

Abstract
AIM
To identify glycosylation-related genes in the HT29 derivative cell line, HT29-MTX-E12, showing differential expression on infection with Helicobacter pylori (H. pylori).
METHODS
Polarised HT29-MTX-E12 cells were infected for 24 h with H. pylori strain 26695. After infection RNA was isolated from both infected and non-infected host cells. Sufficient infections were carried out to provide triplicate samples for microarray analysis and for qRT-PCR analysis. RNA was isolated and hybridised to Affymetrix arrays. Analysis of microarray data identified genes significantly differentially expressed upon infection. Genes were grouped into gene ontology functional categories. Selected genes associated with host glycan structure (glycosyltransferases, hydrolases, lectins, mucins) were validated by real-time qRT-PCR analysis.
RESULTS
Infection of host cells was confirmed by the isolation of live bacteria after 24 h incubation and by PCR amplification of bacteria-specific genes from the host cell RNA. H. pylori do not survive incubation under the adopted culture conditions unless they associate with the adherent mucus layer of the host cell. Microarray analysis identified a total of 276 genes that were significantly differentially expressed (P < 0.05) upon H. pylori infection and where the fold change in expression was greater than 2. Six of these genes are involved in glycosylation-related processes. Real-time qRT-PCR demonstrated significant downregulation (1.8-fold, P < 0.05) of the mucin MUC20. REG4 was heavily expressed and significantly downregulated (3.1-fold, P < 0.05) upon infection. Gene ontology analysis was consistent with previous studies on H. pylori infection.
CONCLUSION
Gene expression data suggest that infection with H. pylori causes a decrease in glycan synthesis, resulting in shorter and simpler glycan structures.
Keywords: Glycosylation, Adherent mucus, Gastric, HT29-MTX-E12, H. pylori strain 26695, Transcriptomics
Core tip: Few studies on Helicobacter pylori (H. pylori) infection focus on glycosylation-related genes in the host cells yet key cell-cell interactions are likely mediated through surface glycoconjugates. We use HT29-MTX-E12 cells, a promising and novel model of the stomach epithelium, to investigate the transcriptomic effects of H. pylori infection. HT29-MTX-E12 cells produce a thick adherent mucus layer and show a level of pluripotency that gastric cells naturally present and which some other model cell lines do not. Furthermore both H. pylori strain 26695 (lacks BabA adhesin) and HT29-MTX-E12 host cells (TLR2-negative) have some features atypical of more common models of H. pylori infection.
INTRODUCTION
Helicobacter pylori (H. pylori) is a major cause of peptic ulcer disease and chronic gastritis, and is a causative agent for both gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma[1-3]. Gastric cancer is the second most common cause of cancer-related deaths and is the fifth greatest killer worldwide after tuberculosis. Eradication of H. pylori infection with antibiotics may cause as much as a 40 % decrease in the cases of gastric cancer[4]. Despite its occurrence in approximately two-thirds of the world’s population, infection with H. pylori is asymptomatic in the majority of cases. How H. pylori manages to establish chronic infection in the hostile environment of the human stomach and to evade a vigorous immune response is not known.
An acute response to H. pylori infection is characteristically marked by the induction of interleukin (IL)-8. This is evident in both human biopsies and cell line studies. An acute response is followed by a chronic response which is marked in humans by the production of IgA autoantibodies to IL-8[5]. A rhesus macaque model of H. pylori infection showed a dramatic change in gastric epithelial morphology over the first few hours of infection but a restoration of normal morphology within 24 h despite the continued presence of the pathogen[6]. These morphological changes were associated with modified O-glycan structures. It has been shown previously that transcript levels of B3GNT5 were upregulated upon H. pylori infection of gastric cell lines MKN45 and AGS and that overexpression of this gene, associated with glycosylation of glycolipids, led to increased sialyl Lewis x presentation and adhesion of H. pylori[7]. Furthermore, host cells in asymptomatic infected human patients appear to modify O-glycans in order to counter the effects of the invading H. pylori[8].
H. pylori resides in the stomach where there is a thick adherent layer of mucus. While most invading pathogens are excluded from epithelial surfaces by the mucous layer, H. pylori survives in the mucous layer and can penetrate it to attach to the underlying cells. The main mucin components of mucus in the stomach are MUC5AC, MUC6, MUC1 and MUC16[9-11] with H. pylori displaying a distinct tropism for MUC5AC[12,13]. Although H. pylori may reside in the mucous layer it is through adhesion to the underlying epithelial cells that the bacteria exert their harmful/inflammatory effects. H. pylori binds to cells through adhesins, such as BabA that binds Lewis b and H-type 1 antigens on epithelial cell glycoproteins or glycolipids. They can also use the SabA adhesin to bind to sialyl Lewis x and sialyl Lewis a antigens, though this is expected to play a greater part in chronic infection. More recently LabA has been identified which binds to LacdiNAc structures present on MUC5AC[14]. Only a small fraction of H. pylori associated with infected cells is found internally[15], instead the bacteria inject the effector protein CagA and subvert host cell signalling[16,17].
Transcriptomic studies, through the use of high density DNA microarrays, have been applied to various aspects of H. pylori infection including analysis of human biopsies[18-23], analysis of human tissue after eradication treatments[24,25], the analysis of animal models[6,26,27] and the analysis of various cell lines[7,28-30]. High variability in the data sets has been attributed to different model systems, different host cells and the diverse nature of the different H. pylori strains used.
Human cell lines, isolated from the gastrointestinal tract (GIT), have been commonly used to study H. pylori infection. The responses of different gastric cell lines to H. pylori infection have been detailed in a recent paper[31]. The HT29 colon cell line and its derivatives have been used frequently as a model of the gastrointestinal tract because the cells are multipotent and can be directed along different paths of selection and differentiation. Some HT29 derivatives have an absorptive (enterocyte-like) phenotype while others have a mucus-secreting (goblet-like) phenotype. This makes HT29 cells particularly suited to the study of glycan structures in the gastrointestinal tract and mucus-associated effects on bacterial infection. The HT29-MTX-E12 (E12) cell line is a HT29 derivative selected on the basis of tight junction formation and adherent mucus production, which has many of the characteristics of a gastric cell[32-36]. In particular, differentiated E12 cells express MUC5AC, MUC1, MUC 6 and only trace amounts of MUC2[35], a composition which more closely resembles the stomach mucin makeup than the intestine.
The aim of this study was to determine the transcriptomic response of the mucus-secreting E12 cells to H. pylori infection with special focus on the glycomic response. Because known H. pylori adhesins bind glycosylated structures on the cell surface and in the mucous layer and glycosylation is known to play a major role in general host-pathogen and cell-cell interactions, it is of interest to know how glycosylated structures may change in response to infection.
MATERIALS AND METHODS
E12 culture
E12 cells, a kind gift from Per Artursson of the University of Uppsala, were maintained in flasks in complete Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal calf serum (Life Technologies), 2 mmol/L L-glutamine (Life Technologies), 1% nonessential amino acids and antibiotics (penicillin (50 U/mL) and streptomycin (50 μg/mL). Cells (2 × 106) were seeded on transwell filters (0.4 μm pore polycarbonate membrane insert) in 6-well plates. Cells were incubated at 37 °C in a humidified 5 % CO2 atmosphere and media was added every second day. Transepithelial electrical resistance (TEER) measurements were taken at 20 d. Antibiotics were removed from the cells at least 24 h before infection assays.
H. pylori culture
Bacteria (H. pylori strain 26695) were plated on Columbia blood agar (Oxoid) and grown for 48 h in a microaerophilic environment using gas packs (Oxoid, BR0038). Bacteria were harvested from plates into Brucella broth, the OD600 adjusted to 0.1 and grown in T25 tissue culture flasks at 37 °C under microaerophilic conditions with shaking at 70-80 rpm until the OD600 reached 0.6. Gram staining of bacterial cultures was used to check coccoid levels were less than 5%. Washed bacteria were resuspended to an OD600 of 1.8 with antibiotic-free RPMI.
Infection protocol
On day 20 E12 cells in transwells were washed with HBSS and 2 mL antibiotic-free complete DMEM media was added to the base of each well and 1 mL of RPMI to the apical surface of each well. H. pylori do not survive in DMEM[37] but will in RPMI. On day 21200 μL bacteria (6 × 108) were added to each well at a multiplicity of infection (MOI) of 300. Plates were incubated at 37 °C for 24 h. Cells were harvested for RNA isolation. Replicate transwells were washed gently with sterile PBS and harvested using trypsin EDTA as described previously[35,38]. Serial dilutions of the trypsinised cells were plated in triplicate on Columbia blood agar plates and incubated under microaerophillic conditions. Colonies were counted after 4-5 d incubation to determine the number of bacteria (CFU/mL) that were associated with the cells. Though some infections resulted in low IL8 transcript levels this did not appear to correlate with low CFU values.
RNA isolation and microarray hybridisation
RNA for microarray hybridisations and qPCR analysis was isolated by TRIzol extraction followed by standard RNA purification protocols (Qiagen). RNA (100 ng) was labelled with biotin using the Affymetrix 3’ IVT Express Protocol. All hybridisations were carried out using the hybridisation and wash solutions as recommended in the Affymetrix 3’ IVT Express Protocol and were carried out by DNA Vision, Charleroi, BE.
