

 Fused tricyclic skeleton in one shot: a RhIII catalyzed formal [4 + 2 + 2] cyclization of N-pivaloyloxybenzamides 1 with 1,6-allene-enes 2 adding two cycles to the benzene ring compatible with ambient air and moisture with a tolerance of many synthetic useful functional groups at room temperature have been developed.
Fused tricyclic skeleton in one shot: a RhIII catalyzed formal [4 + 2 + 2] cyclization of N-pivaloyloxybenzamides 1 with 1,6-allene-enes 2 adding two cycles to the benzene ring compatible with ambient air and moisture with a tolerance of many synthetic useful functional groups at room temperature have been developed.
Abstract
A Rh(iii) catalyzed formal [4 + 2 + 2] cyclization of N-pivaloyloxybenzamides 1 with 1,6-allene-enes 2 by C–H functionalization is reported. The reactions occur at room temperature and are compatible with air and moisture with a tolerance of many synthetically useful functional groups. The follow-up modifications of the products have been demonstrated. After careful mechanistic studies and DFT calculation, a reaction mechanism was proposed.
Introduction
It is well known that medium-sized rings are hard to synthesize due to entropic and enthalpic reasons.1 Among them, eight-membered lactams are found in many natural products and bioactive molecules.2 The normal approach to these molecules often require harsh conditions (such as high temperatures and highly diluted solutions), at least stoichiometric amount of metal reagents, or pre-functionalized starting materials by several steps.3 Therefore, developing a catalytic procedure for the efficient synthesis of eight-membered lactams under mild conditions is still desired.
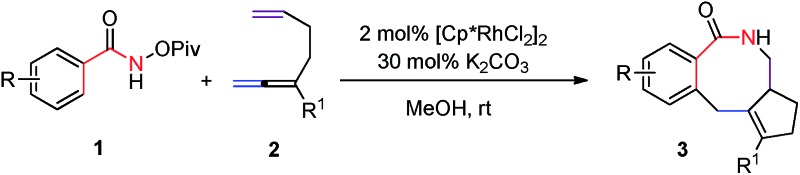
In principle, [2 + 2 + 2 + 2] or [4 + 2 + 2] cyclization would be the most straightforward yet challenging approach to the eight-membered lactams.4 On the other hand, in recent years, directed Rh(iii)-catalyzed C–H functionalization reactions have been extensively studied.5 Among them, the formal [4 + 2]-cyclization between arenes or alkenes with an appropriate directing group and unsaturated compounds such as alkenes, alkynes or allenes facilely affording six-membered products have been well established. 6–12 We envisioned a strategy for the efficient construction of eight-membered rings if a [2 + 2]-atom component, i.e., a molecule contains two unsaturated carbon–carbon bonds, is applied instead of alkenes, alkynes or allenes. The challenge is the highly unfavorable selectivity for the formation of an eight-membered ring since it is much easier to form six-membered rings (A′ vs. B) with the participation of just one C–C π-bond (Scheme 1). Herein, we wish to report the realization of such a concept by applying an allene-ene as the [2 + 2]-atom component catalyzed by [Cp*RhCl2]2 with N-pivaloyloxy benzamides as the [4]-atom component under very mild conditions.
Scheme 1. Rh(iii)-catalyzed cyclization: [4 + 2] vs. [4 + 2 + 2].

