. doi: 10.1152/ajpregu.2000.279.5.R1558. [DOI] [PubMed] [Google Scholar]
- 57.Li G, Barrett EJ, Wang H, Chai W, Liu Z. Insulin at physiological concentrations selectively activates insulin but not insulin-like growth factor I (IGF-I) or insulin/IGF-I hybrid receptors in endothelial cells. Endocrinology. 2005;146:4690–4696. doi: 10.1210/en.2005-0505. [DOI] [PubMed] [Google Scholar]
- 58.Montagnani M, Ravichandran LV, Chen H, Esposito DL, Quon MJ. Insulin receptor substrate-1 and phosphoinositide-dependent kinase-1 are required for insulin-stimulated production of nitric oxide in endothelial cells. Mol Endocrinol. 2002;16:1931–1942. doi: 10.1210/me.2002-0074. [DOI] [PubMed] [Google Scholar]
- 59.Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. 1996;98:894–898. doi: 10.1172/JCI118871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michell BJ, Griffiths JE, Mitchelhill KI, Rodriguez-Crespo I, Tiganis T, Bozinovski S, de Montellano PR, Kemp BE, Pearson RB. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr Biol. 1999;9:845–848. doi: 10.1016/s0960-9822(99)80371-6. [DOI] [PubMed] [Google Scholar]
- 61.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 62.Potenza MA, Marasciulo FL, Chieppa DM, Brigiani GS, Formoso G, Quon MJ, Montagnani M. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol. 2005;289:H813–H822. doi: 10.1152/ajpheart.00092.2005. [DOI] [PubMed] [Google Scholar]
- 63.Cardillo C, Nambi SS, Kilcoyne CM, Choucair WK, Katz A, Quon MJ, Panza JA. Insulin stimulates both endothelin and nitric oxide activity in the human forearm. Circulation. 1999;100:820–825. doi: 10.1161/01.cir.100.8.820. [DOI] [PubMed] [Google Scholar]
- 64.Ijaz S, Yang W, Winslet MC, Seifalian AM. Impairment of hepatic microcirculation in fatty liver. Microcirculation. 2003;10:447–456. doi: 10.1038/sj.mn.7800206. [DOI] [PubMed] [Google Scholar]
- 65.Seifalian AM, Piasecki C, Agarwal A, Davidson BR. The effect of graded steatosis on flow in the hepatic parenchymal microcirculation. Transplantation. 1999;68:780–784. doi: 10.1097/00007890-199909270-00009. [DOI] [PubMed] [Google Scholar]
- 66.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, Mozzi E, Roviaro G, Vanni E, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–1217. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 67.Sookoian S, Castaño GO, Scian R, Mallardi P, Fernández Gianotti T, Burgueño AL, San Martino J, Pirola CJ. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology. 2015;61:515–525. doi: 10.1002/hep.27556. [DOI] [PubMed] [Google Scholar]
- 68.Ijaz S, Yang W, Winslet MC, Seifalian AM. The role of nitric oxide in the modulation of hepatic microcirculation and tissue oxygenation in an experimental model of hepatic steatosis. Microvasc Res. 2005;70:129–136. doi: 10.1016/j.mvr.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 69.Selzner M, Rüdiger HA, Sindram D, Madden J, Clavien PA. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000;32:1280–1288. doi: 10.1053/jhep.2000.20528. [DOI] [PubMed] [Google Scholar]
- 70.Bocca C, Novo E, Miglietta A, Parola M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell Mol Gastroenterol Hepatol. 2015;1:477–488. doi: 10.1016/j.jcmgh.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marrone G, Shah VH, Gracia-Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol. 2016;65:608–617. doi: 10.1016/j.jhep.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, Tamaki K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest. 2015;95:1130–1144. doi: 10.1038/labinvest.2015.95. [DOI] [PubMed] [Google Scholar]
- 73.Francque S, Laleman W, Verbeke L, Van Steenkiste C, Casteleyn C, Kwanten W, Van Dyck C, D’Hondt M, Ramon A, Vermeulen W, et al. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest. 2012;92:1428–1439. doi: 10.1038/labinvest.2012.103. [DOI] [PubMed] [Google Scholar]