Microarray analysis
Spot intensities provided (DNA Vision) were normalised and analysed in GeneSpring (Ver 12.3). Data were log transformed and quantile normalised. Subsequently all normalised data was baseline transformed to the median of all samples. Intensity values for all entities (54675) were initially filtered to exclude values which were less than 50 in all three samples of both conditions (uninfected, infected). Significantly differentially expressed genes were identified based on log values using an unpaired t-test (variance assumed equal)[39], with a FDR cut-off value of 0.05. Multiple test correction was carried using the Benjamini and Hochberg correction. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus[40] and are accessible through GEO Series accession number GSE74492 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74492). Where microarray data is graphed, the raw signals for uninfected samples were the actual spot intensities and for the infected samples the signals were corrected using the fold change determined from normalised data for each gene. Standard deviations are shown for the three samples in each condition using raw spot intensities (uninfected) and raw spot intensities corrected for fold change (infected). Where the microarray data is shown as a log fold change, standard deviations were generated using propagation of errors rules.
For Gene Ontology analysis, DAVID (Database for Annotation, Visualization and Integrated Discovery, v6.7) was used[41] (http://www.david.ncifcrf.gov/ ). The number of significant genes (6034) was reduced further using more stringent parameters (FDR < 0.01, fold change > 1.2). Where directional analysis was carried out, only the top 500 upregulated genes (FDR < 0.005, fold change > 1.2) were included in the GO analysis and this was compared with whatever number of downregulated genes (714) had P-values below the P-value of the 500th upregulated gene (FDR = 0.0046) in order to maintain a balanced comparison. GO analysis also excluded GO terms where the number of genes in the GO term was more than 10% of all the genes on the array; this therefore removed broader gene ontology terms which were not generally informative. These restrictions were not imposed for GO analysis of the glycosylation-related genes because of the lower number of genes involved.
qRT-PCR
qRT-PCR validation was carried out on RNAs from samples collected independently of the microarray samples. The RNA samples for qRT-PCR validation were also from several different infections: this adds some of the variability seen in the data and is an issue that is discussed further in the manuscript. For real time qRT-PCR RNA (1 μg) was reverse transcribed using the VILO cDNA Synthesis Kit (Invitrogen) according to the manufacturer’s instructions. PCR was carried out using diluted template (2 μL) and GoTaq® qPCR Master Mix (18 μL) on a Mx3000P qPCR System (Stratagene). Cycling conditions for all primer pairs was 95 °C for 5 min followed by 40 cycles of 95 °C 10 s and 60 °C 10 s. Dissociation curves were determined using a temperature gradient from 95 °C to 56 °C. Primer sequences were either retrieved from primer repositories (PrimerBank, https://pga.mgh.harvard.edu/primerbank/ and qPrimerDepot, https://primerdepot.nci.nih.gov/), designed using Primer3 (http://primer3plus.com/web_3.0.0/primer3web_input.htm) or taken from relevant papers (Supplementary Table 1 and references therein). Several primer pairs were tested for their potential as housekeeping genes and geNorm analysis[42] was used to select an optimal set of three genes for subsequent normalisation (GAPDH, RPLP0, UAP1). All fold change values were calculated within geNorm which also generated standard deviations based on the rules for the propagation of errors. Standard two-tailed t-tests were used to determine where fold changes were significant. As an approximate measure of gene expression level, cycle threshold (Ct) values determined by qRT-PCR for each gene (uninfected cells) were compared to a Ctmax, in this case IL32 the lowest expressed of our selected genes
RESULTS
This study investigated the effects of H. pylori (strain 26695) colonisation on the transcriptome of E12 cells, a specialised derivative of the HT29 cell line. The cell line forms a functional layer of polarised cells and produces an adherent mucous layer when grown on transwells for 21 d.
Microarray analysis
E12 cells were grown on transwells for 21 d and infected with H. pylori strain 26695 at a MOI of 300. Cells were harvested for RNA isolation after incubation at 37 °C for 24 h. H. pylori maintains viability during infection despite its normal requirement for a microaerophilic environment, possibly as a consequence of close association with the mucous layer surrounding the E12 cells. Microarray analysis of all samples showed separate clustering of infected E12 (n = 3) and uninfected E12 samples (n = 3) (Supplementary Figure 1). The infected samples, originating from triplicate wells in a single infection experiment, clustered very tightly. Although greater variability was seen in the uninfected E12 samples, this is probably because these samples originate from two separate infection experiments.
Confirmation of a global modulation of E12 genes in response to H. pylori infection
Statistical analysis of the microarray data suggested significant differential expression of 6034 genes (FDR < 0.05) upon infection. Of these, 276 unique genes (< 5 %) showed fold changes greater than two (see Table 1 for the top 15 upregulated and downregulated genes; Supplementary Table 2 for all 276 genes). GO analysis of a reduced list (2639 genes) of regulated genes (FDR < 0.01, fold change> 1.2) is presented in Figure 1A. This analysis was sensitive to the selected fold change cut-off therefore we considered it appropriate to choose quite a low value (1.2-fold). To provide some additional perspective, GO analysis was repeated separately on the most significantly upregulated and downregulated genes (data not shown). This analysis gave an idea of the direction in which each significant process (as shown in Figure 1A) was regulated. GO analysis of the top 500 upregulated genes (FDR < 0.005, fold change > 1.2) revealed significant processes related to protein ubiquitination, protein folding, cell redox homeostasis, oxidative phosphorylation and the mitochondrial membrane. GO analysis of the top downregulated genes (FDR = 0.005, fold change > 1.2, 714 genes) revealed significant processes associated with ECM-receptor interaction, focal and cell adhesion, phosphorylation and modulation of the cytoskeleton. Some GO processes included genes with approximately equal numbers of up and downregulated genes: these additional GO terms included Golgi vesicle mediated transport, dolichol-linked oligosaccharide biosynthetic processes, phosphorylation-related signalling, apoptosis, cell junction function, molecular adaptor activity, cell cycle, transcription, antigen presentation and apical junction complex. These terms are consistent with the reported effects of H. pylori on cells and therefore support our supposition that the observed effects on glycosylation, the focus of this study, relate to infection by H. pylori.
Table 1.
Differentially expressed genes associated with Helicobacter pylori infection of E12 cells
| Gene symbol | P-value | Fold change | Gene name |
| Top 15 upregulated | |||
| 1CLIP1 | 0.0025 | 5.42 | CAP-GLY domain containing linker protein 1 |
| C10orf118 | 0.0031 | 5.21 | chromosome 10 open reading frame 118 |
| TOP1 | 0.0352 | 5.09 | topoisomerase (DNA) I |
| LARS | 0.0054 | 5.02 | leucyl-tRNA synthetase |
| CDC27 | 0.0073 | 4.95 | cell division cycle 27 homolog (S. cerevisiae) |
| STIP1 | 0.0042 | 3.92 | stress-induced-phosphoprotein 1 |
| MALAT1 | 0.0077 | 3.82 | metastasis associated lung adenocarcinoma transcript 1 (non-protein coding) |
| IL32 | 0.0021 | 3.54 | interleukin 32 |
| SLC39A6 | 0.0204 | 3.33 | solute carrier family 39 (zinc), member 6 |
| AHNAK2 | 0.0070 | 3.25 | AHNAK nucleoprotein 2 |
| HSP90AB1 | 0.0014 | 3.24 | heat shock protein 90 kDa alpha (cytosolic), B1 |
| ACTR2 | 0.0035 | 3.20 | ARP2 actin-related protein 2 homolog (yeast) |
| EIF4G1 | 0.0349 | 3.16 | eukaryotic translation initiation factor 4 gamma, 1 |
| ACBD3 | 0.0253 | 3.14 | acyl-CoA binding domain containing 3 |
| CCND1 | 0.0222 | 3.09 | cyclin D1 |
| Top 15 downregulated | |||
| PDK4 | 0.0011 | 3.25 | pyruvate dehydrogenase kinase, isozyme 4 |
| BCAT1 | 0.0042 | 3.24 | branched chain amino-acid transaminase 1, cytosolic |
| PELI1 | 0.0059 | 3.24 | Pellino homolog 1 (Drosophila) |
| CYP3A5 | 0.0015 | 3.12 | cytochrome P450, family 3, subfamily A, polypeptide 5 |
| HPGD | 0.0032 | 3.10 | hydroxyprostaglandin dehydrogenase 15-(NAD) |
| LRRC31 | 0.0030 | 3.05 | leucine rich repeat containing 31 |
| KIAA1984 | 0.0082 | 2.97 | KIAA1984 |
| HNRNPD | 0.0054 | 2.86 | Heterogeneous nuclear ribonucleoprotein D (AU-rich element RNA binding protein 1, 37kDa) |
| IL28A | 0.0026 | 2.85 | interleukin 28A (interferon, lambda 2) |
| DDX60 | 0.0026 | 2.84 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 |
| PHF14 | 0.0021 | 2.82 | PHD finger protein 14 |
| LOC100288092 | 0.0025 | 2.82 | Hypothetical protein LOC100288092 |
| ZNF207 | 0.0027 | 2.82 | zinc finger protein 207 |
| NCOA2 | 0.0025 | 2.81 | nuclear receptor coactivator 2 |
| AHSA2 | 0.0064 | 2.72 | AHA1, activator of heat shock 90 kDa protein ATPase homolog 2 (yeast) |
qRT-PCR analysis showed CLIP1 was downregulated.