Results and discussion
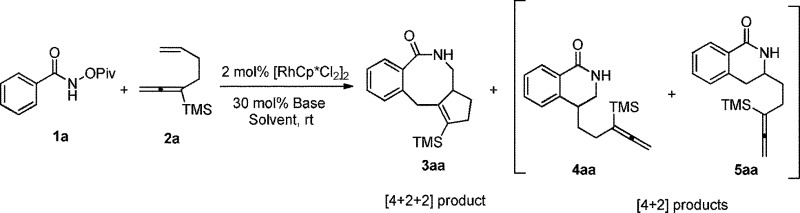
After some trial and error with compounds bearing at least two unsaturated C–C bond, we identified such a reaction, in which 3-(trimethylsilyl)-1,2,6-heptatriene 2a acts as a [2 + 2]-atom component while N-pivaloyloxybenzamide 1a behaves as the [4]-atom component. When 1a was treated with 2a in a mixed solvent of MeOH and water (v/v 20/1) under the catalysis of [Cp*RhCl2]2, the expected [4 + 2 + 2] cyclization product, i.e., the benzo-fused eight-membered lactam 3aa, was indeed formed in 75% NMR yield although together with the [4 + 2] regioisomeric mixture13 of two products 4aa and 5aa with the isolated carbon–carbon double bond being reacted as the minor products (Table 1, entry 1). The structure of 3aa was confirmed by X-ray diffraction study (Scheme 2),14 indicating that the allene unit has been reacted first. After screening the solvent, we found that MeOH was the best (Table 1, entries 1–5). Moisture, air and 4 Å MS had very little influence on the yield (Table 1, entries 6 and 7). Then, the base effect was investigated and 30 mol% of K2CO3 was found to be the optimal (Table 1, entries 2 and 8–13). Control experiments showed that no reaction occurred in the absence of [Cp*RhCl2]2 or bases. Finally, the reaction of 1a and 2a in methanol under the catalysis of [Cp*RhCl2]2 with 30 mol% K2CO3 as base at room temperature was chosen as the optimal reaction conditions for further study (Table 1, entry 11).
Table 1. Optimization of reaction conditions: the Rh-catalyzed cyclization of 1a with 2a a .
| |||||
| Entry | Solvent | Base | t/h | NMR yield
b
(%) |
|
| 3aa | 4aa + 5aa | ||||
| 1 | MeOH/H2O c | NaOAc | 11 | 75 | 12 |
| 2 | MeOH | NaOAc | 21 | 80 | 14 |
| 3 | DCE | NaOAc | 21 | 58 | 17 |
| 4 | DCM | NaOAc | 21 | 65 | 21 |
| 5 | Toluene | NaOAc | 21 | 44 | 17 |
| 6 d | MeOH | NaOAc | 14 | 80 | 16 |
| 7 e | MeOH | NaOAc | 14 | 74 | 15 |
| 8 | MeOH | KOAc | 14 | 68 | 14 |
| 9 | MeOH | CsOAc | 14 | 69 | 14 |
| 10 | MeOH | Na2CO3 | 14 | 69 | 14 |
| 11 | MeOH | K2CO3 | 14 | 81 (72) f | 14 |
| 12 | MeOH | Cs2CO3 | 14 | 76 | 14 |
| 13 | MeOH | K3PO4 | 11 | 73 | 13 |
| 14 g | MeOH | — | 11 | — | — |
| 15 h | MeOH | K2CO3 | 13 | 75 | 14 |
| 16 i | MeOH | K2CO3 | 13 | 72 | 13 |
| 17 j | MeOH | K2CO3 | 13 | 18 | 7 |
| 18 g , k | MeOH | K2CO3 | 13 | — | — |
aThe reaction was conducted with 1a (0.2 mmol), 2a (0.2 mmol), [Cp*RhCl2]2 (0.004 mmol), K2CO3 (0.06 mmol), MeOH (1.2 mL), and monitored by TLC.
bDetermined by 1H NMR using dibromomethane as internal standard.
cThe ratio of MeOH/H2O was 20/1 (1.2 mL/0.06 mL).
dUnder N2 atmosphere.
eUnder N2 atmosphere and 4 Å MS was added.
fIsolated yield in parentheses.
gRecovery of 1a was 98% with 2a disappeared.
h10 mol% K2CO3 was added.
i50 mol% K2CO3 was added.
j1 equiv. K2CO3 was added.
kThe reaction was conducted in the absence of the Rh(iii) catalyst.
Scheme 2. ORTEP representation of 3aa.