- 74.Cerini F, Vilaseca M, Lafoz E, García-Irigoyen O, García-Calderó H, Tripathi DM, Avila M, Reverter JC, Bosch J, Gracia-Sancho J, et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J Hepatol. 2016;64:834–842. doi: 10.1016/j.jhep.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 75.Vilaseca M, García-Calderó H, Lafoz E, García-Irigoyen O, Avila MA, Reverter JC, Bosch J, Hernández-Gea V, Gracia-Sancho J, García-Pagán JC. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology. 2017;65:2031–2044. doi: 10.1002/hep.29084. [DOI] [PubMed] [Google Scholar]
- 76.Simonetto DA, Yang HY, Yin M, de Assuncao TM, Kwon JH, Hilscher M, Pan S, Yang L, Bi Y, Beyder A, et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology. 2015;61:648–659. doi: 10.1002/hep.27387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kopec AK, Joshi N, Towery KL, Kassel KM, Sullivan BP, Flick MJ, Luyendyk JP. Thrombin inhibition with dabigatran protects against high-fat diet-induced fatty liver disease in mice. J Pharmacol Exp Ther. 2014;351:288–297. doi: 10.1124/jpet.114.218545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang XL, Zhang L, Youker K, Zhang MX, Wang J, LeMaire SA, Coselli JS, Shen YH. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes. 2006;55:2301–2310. doi: 10.2337/db05-1574. [DOI] [PubMed] [Google Scholar]
- 79.Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116:1071–1080. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salomone F, Li Volti G, Rosso C, Grosso G, Bugianesi E. Unconjugated bilirubin, a potent endogenous antioxidant, is decreased in patients with non-alcoholic steatohepatitis and advanced fibrosis. J Gastroenterol Hepatol. 2013;28:1202–1208. doi: 10.1111/jgh.12155. [DOI] [PubMed] [Google Scholar]
- 81.Watanabe S, Tagawa T, Yamakawa K, Shimabukuro M, Ueda S. Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. 2005;25:2376–2380. doi: 10.1161/01.ATV.0000187465.55507.85. [DOI] [PubMed] [Google Scholar]
- 82.Stepniakowski KT, Goodfriend TL, Egan BM. Fatty acids enhance vascular alpha-adrenergic sensitivity. Hypertension. 1995;25:774–778. doi: 10.1161/01.hyp.25.4.774. [DOI] [PubMed] [Google Scholar]
- 83.Reusch JE. Diabetes, microvascular complications, and cardiovascular complications: what is it about glucose? J Clin Invest. 2003;112:986–988. doi: 10.1172/JCI19902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goldberg IJ, Dansky HM. Diabetic vascular disease: an experimental objective. Arterioscler Thromb Vasc Biol. 2006;26:1693–1701. doi: 10.1161/01.ATV.0000231521.76545.f6. [DOI] [PubMed] [Google Scholar]
- 85.Fernández-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr Rev. 2003;24:278–301. doi: 10.1210/er.2002-0010. [DOI] [PubMed] [Google Scholar]
- 86.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–949. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 87.La Mura V, Pasarín M, Rodriguez-Vilarrupla A, García-Pagán JC, Bosch J, Abraldes JG. Liver sinusoidal endothelial dysfunction after LPS administration: a role for inducible-nitric oxide synthase. J Hepatol. 2014;61:1321–1327. doi: 10.1016/j.jhep.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 88.Gonzalez-Paredes FJ, Hernández Mesa G, Morales Arraez D, Marcelino Reyes R, Abrante B, Diaz-Flores F, Salido E, Quintero E, Hernández-Guerra M. Contribution of Cyclooxygenase End Products and Oxidative Stress to Intrahepatic Endothelial Dysfunction in Early Non-Alcoholic Fatty Liver Disease. PLoS One. 2016;11:e0156650. doi: 10.1371/journal.pone.0156650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brunt EM, Kleiner DE, Wilson LA, Unalp A, Behling CE, Lavine JE, Neuschwander-Tetri BA; NASH Clinical Research NetworkA list of members of the Nonalcoholic Steatohepatitis Clinical Research Network can be found in the Appendix. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology. 2009;49:809–820. doi: 10.1002/hep.22724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mazzella N, Ricciardi LM, Mazzotti A, Marchesini G. The role of medications for the management of patients with NAFLD. Clin Liver Dis. 2014;18:73–89. doi: 10.1016/j.cld.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 91.Bosch J, Abraldes JG, Berzigotti A, García-Pagan JC. The clinical use of HVPG measurements in chronic liver disease. Nat Rev Gastroenterol Hepatol. 2009;6:573–582. doi: 10.1038/nrgastro.2009.149. [DOI] [PubMed] [Google Scholar]