Figure 1.
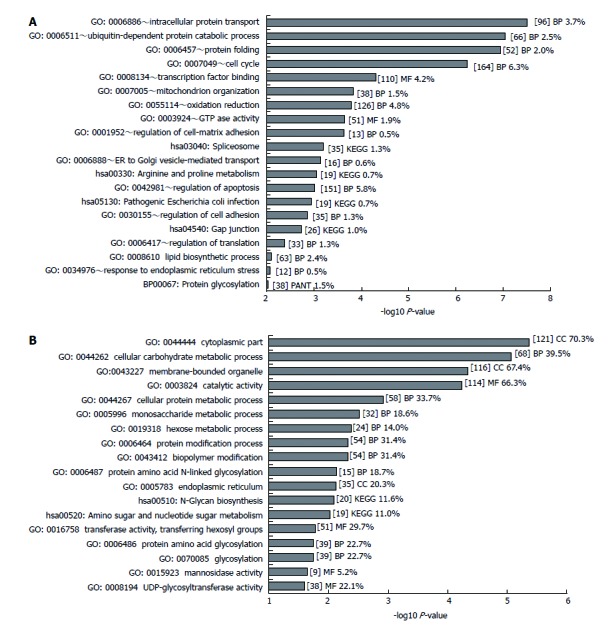
GO analysis of significant differentially expressed genes. A: GO analysis of 2639 genes deemed significantly differentially expressed (P < 0.01, fold change > 1.2) was carried out in DAVID. Only GO terms with a P-value below 0.01 are shown (-log10 P-value > 2). GO terms were excluded where the number of genes belonging to that GO term exceeded 10% of all genes. B: GO analysis of 374 significantly differentially expressed glycosylation-related genes (P < 0.001) was carried out in DAVID. Only GO terms with a P-value below 0.1 are shown (-log10 P-value > 1). Some Panther (PANT) gene ontology and KEGG pathway terms were included from the DAVID analysis. The number of genes included in each term is given within square brackets [ ] together with the percentage of significant genes relative to the total number of genes of that term on the array. BP: Biochemical process; MF: Molecular function; CC: Cellular component.
Modulation of glycosylation-related genes in response to H. pylori infection
In order to study changes in the glycome of E12 cells upon H. pylori infection we examined those gene probes on the Affymetrix U133 Plus 2 Array that were associated with a glycosylation role, selected with reference to the Consortium for Functional Glycomics (CFGv3) array. We identified 170 significantly differentially expressed genes upon H. pylori infection that were glycosylation related (see Table 2 for the top 20 upregulated and downregulated genes; Supplementary Table 3 for all 170 genes). Only 6 (< 4 %) of these genes showed fold changes greater than 2-fold. The most upregulated gene from the microarray analysis was LGALS7/7B, (lectin, galactoside-binding, soluble, 7/7B, 2.3-fold) and the most downregulated gene was REG4 (regenerating islet-derived family, member 4, 2.5-fold) followed by OGT (O-linked N-acetylglucosamine (GlcNAc) transferase, 2.2-fold) and AMIGO2 (adhesion molecule with Ig-like domain 2, 2.2-fold). These small changes in glycosylation-related genes are not uncommon in significant processes that involve glycosylation[7,43]. GO analysis of differentially regulated glycosylation-related genes was referenced against this Affymetrix array-derived glycome to determine which GO terms, relevant to glycosylation, were significant in the H. pylori infected E12 cells (Figure 1B). The most relevant terms were nucleotide sugar metabolism, N-glycan biosynthesis, cell death and galactose metabolic process, all of which generally related to upregulated genes, and hexose metabolic process, mannosidase activity and UDP-glycosyltransferase activity, which related to downregulated genes (Figure 1B).
Table 2.
Glycosylation-related differentially expressed genes associated with Helicobacter pylori infection of E12 cells
| Gene symbol | Fold change | P value | Gene name |
| Top 20 upregulated | |||
| LGALS7/7B | 2.31 | 0.0115 | Lectin, galactoside-binding, soluble, 7 /7B |
| UGGT1 | 1.98 | 0.0059 | UDP-glucose glycoprotein glucosyltransferase 1 |
| ST3GAL5 | 1.90 | 0.0035 | ST3 beta-galactoside alpha-2,3-sialyltransferase 5 |
| PARM1 | 1.87 | 0.0026 | Prostate androgen-regulated mucin-like protein 1 |
| NAGK | 1.79 | 0.0016 | N-acetylglucosamine kinase |
| VSIG1 | 1.71 | 0.0240 | V-set and immunoglobulin domain containing 1 |
| EDEM2 | 1.69 | 0.0032 | ER degradation enhancer, mannosidase alpha-like 2 |
| MPDU1 | 1.69 | 0.0029 | Mannose-P-dolichol utilization defect 1 |
| DPM2 | 1.61 | 0.0015 | Dolichyl-phosphate mannosyltransferase polypeptide 2, reg. s/u |
| LMAN2 | 1.60 | 0.0021 | Lectin, mannose-binding 2 |
| GMPPB | 1.59 | 0.0019 | GDP-mannose pyrophosphorylase B |
| ALG3 | 1.59 | 0.0214 | Asn-linked glycosylation 3, alpha-1,3- mannosyltransferase hom. |
| GALK2 | 1.57 | 0.0027 | Galactokinase 2 |
| DDOST | 1.53 | 0.0094 | Dolichyl-diphosphooligosaccharide--protein glycosyltransferase |
| MCAM | 1.53 | 0.0111 | Melanoma cell adhesion molecule |
| B3GALT6 | 1.53 | 0.0097 | UDP-Gal:betaGal beta 1,3-galactosyltransferase polypeptide 6 |
| SLC35B1 | 1.51 | 0.0025 | Solute carrier family 35, member B1 |
| ST6GALNAC4 | 1.50 | 0.0024 | ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 4 |
| GMPPA | 1.49 | 0.0031 | GDP-mannose pyrophosphorylase A |
| ALDOC | 1.48 | 0.0020 | Aldolase C, fructose-bisphosphate |
| Top 20 downregulated | |||
| REG4 | 2.46 | 0.0018 | Regenerating islet-derived family, member 4 |
| OGT | 2.25 | 0.0037 | O-linked N-acetylglucosamine (GlcNAc) transferase (OGT) |
| AMIGO2 | 2.22 | 0.0039 | Adhesion molecule with Ig-like domain 2 |
| GALNT12 | 2.09 | 0.0033 | UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 12 |
| EXT1 | 1.99 | 0.0256 | Exostosin 1 |
| LY75 | 1.86 | 0.0075 | Lymphocyte antigen 75 (CLEC13B) |
| PIGZ | 1.82 | 0.0072 | Phosphatidylinositol glycan anchor biosynthesis, class Z |
| UGP2 | 1.79 | 0.0320 | UDP-glucose pyrophosphorylase 2 |
| MANEA | 1.79 | 0.0225 | Mannosidase, endo-alpha |
| ALG13 | 1.78 | 0.0138 | Asparagine-linked glycosylation 13 homolog (S. cerevisiae) |
| GBAP1 | 1.75 | 0.0035 | Glucosidase, beta, acid pseudogene 1 |
| SLC35A3 | 1.73 | 0.0071 | Solute carrier family 35 (UDP-N-acetylglucosamine transporter), member A3 |
| ST3GAL4 | 1.71 | 0.0032 | ST3 beta-galactoside alpha-2,3-sialyltransferase 4 |
| B4GALNT3 | 1.71 | 0.0129 | Beta-1,4-N-acetyl-galactosaminyl transferase 3 |
| HMMR | 1.70 | 0.0039 | Hyaluronan-mediated motility receptor (RHAMM) |
| B4GALT6 | 1.69 | 0.0134 | UDP-Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 6 |
| MAN2A1 | 1.60 | 0.0034 | Mannosidase, alpha, class 2A, member 1 |
| LGALS2 | 1.58 | 0.0127 | Lectin, galactoside-binding, soluble, 2 |
| MAN1A1 | 1.53 | 0.0021 | Mannosidase, alpha, class 1A, member 1 |
| MUC13 | 1.53 | 0.0038 | Mucin 13, cell surface associated |
| CD164 | 1.52 | 0.0015 | CD164 molecule, sialomucin |
| B4GALT5 | 1.51 | 0.0071 | UDP-Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 5 |
Changes in terminal sugars of E12 glycoconjugates upon H. pylori infection
Microarray signal strength is not a robust measure of gene expression because probes bind with different affinities to the array. However, fold change measurements are robust because the same probe is used to compare two conditions. In this and the following sections the fold change differences between infected and uninfected gene levels are therefore quantitative. However, this data alone omits useful information about the expression level of one gene relative to another, always bearing in mind that two probes could have very different binding efficiencies for the array. In Figure 2, where three selected groups of glycosylation related genes are presented, a gene has only been included if microarray analysis showed that it was either significantly differentially expressed or that it was represented on the array by more than one probe. In addition a signal strength cut-off was set to 20 to exclude weak genes which were less likely to be relevant (though technically significant in the original analysis). For sialylation (Figure 2A) ST6GALNAC1, NEU1, SLC35A1, ST3GAL5 and NANP were significant but because they were each represented by only one probe their absolute expression levels relative to the other genes shown may be less representative. Where a gene was not significant but represented by multiple probes (e.g., ST3GAL2), the data was considered sufficiently reliable to warrant presenting expression levels relative to the other genes.