When the reaction was conducted on a gram-scale, 3aa was also formed in 62% yield (Table 2, entry 2). The reaction of substrates with electron-donating groups such as methyl, tert-butyl and methoxy or electron-withdrawing groups such as COOMe, Cl, Br, CF3 and NO2 in the para position of the aryl unit all gave corresponding lactams 3ba–3ia in moderate to good yields, showing the potential for further elaboration (Table 2, entries 3–10); when o-methyl N-pivaloyloxybenzamide 1j was used, lactam 3ja could also be formed in a lower yield (23%) with full conversion, most probably due to the steric effect (Table 2, entry 11). It is important to note that silyl group could be replaced with aryl groups with either electron-withdrawing or electron-donating substituents, a hetero-aryl and even an alkyl group, showing the broad substrate scope (Table 2, entries 12–16). The structure of 3ae was further confirmed by X-ray diffraction study (see p. 26 in the ESI†).15 It should be noted that 4- and 5-type by-products were also formed as by-products as judged by 1H NMR analysis (Table 2).
Table 2. The reaction scope a .
| |||||
| Entry | R | R1 | t/h | Yield of 3 (%) | NMR yield of [4 + 5] b (%) |
| 1 | H (1a) | TMS (2a) | 2 | 68 (3aa) | 11 |
| 2 c | H (1a) | TMS (2a) | 2 | 62 (3aa) | N.D. d |
| 3 | 4-Me (1b) | TMS (2a) | 2 | 57 (3ba) | 10 |
| 4 | 4- t Bu (1c) | TMS (2a) | 2 | 47 (3ca) e | 8 |
| 5 | 4-OMe (1d) | TMS (2a) | 2 | 55 (3da) | 10 |
| 6 | 4-CO2Me (1e) | TMS (2a) | 2.5 | 60 (3ea) | 10 |
| 7 | 4-Cl (1f) | TMS (2a) | 2 | 50 (3fa) | 10 |
| 8 | 4-Br (1g) | TMS (2a) | 2 | 50 (3ga) | 11 |
| 9 | 4-CF3 (1h) | TMS (2a) | 3 | 53 (3ha) | 11 |
| 10 | 4-NO2 (1i) | TMS (2a) | 12 | 62 (3ia) | 7 |
| 11 | 2-Me (1j) | TMS (2a) | 48 | 23 (3ja) | 8 |
| 12 | H (1a) | Ph (2b) | 5 | 56 (3ab) f | N.D. |
| 13 | H (1a) | p-ClC6H4 (2c) | 18 | 63 (3ac) g | N.D. |
| 14 | H (1a) | p-MeOC6H4 (2d) | 10 | 45 (3ad) h | N.D. |
| 15 i | H (1a) | 3-Thienyl (2e) | 48 | 47 (3ae) j | N.D. |
| 16 k | H (1a) | Bu (2f) | 14 | 34 (3af) | N.D. |
aThe reaction was conducted with 1 (1 mmol), 2 (1 mmol), [Cp*RhCl2]2 (0.02 mmol), K2CO3 (0.3 mmol) and MeOH (6 mL), and monitored by TLC.
bDetermined by 1H NMR using dibromomethane as internal standard.
cReaction was conducted on 6 mmol scale.
dNot determined.
e97% purity.
f94% purity.
g92% purity.
h91% purity.
iReaction was conducted at 55 °C.
j90% purity.
k 1a (1.5 mmol), 2f (1 mmol) and [Cp*RhCl2]2 (0.04 mmol) were used.
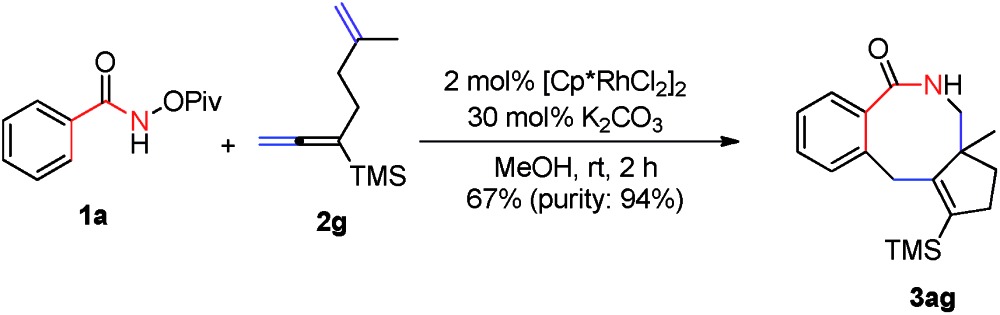
Importantly, N-pivaloyloxy-2-naphthylamide 1k afforded tetracyclic product 3ka in 48% yield with the 3-position C–H bond being exclusively functionalized (eqn (1)). The substituent on the internal side of the alkene moiety was also tolerated affording lactam 3ag with a methyl group on the quaternary carbon center which was hard to form in 67% yield (eqn (2)).
 |
1 |
 |
2 |
Other frequently used directing groups 6b–e 5 failed to yield any expected products with a high recovery of the starting materials (Scheme 3a). The steric effect of the silyl group in the allene moiety was also observed: when TMS was replaced by the bulkier TIPS group, the allene moiety was untouched: only the isolated C–C double incorporated regioisomeric products 4ah and 5ah were formed (Scheme 3b). When N-methoxybenzamide 6a was used to react with 2a under the standard conditions for 96 h, both unsaturated units were involved in the transformation, however, the most commonly observed β-H elimination occurred, affording a five-membered ring in the final product 7aa in 10% isolated yield (Scheme 3c).
Scheme 3. Failed directing groups and steric effect.

Furthermore, when the reaction of 1a was conducted in [D4]-methanol under the standard conditions in the absence or presence of allene-ene 2a, no obvious deuterium incorporation was detected from the recovered 1a and formed 3aa, suggesting that the C–H activation step was irreversible (Scheme 4a and b). Then the kinetic isotope effect (KIE) value of 1a and 1a-d5 was measured by parallel experiments. The high KIE value of 12.3 demonstrated that C–H activation must be involved in the rate-determining step (Scheme 4c).16
Scheme 4. Mechanistic studies.

The orders of each reactant were also measured. A linear relationship was observed for ln([1a]) vs. reaction time, indicating a first-order dependence of the reaction rate with 1a [ln([1a]) = –kt + ln([1a0 ])] (y = a + bx in Scheme 5). A zero-order with the allene moiety 2a ([2a] = –kt + [2a0 ]) (y = a + bx in Scheme 6) was also determined by the linear relationship of [2a] vs. reaction time. Thus, the rate equation for this reaction is d[3aa]/dt = k[1a]1[2a]0.
Scheme 5. The determination of the reaction order with 1a.

Scheme 6. The determination of the reaction order with 2a.

According to the experimental results above, a plausible mechanism has been proposed as shown in Scheme 7. First, [Cp*Rh(CO3)] was formed to be the catalytic species from the [Cp*RhCl2]2 dimer and CO3 2–. Then base-promoted C–H bond rhodation would form rhodacyclic intermediate Int 1, 5–12 which is followed by the insertion of the allene moiety in the 1,6-allene-ene 2a, affording seven-membered rhodacycle Int 2.17 Subsequent cyclic carborhodation of the terminal C–C double bond would yield nine-membered rhodacyclic intermediate Int 3 (Int 3′).17 The N-pivaloyloxy containing intermediate Int 3 (R = Piv) would undergo reductive elimination to form the final [4 + 2 + 2] product 3aa. Alternatively, the N-methoxy intermediate Int 3′ (R = Me) leads exclusively to the methylenecyclopentene product 7aa by β-H elimination (Scheme 3c). Thus, it is concluded that the OPiv group is important since the coordination of the carbonyl oxygen atom in the Piv group of Int 3 with Rh(iii) makes a coordination-saturated stable 18e complex, which must have prevented the β-H elimination, avoiding the formation of 7aa-type product.18
Scheme 7. A possible mechanism.