- 92.Bellis L, Berzigotti A, Abraldes JG, Moitinho E, García-Pagán JC, Bosch J, Rodés J. Low doses of isosorbide mononitrate attenuate the postprandial increase in portal pressure in patients with cirrhosis. Hepatology. 2003;37:378–384. doi: 10.1053/jhep.2003.50053. [DOI] [PubMed] [Google Scholar]
- 93.Francque S, Verrijken A, Mertens I, Hubens G, Van Marck E, Pelckmans P, Van Gaal L, Michielsen P. Noncirrhotic human nonalcoholic fatty liver disease induces portal hypertension in relation to the histological degree of steatosis. Eur J Gastroenterol Hepatol. 2010;22:1449–1457. doi: 10.1097/MEG.0b013e32833f14a1. [DOI] [PubMed] [Google Scholar]
- 94.Erice E, Llop E, Berzigotti A, Abraldes JG, Conget I, Seijo S, Reverter E, Albillos A, Bosch J, García-Pagán JC. Insulin resistance in patients with cirrhosis and portal hypertension. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1458–G1465. doi: 10.1152/ajpgi.00389.2011. [DOI] [PubMed] [Google Scholar]
- 95.Berzigotti A, Albillos A, Villanueva C, Genescá J, Ardevol A, Augustín S, Calleja JL, Bañares R, García-Pagán JC, Mesonero F, et al. Effects of an intensive lifestyle intervention program on portal hypertension in patients with cirrhosis and obesity: The SportDiet study. Hepatology. 2017;65:1293–1305. doi: 10.1002/hep.28992. [DOI] [PubMed] [Google Scholar]
- 96.Seifalian AM, Chidambaram V, Rolles K, Davidson BR. In vivo demonstration of impaired microcirculation in steatotic human liver grafts. Liver Transpl Surg. 1998;4:71–77. doi: 10.1002/lt.500040110. [DOI] [PubMed] [Google Scholar]
- 97.Rijzewijk LJ, van der Meer RW, Lubberink M, Lamb HJ, Romijn JA, de Roos A, Twisk JW, Heine RJ, Lammertsma AA, Smit JW, et al. Liver fat content in type 2 diabetes: relationship with hepatic perfusion and substrate metabolism. Diabetes. 2010;59:2747–2754. doi: 10.2337/db09-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guiu B, Petit JM, Capitan V, Aho S, Masson D, Lefevre PH, Favelier S, Loffroy R, Vergès B, Hillon P, et al. Intravoxel incoherent motion diffusion-weighted imaging in nonalcoholic fatty liver disease: a 3.0-T MR study. Radiology. 2012;265:96–103. doi: 10.1148/radiol.12112478. [DOI] [PubMed] [Google Scholar]
- 99.Constans J, Conri C. Circulating markers of endothelial function in cardiovascular disease. Clin Chim Acta. 2006;368:33–47. doi: 10.1016/j.cca.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 100.Targher G, Bertolini L, Rodella S, Lippi G, Franchini M, Zoppini G, Muggeo M, Day CP. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity (Silver Spring) 2008;16:1394–1399. doi: 10.1038/oby.2008.64. [DOI] [PubMed] [Google Scholar]
- 101.Verrijken A, Francque S, Mertens I, Prawitt J, Caron S, Hubens G, Van Marck E, Staels B, Michielsen P, Van Gaal L. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2014;59:121–129. doi: 10.1002/hep.26510. [DOI] [PubMed] [Google Scholar]
- 102.Perazzo H, Dufour JF. The therapeutic landscape of non-alcoholic steatohepatitis. Liver Int. 2017;37:634–647. doi: 10.1111/liv.13270. [DOI] [PubMed] [Google Scholar]
- 103.de Mesquita FC, Guixé-Muntet S, Fernández-Iglesias A, Maeso-Díaz R, Vila S, Hide D, Ortega-Ribera M, Rosa JL, García-Pagán JC, Bosch J, et al. Liraglutide improves liver microvascular dysfunction in cirrhosis: Evidence from translational studies. Sci Rep. 2017;7:3255. doi: 10.1038/s41598-017-02866-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hernández-Guerra M, García-Pagán JC, Turnes J, Bellot P, Deulofeu R, Abraldes JG, Bosch J. Ascorbic acid improves the intrahepatic endothelial dysfunction of patients with cirrhosis and portal hypertension. Hepatology. 2006;43:485–491. doi: 10.1002/hep.21080. [DOI] [PubMed] [Google Scholar]