Figure 2.

Transcript levels determined by microarray analysis of selected glycosylation related processes. A: Sialylation-related genes; B: Mucin genes; C: O-glycosylation genes. Raw signals reported for each gene were the strongest signals where multiple probes were available (except for MUC2 where the negligible signal from the single probe, indicating very weak expression, was confirmed by qRT-PCR). All signals for uninfected samples were raw values whereas all infected samples were extracted using fold change values of normalised data as detailed in Materials and Methods. Uninfected (dark grey) or Infected (light grey). Error bars show standard deviations (n = 3). (aFDR < 0.05).
Terminal glycosylation of cell surface glycoconjugates directly determines the interaction of bacteria with the host cell. Therefore, sialylation and fucosylation were expected to be key processes affected by H. pylori infection. Terminal sialylation of glycoconjugates is a consequence of sialyltransferase expression and therefore can be predicted based on the transcript expression levels of the sialyltransferase enzymes from the microarray data. ST3GAL4, which can sialylate most glycoconjugate types and is believed to be involved in sialyl Lewis x and sialyl T antigen formation, was significantly downregulated (1.7-fold) whereas ST3GAL5, which is involved in ganglioside GM3 synthesis in glycolipids, was significantly upregulated (1.9-fold) on H. pylori infection (Figure 2A). ST6GALNAC1 (down, 1.3-fold) and ST6GALNAC4 (up, 1.8-fold) were also significantly differentially expressed upon H. pylori infection. ST6GALNAC4 is involved in the biosynthesis of ganglioside (GD1A from GM1B) and the transfer of NeuAc to GalNAc residues of O-glycans to form the stunted sialyl Tn antigen. Based on signal intensities, ST3GALs 2-5 were all moderately expressed (raw control intensity < 500), ST3GAL1 was highly expressed (> 500) and ST3GAL6 was negligibly expressed (< 10). ST6GALs were lowly (< 50) (ST6GAL1) to negligibly expressed (ST6GAL2) and all ST8SIAs (1-5) were also negligibly expressed. This is in line with the predominance of NeuAcα2-3Gal linkages over NeuAcα2-6Gal in HT29 cells[44] and the predominance of O-glycans over N-glycans. The overall effect to be expected is therefore one of increased sialyl Tn antigen, decreased sialyl Lewis x/a determinants on glycoproteins with potential effects on glycolipids. This may result in shorter glycan chains and may affect packing density because some GALNTs require recognition of neighbouring GalNAc residues, now increasingly masked by sialylation, to initiate new glycan chains.
Although a number of fucosyltransferases (FUT1, FUT3, FUT4, FUT5, FUT6 and FUT11) were moderately expressed, only FUT1 was upregulated significantly (1.2-fold) upon H. pylori infection (data not shown).This would suggest no major role for fucosylation in the E12 response to H. pylori infection. FUCA1 is a lysosomal fucose hydrolase. Its strong expression and upregulation on infection (1.2-fold) might suggest net removal of surface expressed fucose residues.
Changes in O-glycosylation upon H. pylori infection
As stated above, E12 cells produce a copious adherent mucous layer of which the main components are O-glycans. The data presented here show expression of MUC5AC, MUC1, MUC20 and MUC13. Only MUC13 and MUC20 were significantly downregulated (Figure 2B). MUC5AC was represented by two high signal probes on the array but neither showed any change upon H. pylori infection. (It is noted that MUC5AC appears as a significantly upregulated gene (1.2-fold) in Supplementary Table 3, however, this probe produced only a weak signal).
Mucin O-glycosylation initiates with the transfer of N-acetylgalactosamine (GalNAc) to a Ser/Thr residue on the protein catalysed by the polypeptide N-acetylgalactosamine transferases (GALNTs). Of 14 GALNTs represented on the array all 5 that were significant were downregulated upon H. pylori infection - GALNT1 (1.5-fold), GALNT2 (1.3-fold), GALNT7 (1.4-fold), GALNT10 (1.4-fold) and GALNT12 (2.1-fold) (Figure 2C). GALNT12 is believed to be involved in the initial glycosylation of MUC5AC and MUC1 but not of some other mucins[45]. It has different activities towards already glycosylated MUC5AC and MUC1 peptides suggesting that its activity may determine packing density of multiple O-linked glycans and that specifically glycan density may be reduced on H. pylori infection where GALNT12 appears downregulated. Sialylation of the polypeptide-linked GalNAc by ST6GALNACs may also change the recognition of initiation sites by specific polypeptide GalNAc transferases. Changes in O-glycan initiation have been reported previously in serum proteins of patients with gastric carcinoma[46]. Differential expression of GalNAc transferases in gastric cancer cell lines and gastric carcinoma may be associated with occupancy of potential O-glycosylation sites in mucins[47,48]. GalNAc transferase expression has been reported to be variable among gastric cell lines with GALNTs 1, 2 and 3 showing ubiquitous expression while other GalNAc transferases show more restricted expression[49].
No genes coding enzymes that have the potential to produce core 1 to core 8 O-linked glycan structures were significantly regulated upon H. pylori infection. C1GALT1 and C1GALT1C1 (COSMC) were expressed at moderate and high levels respectively based on signal intensities suggesting the likely presence of core 1 structures on E12 O-glycans. Extension of the core structures by B4GALTs and B3GNTs seemed likely with moderate expression of several B4GALTs and B3GNTs. The galactose transferases and N-acetylglucosamine transferases that were significantly downregulated were those more often associated with N-glycan (B4GALT5, 1.5-fold; B4GALT1, 1.2-fold) or glycosphingolipid synthesis (B4GALT5, B4GALT1 and the strongly expressed B3GNT5, 1.5-fold). We detected downregulation of B3GNT5 and B3GALT5 whereas a gastric biopsy study showed upregulation of the same genes[50]. The downregulated B3GNT2 (1.4-fold) is commonly involved in polylactosamine synthesis so may be involved in N-glycans, O-glycans and/or glycosphingolipids. The other common form of non-mucin O-glycosylation in E12 cell glycoconjugates initiated by O-GlcNAc protein glycosyltransferase was significantly downregulated on H. pylori infection (OGT, 2.3-fold) according to microarray data (Supplementary Table 3).
Changes in N-glycosylation upon H. pylori infection
N-glycosylation can be split into the lipid-linked oligosaccharide (LLO) precursor synthesis process generating Glc3Man9GlcNAc2 and the processes of trimming and extension. There was a general upregulation of the genes involved in the LLO step except for ALG13 which instead showed downregulation. ALG13 showed the largest fold change (1.8-fold, Table 2) of all the genes involved in LLO precursor synthesis. There was a general downregulation of the genes involved in trimming and extension. All mannosidases, most moderately expressed, were downregulated (Supplementary Table 3) and one, MAN2A2, was consequently confirmed by qPCR (below).
The significance of changes in N-glycosylation in an O-glycan-dominated environment is unclear. Also, whether the altered N-glycans are on mucins, other transmembrane glycoproteins, or both, cannot be determined from the available data. It has been suggested previously that LLO-precursor synthesis could be a constitutive process where precursors are available in excess and control is elsewhere[51]. ALG13 may, however, play a key role because it showed the largest fold change of all the genes in the precursor synthesis pathway, it was downregulated unlike other genes in the LLO-precursor synthesis pathway and interestingly it adds on the first GlcNAc of the LLO-precursor. Downregulated mannosidases (MAN2A1, 1.9-fold; MAN1A1, 1.5-fold; MAN2B2, 1.3-fold) are suggestive of an overall reduction of trimming and an increased level of the Man9GlcNAc2 glycan. Overall the effects suggest shorter and less complex N-glycan chains following H. pylori infection.
Changes in availability of activated sugars upon H. pylori infection
Glycan synthesis could be limited by changes in the abundance and availability of activated sugars. The UDP-galactose (SLC35A2, 1.3-fold; SLC35B1, 1.5-fold) and GDP-fucose (SLC35C2, 1.3-fold) transporters were significantly upregulated while the CMP-sialic acid (SLC35A1, 1.3-fold) and UDP-GlcNAc (SLC35A3, 2.1-fold) transporters were downregulated upon infection (Supplementary Table 3). The UDP-Gal, UDP-GlcNAc and CMP-sialic acid transporters were all strongly expressed while the GDP-fucose transporter was moderately expressed. N-Acetylneuraminic acid synthase (NANS) was very strongly expressed and significantly upregulated on infection (Figure 2A). This suggests the availability of Neu5Ac, however, the downregulation of both SLC35A1 and CMAS expression (though marginal) (Figure 2A) would possibly make a subsequent rise in activated Neu5Ac (CMP-Neu5Ac) levels seem less likely. Increased levels of sialidase (NEU1) may lead to cell shedding of sialylated epitopes with a compensatory switch on of Neu5Ac biosynthesis (Figure 2A). GO analysis had indicated the significance of hexose and, more specifically, galactose metabolism. All significant galactose metabolism enzymes were upregulated as were all significant mannose metabolism enzymes. All significant fucose metabolism enzymes were downregulated as were all sialic acid metabolic enzymes.