Follow-up modifications of the products
N-Protected eight-membered benzolactams have been found to be bioactive as dopamine D3 receptor ligands.2i Thus, product 3aa has been treated with 1-(3-chloropropyl)-4-(p-methoxyphenyl)piperazine to afford the analogue 9 in 75% yield (Scheme 8a). To further demonstrate the synthetic applications of the products, the functionalization of the TMS group in 3aa had been conducted: (a) desilylation product 10 could be formed in 63% yield by treating 3aa with TiCl4 in DCM for 16 h. 17c,19 (b) The TMS group of 3aa could be transformed to synthetically useful vinyl iodide 11,20 which will be useful for any elaboration based on cross coupling reactions (Scheme 8b).
Scheme 8. Follow-up modifications of the products.

Theoretical studies on mechanism
DFT calculations have been performed, using 3-methyl-1,2,6-heptatriene (2a′) as the model of substrate 2a and N-acetoxybenzamide (1a′) as the model of N-pivaloyloxybenzamide 1a, in which the TMS group in 2a and the tertiary butyl group in 1a have both been replaced with the methyl group (eqn (3)). We believe that the catalytic species involved in this reaction is a carbonate-ligated species, Cp*Rh(CO3), which may be generated via Rh dimer dissociation and ligand exchange from [Cp*RhCl2]2 and K2CO3. The energetic profiles for the initial steps of the C–H activation and the subsequent carborhodations are provided in Fig. 1. The initial formation of the complex IN1 (selected as the free energy reference) involves the coordination of the nitrogen atom in the N-acetoxybenzamide substrate (2a′) with the Rh center of Cp*Rh(CO3), which would be followed by the deprotonation of the N–H unit by the CO3 2–. This N–H deprotonation step is computed to require a 12.2 kcal mol–1 activation barrier (TS1) to afford the N-metalated intermediate IN2, which is 4.5 kcal mol–1 less stable than the precursor IN1. Subsequently, IN2 isomerizes to a more stable intermediate IN3, in which the Rh···Ohydroxyl coordination in IN2 is replaced with the Rh···Ocarbonyl coordination, with a free energy of 6.6 kcal mol–1 lower than the starting point (IN1).
 |
3 |
Fig. 1. The energetic profiles for the C–H activation and the carborhodations of IN1.

The following aryl C–H activation step is realized by a concerted metalation/deprotonation (CMD) process 21a–k (also termed as ambiphilic metal ligand activation (AMLA))21l via a six-membered cyclic transition state TS2, in which the Rh–C bond formation occurs simultaneously with the deprotonation of the ortho aromatic proton to afford the rhodacyclic intermediate IN4. This CMD step is computed to feature the highest relative energy barrier of the whole profile (20.8 kcal mol–1, TS2, Fig. 1) and may, thus, be rate-determining. This calculated prediction is in consistent with the results of the above mentioned kinetics and deuterium exchange experiments (Schemes 4–6).
The dissociation of H2CO3 from IN4 and the coordination of the allene moiety in the 1,6-allene-ene 2a′ affords IN5 with a relative energy of 4.0 kcal mol–1 lower than the starting point. Then the coordinated distal allenic double bond undergoes an insertion into the Rh–C bond via TS3 to provide the seven-membered rhodacycle intermediate IN6, in which the N-acetoxy moiety coordinates to Rh center with the pendant carbonyl oxygen. This cyclic carborhodation step is computed to be exergonic (ΔG sol = –11.4 kcal mol–1) and requires a 14.7 kcal mol–1 activation barrier (TS3, Fig. 1).
According to the experimental results, the [4 + 2] regioisomeric products 4aa and 5aa are obtained as the minor by-products simultaneously with the desired [4 + 2 + 2] product 3aa. To validate the selectivity, the pathways for the competing insertion of the isolated carbon–carbon double bond with either direction into the Rh–C bond have also been calculated, which are depicted in Scheme 9. The two competing insertion pathways are realized via TS3′ and TS3′′, respectively, with similar activation energy barriers of over 17 kcal mol–1, which are about 2.5 kcal mol–1 higher than TS3, indicating that the terminal allenic double bond is more reactive than the isolated double bond in this insertion step. So the terminal allenic double bond is calculated to carborhodate preferentially, which is in accordance with experimental observations of 4aa and 5aa as minor by-products.
Scheme 9. The activation free energies of forming 4aa′ and 5aa′ (ΔG sol (ΔG 298 K)).

From the seven-membered rhodacycle IN6, subsequent second insertion of the terminal C–C double bond is a key step for the formation of the final eight-membered lactam 3aa product. Besides the second cyclic carborhodation pathway, the possibility of other competitive pathways, such as C–N bond formation and β-H elimination from IN6, are also be investigated. The energetic profiles for possible reactions from IN6 are presented in Fig. 2. To facilitate the second cyclic carborhodation, the seven-membered rhodacycle IN6 should first isomerize to the more stable intermediate IN7 (11.7 kcal mol–1 lower in energy than IN6), in which the Rh center coordinates with the terminal C–C double bond instead of the carbonyl moiety in IN6. Then the subsequent insertion of the terminal C–C double bond into the Rh–C bond occurs via TS4 with an activation barrier of 9.1 kcal mol–1. This step is exergonic with the generation of the nine-membered rhodacyclic intermediate IN8, which is also stabilized by the coordination of the pendant carbonyl oxygen in the N-acetoxy moiety with the Rh center.
Fig. 2. The energetic profiles for possible cyclic carborhodation, C–N bond formation and β-H elimination pathways from IN6.

The activation free energy of direct reductive elimination from IN6 is computed to be 35.5 kcal mol–1 (TS5, Fig. 2), indicating that this direct C–N formation step is not favorable kinetically. Based on the mechanistic investigations of a closely related systems,22 a more kinetically favorable pathway of C–N formation from IN6 is discovered through a high oxidation state Rh(v) nitrene species IN9,23 generated by the migration of the acetoxy moiety from N to Rh via a five-membered transition state (TS6, Fig. 2). This migration step is computed to be slightly endergonic (ΔG sol = 6.8 kcal mol–1) and requires a 13.8 kcal mol–1 activation barrier (TS6). The nitrene Rh(v) species IN9 has a Rh N distance of only 1.867 Å, which is much shorter than the Rh–N single bond distance of 2.162 Å in IN8. The following reductive elimination of nitrene Rh(v) species IN9 is much easier with a barrier of only 7.7 kcal mol–1 via TS7, affording IN10 irreversibly with high exergonicity. Overall, the activation energy for the C–N bond formation pathway from IN6 is 14.5 kcal mol–1, as defined by the energy gap between TS7 and IN6.
Another competitive pathway from IN6 is the β-H elimination process, affording the allenylation product. In this step, IN6 should first isomerize into IN11 (Fig. 2), in which the β-H coordinates with the Rh center to be perfectly oriented for elimination, with the free energy increased by about 2.5 kcal mol–1. The β-H elimination occurs subsequently via TS8 with an activation barrier of 5.8 kcal mol–1, leading to rhodium hydride species IN12, which is slightly higher in energy by 4.4 kcal mol–1 than the corresponding precursor (IN11). However, the following reductive elimination of the rhodium hydride species IN12 is not as facile as the β-H elimination. This reductive elimination step is computed to require an activation barrier of 11.8 kcal mol–1 (TS9, Fig. 2). Hence, it should overcome an activation energy of 18.7 kcal mol–1 totally for the allenation product generation, which is the free energy gap between TS9 and IN6.
Thus, as can be deduced from the energetic data mentioned above, the second cyclic carborhodation pathway forming the nine-membered rhodacyclic intermediate IN8 (ΔG≠sol = 9.1 kcal mol–1) is the most kinetically favorable among the several alternative pathways from IN6, which is in good agreement with the experimental observations.
Continuing with the favorable pathway from the nine-membered rhodacyclic intermediate IN8, there are also two competitive pathways, forming the final [4 + 2 + 2] product 3aa′ by reductive elimination and the methylenecyclopentene product 7aa′ by β-H elimination, respectively. The energetic profiles of both possible pathways from IN8 are presented in Fig. 3 for comparison.
Fig. 3. The energetic profiles for possible C–N bond formation and β-H elimination pathways from IN8.

Similar with the about mentioned C–N formation pathway from IN6 (Fig. 2), the migration of the acetoxy moiety from N to Rh occurs prior to the C–N bond formation, generating a high oxidation state Rh(v) nitrene intermediate IN14, which is 4.8 kcal mol–1 higher in energy than the precursor (IN8, Fig. 3). This migration step is realized via a five-membered transition sate TS11, overcoming an activation barrier of 14.2 kcal mol–1. Then subsequent reductive elimination of the nitrene Rh(v) species IN14 is very facile via TS12 with a barrier of only 0.8 kcal mol–1, exergonic largely by over 68 kcal mol–1, to form the eight-membered ring in IN15. Then the protonolysis of the N atom of IN15 by HCO3 – would regenerate the catalytically active species Rh(iii) and yield the final product 3aa′, with an overall exergonicity of 130 kcal mol–1.
For the β-H elimination process from IN8 (Fig. 3), isomerization of IN8 into IN16 should occur first, with the free energy increased by 5.2 kcal mol–1, which is caused by the lower stability of C–H···Rh coordination in IN16 than Ocarbonyl···Rh coordination in IN8. In the following step, rhodium hydride species IN17 would be formed by β-H elimination of IN16 via TS13 with an activation barrier of only 3.1 kcal mol–1. The subsequent reductive elimination of the rhodium hydride species IN17 is highly energy demanding via TS14, hence, raising the barrier of the whole β-H elimination process of IN8 to 27.1 kcal mol–1, which is 12.9 kcal mol–1 higher than the alternative C–N bond formation pathway. Therefore, the final [4 + 2 + 2] product is obtained experimentally, instead of the 7-type methylenecyclopentene product 7aa′.
Thus, the whole catalytic cycle for the [4 + 2 + 2] cyclization mechanism is completed by DFT calculations. Shown in Fig. 4 is the reaction coordinate of the most kinetically favorable pathway involving N–H deprotonation/CMD activation of C–H bond/first carborhodation of the distal allenic double bond/second cyclic carborhodation of the isolated carbon–carbon double/C–N bond formation steps. As can be deduced from the energetic data in Fig. 4, the [4 + 2 + 2] cyclization reaction is calculated to occur irreversibly, as the final eight-membered lactam product lies over 130 kcal mol–1 below the starting point, and the CMD C–H bond activation is the rate-limiting step, with the highest relative energy barrier of 20.8 kcal mol–1 (TS2), which agrees well with the observations of the kinetics and deuterium exchange experiments (Schemes 3–6).
Fig. 4. The summary of the free energy profile for the whole [4 + 2 + 2] cyclization reaction.