Modulation of lectin levels upon H. pylori infection
The two LMAN lectins LMAN1 (1.5-fold down) and LMAN2 (1.6-fold up) were expressed strongly and differentially (Supplementary Table 3). Both lectins are expressed in the endoplasmic reticulum and Golgi body and are believed to be involved in N-glycan sorting and recycling. Of the Gal binding lectins, LGALS1 (1.4-fold) and LGALS7 (2.3-fold) were both upregulated significantly, and LGALS2 (1.6-fold) and LGALS8 (1.4-fold) were both downregulated significantly, all being expressed at moderate levels (Supplementary Table 3). LGALS7 is a myoepithelial cell marker, associated positively with cancer progression and metastasis, while LGALS8 is involved in integrin-like cell interactions and is negatively associated with malignancy. The opposite regulation of LGALS7 and LGALS8, therefore, is consistent with an altered cell phenotype. LGALS3 and LGALS4 were expressed very strongly (raw signal >5000) but not differentially upon infection according to microarray data. Where oligosaccharide chains are not terminated with sialic acid, the major exposed sugar is galactose. It might therefore be relevant that two Gal binding lectins (LGALs) were expressed very strongly (LGALS3 and LGALS4) and four others were significantly regulated upon infection. Most other lectins (CLECs, collectins, selectins, siglecs) were either weakly or negligibly, but not differentially, expressed.
qRT-PCR validation of prediction changes in gene expression level
qRT-PCR validation was carried out on RNA from samples collected independently of the microarray samples and from several different infection experiments in an effort to identify variability between infections. qRT-PCR analysis was carried out on a selected set of genes. Initially two non-glycogenes (PDK4, CLIP1) were selected based on the maximum fold changes observed on the microarray. PDK4 expression was confirmed as downregulated though with a smaller fold change. It was shown subsequently that CLIP1 was not upregulated though initial microarray analysis had suggested upregulation. Only one (1558924_s_at) of six CLIP1 probes on the array suggested upregulation: a probe producing a much higher intensity signal (201975_at) instead suggested 1.3-fold significant downregulation and it is this probe that correlated with the qRT-PCR analysis (Figure 3). In general H. pylori-infected samples showed qRT-PCR-corroborated upregulation of IL8 and downregulation of REG4 (Figure 3A), though the upregulation of IL8 (and IL32) was highly variable across samples (see Supplementary Figure 2) resulting in an absence of significance. The stomach mucin MUC1 showed no significant change in expression by either microarray analysis or qRT-PCR. This was true also for MUC13, a gene found at low levels in normal gastric mucosa but often expressed in metaplasia[52,53]. These two genes did however show consistent (not significant) downregulation in both microarray analysis and qRT-PCR. A third mucin gene, MUC20, was shown to be significantly downregulated (1.4-fold). MUC5AC showed no significant expression change based on microarray data (Figure 2B) or in a preliminary screen by qRT-PCR (data not shown). qRT-PCR did not corroborate regulation of a number of genes where the expected fold changes by microarray analysis were less than 2 (LGALS2, ST3GAL5, MGAT2, FPGT and SLC35A2).
Figure 3.
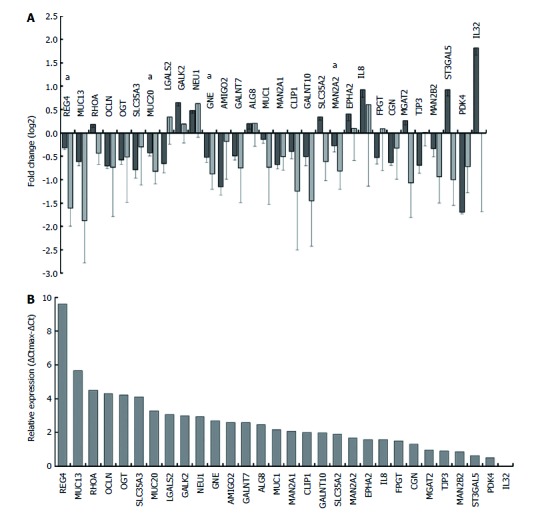
qRT-PCR validation of selected genes. Differential expression of selected genes upon Helicobacter pylori (H. pylori) infection of E12 cells was compared between microarray (dark grey) and qRT-PCR (light grey) analyses. qRT-PCR validation was carried out on RNAs from samples collected independently of the microarray samples. A: Microarray data (n = 3) was normalised across samples with GeneSpring. qRT-PCR data (n = 3) were normalised using geNorm and a panel of three housekeeping genes. Error bars show standard deviations determined using propagation of error rules; B: Relative expression of each gene in uninfected E12 cells was estimated by qRT-PCR using ΔCt determinations. ΔCtmax was the ΔCt for the lowest expressing gene (IL32) after correcting Ct values across samples with the housekeeping gene GAPDH. Further details are given in Materials and Methods. All genes were significantly differentially expressed in microarray analysis and genes marked with asterisks (a) were confirmed significant by qRT-PCR.
Four genes (REG4, MUC20, GNE and MAN2A2) showed significant differential regulation by qRT-PCR (P < 0.05), and others approached significance (P < 0.1 - MAN2B2, SLC35A2, RHOA; P < 0.2 - TFF1, ST3GAL5 and PDK4) (Table 3). The absence of significance in some cases was partly due to the small fold changes detected both by microarray and qRT-PCR analyses, but it was also due to variability in the samples used for qRT-PCR. In several cases one of the three uninfected samples behaved differently from the other two. This was very striking for the interleukins IL8 and IL32, and for TFF1 which showed high downregulation (10.4-fold) on infection, but was not significant because one uninfected sample (3 in Supplementary Figure 2) showed much lower levels than the other two uninfected samples (1 and 2, Supplementary Figure 2). Hierarchical clustering (Pearson correlation with average linkage clustering) clearly shows the inconsistency across the qRT-PCR samples (Supplementary Figure 2) and how inconsistent genes cluster with other inconsistent genes suggesting a biological rather than a technical reason for this variability. This anomalous behaviour in the uninfected samples had also been seen in the microarray analysis. A small cluster of seven genes including all of the MUC genes assayed and GALNT10, suggests some common regulatory feature. REG4, TFF1 and ST3GAL5 also formed a tight cluster (Supplementary Figure 2).
Table 3.
qRT-PCR significance of selected genes
| Gene | Fold Change | Regulation | Significance |
| GNE | 1.84 | Down | 1 |
| MUC20 | 1.77 | Down | 1 |
| REG4 | 3.06 | Down | 1 |
| MAN2A2 | 1.76 | Down | 1 |
| MAN2B2 | 1.91 | Down | NS P < 0.1 |
| SLC35A2 | 1.53 | Down | NS P < 0.1 |
| RHOA | 1.35 | Down | NS P < 0.1 |
| 1TFF1 | 10.37 | Down | NS P < 0.2 |
| MAN2A1 | 1.42 | Down | NS P < 0.2 |
| ST3GAL5 | 2.00 | Down | NS P < 0.2 |
| MUC13 | 3.66 | Down | NS |
| PDK4 | 1.65 | Down | NS |
| GALNT10 | 2.74 | Down | NS |
| GALNT7 | 1.68 | Down | NS |
| MGAT2 | 2.09 | Down | NS |
| MUC1 | 1.66 | Down | NS |
| NEU1 | 1.54 | Up | NS |
| CLIP1 | 2.37 | Down | NS |
| LGALS2 | 1.27 | Up | NS |
| OGT | 1.43 | Down | NS |
| AMIGO2 | 1.14 | - | NS |
| ALG8 | 1.15 | - | NS |
| GALK2 | 1.14 | - | NS |
| SLC35A3 | 1.23 | Down | NS |
| FPGT | 1.07 | - | NS |
| CGN | 1.26 | Down | NS |
| TJP3 | 1.01 | - | NS |
| OCLN | 1.66 | Down | NS |
| EPHA2 | 1.07 | - | NS |
| IL32 | 1.00 | - | NS |
| IL8 | 1.53 | Up | NS |
TFF1 is not shown in Figure 3, and four further genes (MGAT1, LGALS7, OSTC and LGALS7) are only shown in Supplementary Figure 2.
DISCUSSION
A recent study of gastric cell lines has shown a wide range of responses to H. pylori[31]. Commonly used cell lines differ in their ability to polarise, to maintain a stomach-like epithelial morphology and differ in their pattern recognition receptors. H. pylori is, for the most part, a non-invasive bacterium that colonises the mucous layer surrounding the stomach and does not survive without this mucous layer. We have chosen for our study the HT29 derivative E12 cells because they produce an adherent mucous layer and the cells support H. pylori infection while the parent HT29 cells do not support H. pylori infection[36]. The main mucin components of the stomach mucous layer in vivo are the secreted gel-forming mucin, MUC5AC, and the cell surface mucin, MUC1. Other mucins include MUC4, MUC12, MUC13 and MUC6[54]. E12 cells have been shown by immunofluorescent methods to express high levels of MUC5AC and MUC1, and to a lesser extent MUC2[35]. The transcriptomic data presented here show expression of MUC5AC, MUC13, MUC1 and MUC20 with little or no expression of either MUC2 or MUC6. E12 cells have therefore a mucin profile approximating that of the stomach and provide a good model for H. pylori infection studies.