To further elucidate the influence of the N–OR moiety on the reaction, we also investigated the possible pathways from the N-methoxy containing intermediate IN8_Me. Fig. 5 presents the free energy profiles for the favored pathway of β-H elimination and other two competitive possibilities. IN8_Me should first isomerize to IN16_Me, in which the β-H atom has an agostic interaction with the Rh center and be well oriented for the following elimination. The subsequent β-H elimination step leads to the rhodium hydride species IN17_Me via TS13_Me, with a barrier of only 5.1 kcal mol–1. The subsequent reductive elimination breaks the Rh–H bond and forms the N–H bond simultaneously by overcoming a free energy barrier of 21.5 kcal mol–1 (TS14_Me) to afford IN18_Me. Thus, the overall β-H elimination process of IN8_Me is facile, with a 24.6 kcal mol–1 activation barrier. What's more, the direct C–N bond forming process from IN8_Me via TS10_Me with an even higher activation barrier of 53.8 kcal mol–1, could be ruled out safely. And the similar intramolecular oxidation of the Rh(iii) species IN8_Me to Rh(v) nitrene intermediate by the migration of the N–OMe moiety via a three-center TS is also highly energy demanding (39.7 kcal mol–1, TS11_Me). Therefore, the N-methoxy containing intermediate IN8_Me leads to the 7-type methylenecyclopentene product by β-H elimination exclusively (Scheme 3c).
Fig. 5. The energetic profiles for the β-H elimination pathway from IN8_Me.

We concluded that the key factor of the influence of the N–OR moiety on the reaction selectivity could be attributed to the extra coordination of the carbonyl oxygen in the N-acetoxy moiety with the Rh center in IN8. First of all, this extra coordination in IN8 facilitates the migration of the acyloxy from N to Rh, thus enable the formation of the high oxidation state Rh(v) nitrene intermediate IN14 (Fig. 3), which is the C–N bond formation precursor. Secondly, the extra stabilization of IN8 by this kind of coordination increases the barrier of the whole β-H elimination process in the N-acetoxy containing system, avoiding the formation of the 7-type methylenecyclopentene product.
Conclusions
In conclusion, we have developed the first [Cp*RhCl2]2-catalyzed [4 + 2 + 2] cyclization between N-pivaloyloxy benzamides and 1,6-allene-enes forming eight-membered lactams. These reactions proceed at room temperature and are compatible with ambient air and moisture. Many arenes and allene-enes with many synthetically useful functional groups are compatible in the reactions. After careful mechanistic studies and DFT calculation, the reaction mechanism was presented in detail and the role of the N–OR moiety on the selectivity for the formation of the final products is also unveiled. The formed lactams have been transformed to cyclic vinylic iodide or desilylated product as well as the analogue of dopamine D3 receptor. With easy availability of both starting materials and new concept to readily synthesize the 8-membered rings, which are otherwise hard to construct by other means, this [4 + 2 + 2] cyclization protocol will be of high interest in organic chemistry and related disciplines. Further studies involving three or more components and chiral [4 + 2 + 2] reaction are being carried out in our laboratory.
Experimental section
Materials
[Cp*RhCl2]2 was purchased from Strem. N-Pivaloyloxybenzamides were prepared according to the literature procedures. 7c,o Grignard reagents were prepared from the corresponding aryl halides and magnesium. Other commercially available chemicals were purchased and used without additional purification unless noted otherwise. 1H NMR spectra were recorded on a Bruker-300 MHz spectrometer (except for 2h, a Bruker-700 MHz spectrometer was used) and 13C NMR spectra were recorded at 75 MHz. All 1H NMR experiments were measured with tetramethylsilane (0 ppm) or the signal of residual CHCl3 (7.26 ppm) in CDCl3 as the internal reference, 13C NMR experiments were measured in relative to the signal of CDCl3 (77.0 ppm), and 19F NMR experiments were measured in relative to the signal of residual CFCl3 (0 ppm) in CDCl3.
Typical procedure for the [4 + 2 + 2] cyclization reactions
To a dried Schlenk tube equipped with a Teflon-coated magnetic stirring bar were added N-pivaloyloxybenzamide 1a (221.6 mg, 1 mmol), [Cp*RhCl2]2 (12.7 mg, 0.02 mmol), K2CO3 (41.3 mg, 0.3 mmol), 3-(trimethylsilyl)-1,2,6-heptatriene 2a (166.0 mg, 1 mmol) and MeOH (6 mL) sequentially at rt. After being stirred for 2 h, the reaction was complete as monitored by TLC. Filtration through a short column of silica gel (eluent: MeOH) and evaporation afforded the crude product, which was purified by flash column chromatography on silica gel (eluent: dichloromethane/ethyl acetate = 4/1) to afford 3aa (194.1 mg, 68%): solid; mp 188.2–188.9 °C (hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.77–7.65 (m, 1H, NH), 7.48–7.33 (m, 2H, Ar–H), 7.29 (t, J = 6.2 Hz, 2H, Ar–H), 3.80 (d, J = 13.5 Hz, 1H, one proton of CH2), 3.46 (d, J = 13.2 Hz, 1H, one proton of CH2), 3.26–3.10 (m, 1H, one proton of NCH2), 2.96–2.75 (m, 2H, one proton of NCH2 + CH), 2.39–2.16 (m, 2H, CH2), 1.98–1.85 (m, 1H, one proton of CH2), 1.14–0.94 (m, 1H, one proton of CH2), 0.26 (s, 9H, 3 × CH3); 13C NMR (75 MHz, CDCl3) δ 173.9, 150.6, 137.6, 137.2, 134.6, 130.3, 129.2, 127.4, 126.8, 52.0, 46.5, 35.7, 34.9, 28.9, –0.07; IR (neat, cm–1) 3291, 3187, 3063, 2945, 2852, 1660, 1604, 1460, 1446, 1404, 1353, 1338, 1248, 1048, 1038, 1003; MS (EI, 70 eV) m/z (%) 286 (M+ + 1, 18.79), 285 (M+, 62.45), 256 (100), 255 (100), 73 (100); anal. calc. for C17H23NOSi (M W = 285.5): C 71.53, H 8.12, N 4.91; found: C 71.50, H 8.14, N 4.73%.
Computational methodology
All calculations were performed with the Gaussian 09 program.24 Geometries have been fully optimized with the density functional theory of M06 method. 25,26 The standard 6-31G(d,p)27 basis set was used for carbon, hydrogen, nitrogen and oxygen atoms and LANL2DZ basis set28 with effective core potential (ECP) for rhodium. Harmonic vibration frequency calculations were carried out for all the stationary points to confirm each structure being either a minimum (no imaginary frequency) or a transition structure (one imaginary frequency). Solvent effect has been considered by using the CPCM29 (UAHF atomic radii) model based on the gas-phase-optimized structures. The reported relative energies are free energies at 298 K (ΔG 298 K) in the gas phase and the Gibbs free energies (ΔG sol) in methanol (dielectric constant ε = 32.61), unless otherwise specified.
Acknowledgments
Financial support is acknowledged from National Basic Research Program (2015CB856600) of China and National Natural Science Foundation (21232006 and 21472224). S. M. is a Qiu Shi Adjunct Professor at Zhejiang University. We thank Dr Weiming Yuan in this group for reproducing the results for synthesis of 3ea, 3ac and 3ka.
Footnotes
References
- For related reviews, see: ; (a) Shiina I. Chem. Rev. 2007;107:239. doi: 10.1021/cr050045o. [DOI] [PubMed] [Google Scholar]; (b) Illuminati G., Mandolini L. Acc. Chem. Res. 1981;14:95. [Google Scholar]