The cellular response to pathogenic organisms is orchestrated by Toll-like receptors (TLRs). Strong evidence suggests that H. pylori infection is TLR2-mediated[55-57] yet there is some evidence that TLR4 is involved[58,59]. E12 cells however produce the hallmark response of IL-8 production upon H. pylori infection despite being TLR2 negative. The cellular response will also be determined by the receptor/adhesin characteristics of the host-pathogen interaction. Some H. pylori strains can bind to cells through the BabA adhesin which binds to the fucosylated Lewis b antigens on epithelial cell surface glycoconjugates[60,61]. Strain 26695, used throughout this study, possess an active cag pathogenicity island (PAI) associated with virulence[62] yet it has an inactive BabA adhesin[36]. Furthermore, fucosylated structures are relatively scarce on mucins secreted by E12 cells[36]. Therefore in this infection system the BabA adhesin interaction with Lewis antigens is not a factor in the IL-8 response to H. pylori and other H. pylori adhesin-host cell receptor combinations[63] are likely to play a role in mediating these responses.
Expression patterns of H. pylori-infected and uninfected E12 cells indicated variability especially in the uninfected E12 cells. The reasons for this are unclear. It might suggest that some E12 cells did not fully differentiate to form a fully polarised cell layer. However, TEER measurements were consistently high (about 600 Ω/cm2) across all uninfected samples and could not differentiate E12 subpopulations. E12 cells, like the parent HT29 cells, are not a clonal population[64,65]. It has been shown that E12 cells show the occasional MUC2-expressing cell and that these cells can be expanded under the appropriate conditions[34]. It may therefore be that random samples contain differing proportions of cell types and so produce different expression profiles. It is likely that clonal cell lines would give more consistent results, however, we would argue that in vivo cells lining the stomach are not clonal, and therefore that non-clonal E12 cells may better reflect variation that is present in vivo.
Infection of the E12 cells was confirmed in this work by the presence of cagA RNA in exposed cells (data not shown). All significant expression changes on H. pylori infection were small with only 4%-5% being greater than 2-fold. This has been noted previously in similar experiments with H. pylori infection. The altered expression of glycosyltransferases and other glycosylation-related genes identified in this study is different in some specifics to that seen in related studies. This could be because E12 cells are derived from the colon where similar studies of H. pylori infection have used gastric cell lines[7,28,29]. But variability in gene expression response is also seen across gastric cell lines possibly as a result of differences in mucin presentation. We would therefore argue that E12 cells, with their gastric-like mucin composition, constitute a valid model of H. pylori infection. Although the expression of specific genes did not correspond with that seen in similar studies, gene ontology terms were in general agreement suggesting that, in the broader functional picture, there are strong similarities across the different H. pylori infection studies. Variability in gene response could equally be due to the choice of H. pylori strain.
One consistently downregulated gene was REG4, a 17 kD secreted C-type lectin, expressed in selected enteroendocrine cells throughout the GIT and some goblet cells[66]. The functional role of REG4 is still unclear though it appears to be coexpressed with various enteroendocrine hormones. Although it is reportedly not expressed in normal stomach tissue, it is expressed, and has potential as a biomarker, in gastric carcinoma[67]. qPCR confirmed microarray data that expression of the REG4 transcript was very high. Why this should be in E12 cells is unclear. It could be a manifestation of the colonic origins of HT29/E12 cells, or of the selection process for generating the E12 derivative, or it may be related to the high mucin production of this cell line. Another member of the REG family, REG3γ, has been shown to be intimately involved in the mucous layer protecting the underlying epithelial cells from bacterial colonisation[68] but there is no evidence yet that REG4 has any anti-bacterial activity.
The overall shortening of O-glycans and the decreased complexity of both N- and O-glycans that this study suggests follow H. pylori infection may be driven by the infecting bacteria in their efforts to ease access through the mucus to the underlying enterocytes. It could equally be a host cell response to reduce potential bacterial adhesion sites or a way to facilitate shedding of the mucous layer and the attached bacteria. The results presented from the E12 model will require confirmation by detailed oligosaccharide analysis. Alternative approaches, such as lectin histochemistry or direct biophysical analysis of released glycans from glycoproteins and glycolipids, may not be able to detect the responses seen at the transcriptomic level because of overlapping specificities of lectins. However, it is of interest that the results presented are consistent with the Rhesus Macaque in vivo model of H. pylori infection[6] and with the analysis of adherent mucins isolated from asymptomatic H. pylori infected patients[8].
In conclusion, E12 cells are a promising model of the stomach epithelium; the cells form tight junctions, they polarise, they produce a thick adherent layer of mainly MUC5AC and have a mucin composition similar to that of the stomach. The glycosyl transferase gene expression profiles suggested core 1 and core 2 extended structures on O-glycans, with little or no core 3-8 structures, which is consistent with mass spectroscopy studies of isolated E12 glycoproteins. This study has shown that H. pylori infection increased expression of some sialyltransferases which may lead to shortened O-glycan chains. Furthermore, differential changes to various ppGalNAc transferase transcripts (mainly downregulation) suggested effects on O-glycan packing density and O-glycan site occupancy. The enzyme profiles would also suggest a change in N-glycan structures upon infection to more high-mannose and less complex oligosaccharides. Such changes may not affect the overall glycocalyx since it is so O-glycan dominated but could affect specific N-glycoproteins. Many of the transferase enzyme transcripts that are regulated upon H. pylori infection have a noticeable preference for glycolipids which is consistent with the presence of Lewis b ligands on glycolipids rather than glycoproteins in E12 cells[36]. While the overall changes to glycosyltransferase transcripts upon infection were small, small changes in enzyme activities that determine physicochemical properties of the mucus and the range of exposed epitopes could lead to profound effects on the phenotype of these infected cells.
ACKNOWLEDGMENTS
We would like to thank Aislinn Slater for technical assistance and also acknowledge the late Prof Anthony Moran for his early input into this project.
COMMENTS
Background
Key pathogen-host cell interactions are likely mediated through the glycan components of surface glycoproteins and glycolipids. Helicobacter pylori (H. pylori) is protected in the harsh environment of the stomach by the mucus layer of the gastric epithelium. Here we have used a HT29-derivative cell line that secretes a thick adherent mucus layer. This study explores changes in host cell gene transcription, specifically the genes that code mucins, glycosyltransferases, glycosyl hydrolases and lectins that may be associated with H. pylori infection.
Research frontiers
H. pylori infections are currently effectively cleared by antibiotics, however, the occurrence of antibiotic resistant strains is increasing and it is inevitable that new treatments will need to be developed. Understanding the key interactions between host and pathogen will drive the development of new drugs possibly analogues of surface sugars such as sialic acid and fucose.
Innovations and breakthroughs
Different strains of H. pylori display different levels of virulence. Some strains of H. pylori bind via BabA adhesins to Lewis antigens on the host cells and the host response is mediated through the TLR2 receptor. Here they use a common virulent strain (26695) that does not present one of the typical adhesins (BabA) commonly involved in H. pylori adhesion.
Applications
Changes in glycan structure on host cells in response to H. pylori infection will be changes initiated by the host to fight off infection and changes in the host induced by the infecting bacteria. By understanding which enzymes are involved in these host and pathogen modified glycan structure changes, it may be possible to design specific inhibitors of these enzymes.
Terminology
Transepithelial electrical resistance is a measure of the integrity of a cell layer and indicates that tight junctions have formed between cells.
Peer-review
This is a well written manuscript that attempts to identify genes significantly differentially expressed upon H. pylori infected in the HT29 derivative cell line by using microarray analysis and qRT-PCR analysis.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Ireland
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study did not involve either human or animal subjects.
Institutional animal care and use committee statement: This study did not involve animal subjects.
Conflict-of-interest statement: The authors declare no conflicts-of-interest.
Data sharing statement: All microarray data is available from the Gene Expression Omnibus database as dataset GSE74492. Other data is available from the corresponding author at michael.cairns@nuigalway.ie.
Peer-review started: February 20, 2017
First decision: April 7, 2017
Article in press: July 12, 2017
P- Reviewer: Tongtawee T, Yamaoka Y S- Editor: Qi Y L- Editor: A E- Editor: Huang Y
Contributor Information
Michael T Cairns, Glycoscience Group, National Centre for Biomedical Engineering Science, National University of Ireland Galway, H91 CF50 Galway, Ireland. michael.cairns@nuigalway.ie.
Ananya Gupta, School of Natural Sciences, National University of Ireland Galway, H91 CF50 Galway, Ireland.
Julie A Naughton, Conway Institute of Biomolecular and Biomedical Research, School of Medicine and Medical Sciences, University College Dublin, Dublin 4, Ireland.
Marian Kane, Glycoscience Group, National Centre for Biomedical Engineering Science, National University of Ireland Galway, H91 CF50 Galway, Ireland.
Marguerite Clyne, Conway Institute of Biomolecular and Biomedical Research, School of Medicine and Medical Sciences, University College Dublin, Dublin 4, Ireland.
Lokesh Joshi, Glycoscience Group, National Centre for Biomedical Engineering Science, National University of Ireland Galway, H91 CF50 Galway, Ireland.
References
- 1.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peek RM Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–248. doi: 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 3.Segal ED, Lange C, Covacci A, Tompkins LS, Falkow S. Induction of host signal transduction pathways by Helicobacter pylori. Proc Natl Acad Sci USA. 1997;94:7595–7599. doi: 10.1073/pnas.94.14.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma JL, Zhang L, Brown LM, Li JY, Shen L, Pan KF, Liu WD, Hu Y, Han ZX, Crystal-Mansour S, et al. Fifteen-year effects of Helicobacter pylori, garlic, and vitamin treatments on gastric cancer incidence and mortality. J Natl Cancer Inst. 2012;104:488–492. doi: 10.1093/jnci/djs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crabtree JE, Peichl P, Wyatt JI, Stachl U, Lindley IJ. Gastric interleukin-8 and IgA IL-8 autoantibodies in Helicobacter pylori infection. Scand J Immunol. 1993;37:65–70. doi: 10.1111/j.1365-3083.1993.tb01666.x. [DOI] [PubMed] [Google Scholar]
- 6.Cooke CL, An HJ, Kim J, Canfield DR, Torres J, Lebrilla CB, Solnick JV. Modification of gastric mucin oligosaccharide expression in rhesus macaques after infection with Helicobacter pylori. Gastroenterology. 2009;137:1061–1071, 1071.e1-1071.e8. doi: 10.1053/j.gastro.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Marcos NT, Magalhães A, Ferreira B, Oliveira MJ, Carvalho AS, Mendes N, Gilmartin T, Head SR, Figueiredo C, David L, et al. Helicobacter pylori induces beta3GnT5 in human gastric cell lines, modulating expression of the SabA ligand sialyl-Lewis x. J Clin Invest. 2008;118:2325–2336. [Google Scholar]
- 8.Curt MJC, Lecointe K, Mihalache A, Rossez Y, Gosset P, Leonard R, Robbe-Masselot C. Alteration or adaptation, the two roads for human gastric mucin glycosylation infected by Helicobacter pylori. Glycobiology. 2015;25:617–631. doi: 10.1093/glycob/cwv004. [DOI] [PubMed] [Google Scholar]
- 9.vanKlinken BJW, Dekker J, Buller HA, DeBolos C, Einerhand AWC. Biosynthesis of mucins (MUC2-6) along the longitudinal axis of the human gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 1997;273:G296–G302. doi: 10.1152/ajpgi.1997.273.2.G296. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Cheon DJ, Lu Z, Cunningham SL, Chen CM, Luo RZ, Xing D, Orsulic S, Bast RC Jr, Behringer RR. MUC16 expression during embryogenesis, in adult tissues, and ovarian cancer in the mouse. Differentiation. 2008;76:1081–1092. doi: 10.1111/j.1432-0436.2008.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGuckin MA, Lindén SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9:265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 12.Van de Bovenkamp JH, Mahdavi J, Korteland-Van Male AM, Büller HA, Einerhand AW, Borén T, Dekker J. The MUC5AC glycoprotein is the primary receptor for Helicobacter pylori in the human stomach. Helicobacter. 2003;8:521–532. doi: 10.1046/j.1523-5378.2003.00173.x. [DOI] [PubMed] [Google Scholar]
- 13.Van den Brink GR, Tytgat KM, Van der Hulst RW, Van der Loos CM, Einerhand AW, Büller HA, Dekker J. H pylori colocalises with MUC5AC in the human stomach. Gut. 2000;46:601–607. doi: 10.1136/gut.46.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossez Y, Gosset P, Boneca IG, Magalhães A, Ecobichon C, Reis CA, Cieniewski-Bernard C, Joncquel Chevalier Curt M, Léonard R, Maes E, et al. The lacdiNAc-specific adhesin LabA mediates adhesion of Helicobacter pylori to human gastric mucosa. J Infect Dis. 2014;210:1286–1295. doi: 10.1093/infdis/jiu239. [DOI] [PubMed] [Google Scholar]
- 15.Petersen AM, Krogfelt KA. Helicobacter pylori: an invading microorganism? A review. FEMS Immunol Med Microbiol. 2003;36:117–126. doi: 10.1016/S0928-8244(03)00020-8. [DOI] [PubMed] [Google Scholar]
- 16.Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 17.Backert S, Clyne M, Tegtmeyer N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun Signal. 2011;9:28. doi: 10.1186/1478-811X-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Comelli EM, Lariani S, Zwahlen MC, Fotopoulos G, Holzwarth JA, Cherbut C, Dorta G, Corthésy-Theulaz I, Grigorov M. Biomarkers of human gastrointestinal tract regions. Mamm Genome. 2009;20:516–527. doi: 10.1007/s00335-009-9212-7. [DOI] [PubMed] [Google Scholar]
- 19.Galamb O, Gyõrffy B, Sipos F, Dinya E, Krenács T, Berczi L, Szõke D, Spisák S, Solymosi N, Németh AM, et al. Helicobacter pylori and antrum erosion-specific gene expression patterns: the discriminative role of CXCL13 and VCAM1 transcripts. Helicobacter. 2008;13:112–126. doi: 10.1111/j.1523-5378.2008.00584.x. [DOI] [PubMed] [Google Scholar]
- 20.Galamb O, Sipos F, Gyorffy B, Molnar B, Tulassay Z. Identification of helicobacter pylori and antrum erosion-specific gene expression patterns in gastric biopsy samples by whole genomic microarray analysis. Helicobacter. 2005;10:465–466. [Google Scholar]
- 21.Galamb O, Sipos F, Molnar B, Szoke D, Spisak S, Tulassay Z. Evaluation of malignant and benign gastric biopsy specimens by mRNA expression profile and multivariate statistical methods. Cytometry B Clin Cytom. 2007;72:299–309. doi: 10.1002/cyto.b.20189. [DOI] [PubMed] [Google Scholar]
- 22.Galamb O, Sipos F, Molnar B, Tulassay Z. Diagnosis of malignancy and benign alterations in gastric biopsy specimen by mRNA expression profiling and multivariate statistical methods. Gastroenterology. 2004;126:A408–A408. [Google Scholar]
- 23.Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island-dependent manner. Gastroenterology. 2008;134:1049–1057. doi: 10.1053/j.gastro.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Resnick MB, Sabo E, Meitner PA, Kim SS, Cho Y, Kim HK, Tavares R, Moss SF. Global analysis of the human gastric epithelial transcriptome altered by Helicobacter pylori eradication in vivo. Gut. 2006;55:1717–1724. doi: 10.1136/gut.2006.095646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai CJ, Herrera-Goepfert R, Tibshirani RJ, Yang SF, Mohar A, Guarner J, Parsonnet J. Changes of gene expression in gastric preneoplasia following Helicobacter pylori eradication therapy. Cancer Epidemiol Biomarkers Prev. 2006;15:272–280. doi: 10.1158/1055-9965.EPI-05-0632. [DOI] [PubMed] [Google Scholar]
- 26.Toyoda T, Tsukamoto T, Yamamoto M, Ban H, Saito N, Takasu S, Shi L, Saito A, Ito S, Yamamura Y, et al. Gene expression analysis of a Helicobacter pylori-infected and high-salt diet-treated mouse gastric tumor model: identification of CD177 as a novel prognostic factor in patients with gastric cancer. BMC Gastroenterol. 2013;13:122. doi: 10.1186/1471-230X-13-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeno T, Ota H, Sugiyama A, Ishida K, Katsuyama T, Genta RM, Kawasaki S. Helicobacter pylori-induced chronic active gastritis, intestinal metaplasia, and gastric ulcer in Mongolian gerbils. Am J Pathol. 1999;154:951–960. doi: 10.1016/S0002-9440(10)65343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Etr SH, Mueller A, Tompkins LS, Falkow S, Merrell DS. Phosphorylation-independent effects of CagA during interaction between Helicobacter pylori and T84 polarized monolayers. J Infect Dis. 2004;190:1516–1523. doi: 10.1086/424526. [DOI] [PubMed] [Google Scholar]
- 29.You YH, Song YY, Meng FL, He LH, Zhang MJ, Yan XM, Zhang JZ. Time-series gene expression profiles in AGS cells stimulated with Helicobacter pylori. World J Gastroenterol. 2010;16:1385–1396. doi: 10.3748/wjg.v16.i11.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eftang LL, Esbensen Y, Tannæs TM, Bukholm IR, Bukholm G. Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiol. 2012;12:9. doi: 10.1186/1471-2180-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider S, Carra G, Sahin U, Hoy B, Rieder G, Wessler S. Complex Cellular Responses of Helicobacter pylori-Colonized Gastric Adenocarcinoma Cells. Infect Immun. 2011;79:2362–2371. doi: 10.1128/IAI.01350-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behrens I, Stenberg P, Artursson P, Kissel T. Transport of lipophilic drug molecules in a new mucus-secreting cell culture model based on HT29-MTX cells. Pharm Res. 2001;18:1138–1145. doi: 10.1023/a:1010974909998. [DOI] [PubMed] [Google Scholar]
- 33.Alemka A, Clyne M, Shanahan F, Tompkins T, Corcionivoschi N, Bourke B. Probiotic colonization of the adherent mucus layer of HT29MTXE12 cells attenuates Campylobacter jejuni virulence properties. Infect Immun. 2010;78:2812–2822. doi: 10.1128/IAI.01249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navabi N, McGuckin MA, Lindén SK. Gastrointestinal cell lines form polarized epithelia with an adherent mucus layer when cultured in semi-wet interfaces with mechanical stimulation. PLoS One. 2013;8:e68761. doi: 10.1371/journal.pone.0068761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolan B, Naughton J, Tegtmeyer N, May FE, Clyne M. The interaction of Helicobacter pylori with the adherent mucus gel layer secreted by polarized HT29-MTX-E12 cells. PLoS One. 2012;7:e47300. doi: 10.1371/journal.pone.0047300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naughton JA, Mariño K, Dolan B, Reid C, Gough R, Gallagher ME, Kilcoyne M, Gerlach JQ, Joshi L, Rudd P, et al. Divergent mechanisms of interaction of Helicobacter pylori and Campylobacter jejuni with mucus and mucins. Infect Immun. 2013;81:2838–2850. doi: 10.1128/IAI.00415-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Amsterdam K, van der Ende A. Nutrients released by gastric epithelial cells enhance Helicobacter pylori growth. Helicobacter. 2004;9:614–621. doi: 10.1111/j.1083-4389.2004.00272.x. [DOI] [PubMed] [Google Scholar]
- 38.Cottet S, Corthésy-Theulaz I, Spertini F, Corthésy B. Microaerophilic conditions permit to mimic in vitro events occurring during in vivo Helicobacter pylori infection and to identify Rho/Ras-associated proteins in cellular signaling. J Biol Chem. 2002;277:33978–33986. doi: 10.1074/jbc.M201726200. [DOI] [PubMed] [Google Scholar]
- 39.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 40.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 42.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaudier E, Forestier L, Gouyer V, Huet G, Julien R, Hoebler C. Butyrate regulation of glycosylation-related gene expression: evidence for galectin-1 upregulation in human intestinal epithelial goblet cells. Biochem Biophys Res Commun. 2004;325:1044–1051. doi: 10.1016/j.bbrc.2004.10.141. [DOI] [PubMed] [Google Scholar]
- 44.Hennebicq-Reig S, Lesuffleur T, Capon C, De Bolos C, Kim I, Moreau O, Richet C, Hémon B, Recchi MA, Maës E, et al. Permanent exposure of mucin-secreting HT-29 cells to benzyl-N-acetyl-alpha-D-galactosaminide induces abnormal O-glycosylation of mucins and inhibits constitutive and stimulated MUC5AC secretion. Biochem J. 1998;334(Pt 1):283–295. doi: 10.1042/bj3340283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo JM, Zhang Y, Cheng L, Iwasaki H, Wang H, Kubota T, Tachibana K, Narimatsu H. Molecular cloning and characterization of a novel member of the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase family, pp-GalNAc-T12. FEBS Lett. 2002;524:211–218. doi: 10.1016/s0014-5793(02)03007-7. [DOI] [PubMed] [Google Scholar]
- 46.Gomes C, Almeida A, Ferreira JA, Silva L, Santos-Sousa H, Pinto-de-Sousa J, Santos LL, Amado F, Schwientek T, Levery SB, et al. Glycoproteomic analysis of serum from patients with gastric precancerous lesions. J Proteome Res. 2013;12:1454–1466. doi: 10.1021/pr301112x. [DOI] [PubMed] [Google Scholar]
- 47.Campos D, Freitas D, Gomes J, Magalhães A, Steentoft C, Gomes C, Vester-Christensen MB, Ferreira JA, Afonso LP, Santos LL, et al. Probing the O-glycoproteome of gastric cancer cell lines for biomarker discovery. Mol Cell Proteomics. 2015;14:1616–1629. doi: 10.1074/mcp.M114.046862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomes J, Marcos NT, Berois N, Osinaga E, Magalhães A, Pinto-de-Sousa J, Almeida R, Gärtner F, Reis CA. Expression of UDP-N-acetyl-D-galactosamine: Polypeptide N-acetylgalactosaminyltransferase-6 in gastric mucosa, intestinal metaplasia, and gastric carcinoma. J Histochem Cytochem. 2009;57:79–86. doi: 10.1369/jhc.2008.952283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcos NT, Cruz A, Silva F, Almeida R, David L, Mandel U, Clausen H, Von Mensdorff-Pouilly S, Reis CA. Polypeptide GalNAc-transferases, ST6GalNAc-transferase I, and ST3Gal-transferase I expression in gastric carcinoma cell lines. J Histochem Cytochem. 2003;51:761–771. doi: 10.1177/002215540305100607. [DOI] [PubMed] [Google Scholar]
- 50.Magalhaes A, Marcos-Pinto R, Nairn AV, dela Rosa M, Ferreira RM, Junqueira-Neto S, Freitas D, Gomes J, Oliveira P, Santos MR, et al. Helicobacter pylori chronic infection and mucosal inflammation switches the human gastric glycosylation pathways. Biochimica Et Biophys Acta-Molecular Basis Dis. 2015;1852:1928–1939. doi: 10.1016/j.bbadis.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nairn AV, Aoki K, dela Rosa M, Porterfield M, Lim JM, Kulik M, Pierce JM, Wells L, Dalton S, Tiemeyer M, et al. Regulation of glycan structures in murine embryonic stem cells: combined transcript profiling of glycan-related genes and glycan structural analysis. J Biol Chem. 2012;287:37835–37856. doi: 10.1074/jbc.M112.405233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams SJ, Wreschner DH, Tran M, Eyre HJ, Sutherland GR, McGuckin MA. Muc13, a novel human cell surface mucin expressed by epithelial and hemopoietic cells. J Biol Chem. 2001;276:18327–18336. doi: 10.1074/jbc.M008850200. [DOI] [PubMed] [Google Scholar]
- 53.Shimamura T, Ito H, Shibahara J, Watanabe A, Hippo Y, Taniguchi H, Chen Y, Kashima T, Ohtomo T, Tanioka F, et al. Overexpression of MUC13 is associated with intestinal-type gastric cancer. Cancer Sci. 2005;96:265–273. doi: 10.1111/j.1349-7006.2005.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008;1:183–197. doi: 10.1038/mi.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith SM, Moran AP, Duggan SP, Ahmed SE, Mohamed AS, Windle HJ, O’Neill LA, Kelleher DP. Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol. 2011;186:2462–2471. doi: 10.4049/jimmunol.1000864. [DOI] [PubMed] [Google Scholar]
- 56.Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, Fox JG, Wang TC, Kurt-Jones EA. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun. 2004;72:6446–6454. doi: 10.1128/IAI.72.11.6446-6454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yokota S, Ohnishi T, Muroi M, Tanamoto K, Fujii N, Amano K. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Microbiol. 2007;51:140–148. doi: 10.1111/j.1574-695X.2007.00288.x. [DOI] [PubMed] [Google Scholar]
- 58.Uno K, Kato K, Atsumi T, Suzuki T, Yoshitake J, Morita H, Ohara S, Kotake Y, Shimosegawa T, Yoshimura T. Toll-like receptor (TLR) 2 induced through TLR4 signaling initiated by Helicobacter pylori cooperatively amplifies iNOS induction in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1004–G1012. doi: 10.1152/ajpgi.00096.2007. [DOI] [PubMed] [Google Scholar]
- 59.Ishihara S, Rumi MA, Kadowaki Y, Ortega-Cava CF, Yuki T, Yoshino N, Miyaoka Y, Kazumori H, Ishimura N, Amano Y, et al. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J Immunol. 2004;173:1406–1416. doi: 10.4049/jimmunol.173.2.1406. [DOI] [PubMed] [Google Scholar]
- 60.Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 61.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 62.Odenbreit S, Swoboda K, Barwig I, Ruhl S, Borén T, Koletzko S, Haas R. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect Immun. 2009;77:3782–3790. doi: 10.1128/IAI.00364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pachathundikandi SK, Tegtmeyer N, Backert S. Signal transduction of Helicobacter pylori during interaction with host cell protein receptors of epithelial and immune cells. Gut Microbes. 2013;4:454–474. doi: 10.4161/gmic.27001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pinto M, Appay MD, Simonassmann P, Chevalier G, Dracopoli N, Fogh J, Zweibaum A. Enterocytic differentiation of cultured human-colon cancer-cells by replacement of glucose by galactose in the medium. Biol Cell. 1982;44:193–196. [Google Scholar]
- 65.Huet C, Sahuquillo-Merino C, Coudrier E, Louvard D. Absorptive and mucus-secreting subclones isolated from a multipotent intestinal cell line (HT-29) provide new models for cell polarity and terminal differentiation. J Cell Biol. 1987;105:345–357. doi: 10.1083/jcb.105.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heiskala K, Andersson LC. Reg IV is differently expressed in enteroendocrine cells of human small intestine and colon. Regul Pept. 2013;183:27–34. doi: 10.1016/j.regpep.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 67.Mitani Y, Oue N, Matsumura S, Yoshida K, Noguchi T, Ito M, Tanaka S, Kuniyasu H, Kamata N, Yasui W. Reg IV is a serum biomarker for gastric cancer patients and predicts response to 5-fluorouracil-based chemotherapy. Oncogene. 2007;26:4383–4393. doi: 10.1038/sj.onc.1210215. [DOI] [PubMed] [Google Scholar]
- 68.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]