- (a) Arndt R. R., Eggers S. H., Jordaan A. Tetrahedron. 1969;25:2767. [Google Scholar]; (b) Sato H., Kawagishi H., Nishimura T., Yoneyama S., Yoshimoto Y., Sakamura S., Furusaki A., Katsuragi S., Matsumoto T. Agric. Biol. Chem. 1985;49:2969. [Google Scholar]; (c) Laakso J. A., Gloer J. B., Wicklow D. T., Dowd P. F. J. Org. Chem. 1992;57:2066. [Google Scholar]; (d) Belofsky G. N., Gloer J. B., Wicklow D. T., Dowd P. F. Tetrahedron. 1995;51:3959. [Google Scholar]; (e) Zintchema A. A., Bikoboa D. N., Atchadé A. T., Mbing J. N., Gangoue-Pieboji J., Tih R. G., Blond A., Pegnyemb D. E., Bodo B. Phytochemistry. 2008;69:2209. doi: 10.1016/j.phytochem.2008.04.013. [DOI] [PubMed] [Google Scholar]; (f) Qiao M., Ji N., Liu X., Li K., Zhu Q., Xue Q. Bioorg. Med. Chem. Lett. 2010;20:5677. doi: 10.1016/j.bmcl.2010.08.024. [DOI] [PubMed] [Google Scholar]; (g) Ndongo J. T., Shaaban M., Mbing J. N., Bikobo D. N., Théodore Atchadé A., Pegnyemb D. E., Laatsch H. Phytochemistry. 2010;71:1872. doi: 10.1016/j.phytochem.2010.08.006. [DOI] [PubMed] [Google Scholar]; (h) Njocka G. B., Bartholomeusz T. A., Bikoboc D. N., Foroozandeh M., Shivapurkar R., Christen P., Pegnyemb D. E., Jeannerat D. Helv. Chim. Acta. 2013;96:1298. [Google Scholar]; (i) Ortega R., Raviña E., Masaguer C. F., Areias F., Brea J., Loza M. I., López L., Selent J., Pastor M., Sanz F. Bioorg. Med. Chem. Lett. 2009;19:1773. doi: 10.1016/j.bmcl.2009.01.067. [DOI] [PubMed] [Google Scholar]
- For reviews, see: ; (a) Gritsch P. J., Leitner C., Pfaffenbach M., Gaich T. Angew. Chem., Int. Ed. 2014;53:1208. doi: 10.1002/anie.201307391. [DOI] [PubMed] [Google Scholar]; (b) Maier M. E. Angew. Chem., Int. Ed. 2000;39:2073. doi: 10.1002/1521-3773(20000616)39:12<2073::aid-anie2073>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]; (c) Molander G. A. Acc. Chem. Res. 1998;31:603. [Google Scholar]; (d) Parker D., Macrocycle synthesis: a practical approach, Oxford University Press, Oxford, 1996. [Google Scholar]; (e) Kitagaki S., Teramoto S., Mukai C. Org. Lett. 2007;9:2549. doi: 10.1021/ol0709329. [DOI] [PubMed] [Google Scholar]; (f) Ohno H., Hamaguchi H., Ohata M., Tanaka T. Angew. Chem., Int. Ed. 2003;42:1749. doi: 10.1002/anie.200250589. [DOI] [PubMed] [Google Scholar]; (g) Klapars A., Parris S., Anderson K. W., Buchwald S. L. J. Am. Chem. Soc. 2004;126:3529. doi: 10.1021/ja038565t. [DOI] [PubMed] [Google Scholar]; (h) Spring D. R., Krishnan S., Schreiber S. L. J. Am. Chem. Soc. 2000;122:5656. [Google Scholar]; (i) Ma S., Gu Z. J. Am. Chem. Soc. 2006;128:4942. doi: 10.1021/ja057985a. [DOI] [PubMed] [Google Scholar]; (j) Zhu C., Zhang X., Lian X., Ma S. Angew. Chem., Int. Ed. 2012;51:7817. doi: 10.1002/anie.201202971. [DOI] [PubMed] [Google Scholar]
- For a [4 + 2 + 2] of 4,6-heptatrienyl isocyanates with terminal alkynes for the formation of similar [6.3.0]-bicyclic products, see: ; (a) Yu R. T., Friedman R. K., Rovis T. J. Am. Chem. Soc. 2009;131:13250. doi: 10.1021/ja906641d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miege F., Meyer C., Sossy J. Angew. Chem., Int. Ed. 2011;50:5932. doi: 10.1002/anie.201101220. [DOI] [PubMed] [Google Scholar]
- For reviews of Rh(iii)-catalyzed C–H activations, see: ; (a) Satoh T., Miura M. Chem.–Eur. J. 2010;16:11212. doi: 10.1002/chem.201001363. [DOI] [PubMed] [Google Scholar]; (b) Colby D. A., Bergman R. G., Ellman J. A. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Song G., Wang F., Li X. Chem. Soc. Rev. 2012;41:3651. doi: 10.1039/c2cs15281a. [DOI] [PubMed] [Google Scholar]; (d) Patureau F. W., WencelDelord J., Glorius F. Aldrichimica Acta. 2012;45:31. [Google Scholar]; (e) Colby D. A., Tsai A. S., Bergman R. G., Ellman J. A. Acc. Chem. Res. 2012;45:814. doi: 10.1021/ar200190g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhu C., Wang R., Falck J. R. Chem.–Asian J. 2012;7:1502. doi: 10.1002/asia.201200035. [DOI] [PubMed] [Google Scholar]; (g) Kuhl N., Schröder N., Glorius F. Adv. Synth. Catal. 2014;356:1443. [Google Scholar]; (h) Ackermann L. Acc. Chem. Res. 2014;47:281. doi: 10.1021/ar3002798. [DOI] [PubMed] [Google Scholar]
- For Rh(iii)-catalyzed formal [4 + 1]-cyclization, see: ; (a) Zhu C., Xie W., Falck J. R. Chem.–Eur. J. 2011;17:12591. doi: 10.1002/chem.201102475. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Du Y., Hyster T. K., Rovis T. Chem. Commun. 2011;47:12074. doi: 10.1039/c1cc15843k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu C., Falck J. R. Chem. Commun. 2012;48:1674. doi: 10.1039/c2cc16963k. [DOI] [PubMed] [Google Scholar]; (d) Hyster T. K., Ruhl K. E., Rovis T. J. Am. Chem. Soc. 2013;135:5364. doi: 10.1021/ja402274g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yu D., Suri M., Glorius F. J. Am. Chem. Soc. 2013;135:8802. doi: 10.1021/ja4033555. [DOI] [PubMed] [Google Scholar]; (f) Ryu T., Kim J., Park Y., Kim S., Lee P. H. Org. Lett. 2013;15:3986. doi: 10.1021/ol401775t. [DOI] [PubMed] [Google Scholar]; (g) Ye B., Cramer N. Angew. Chem., Int. Ed. 2014;53:7896. doi: 10.1002/anie.201404895. [DOI] [PubMed] [Google Scholar]
- For Rh(iii)-catalyzed formal [4 + 2]-cyclization of amides and their derivates with alkynes, see: ; (a) Hyster T. K., Rovis T. J. Am. Chem. Soc. 2010;132:10565. doi: 10.1021/ja103776u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Song G., Chen D., Pan C., Crabtree R. H., Li X. J. Org. Chem. 2010;75:7487. doi: 10.1021/jo101596d. [DOI] [PubMed] [Google Scholar]; (c) Guimond N., Gorelsky S. I., Fagnou K. J. Am. Chem. Soc. 2011;133:6449. doi: 10.1021/ja201143v. [DOI] [PubMed] [Google Scholar]; (d) Hyster T. K., Rovis T. Chem. Sci. 2011;2:1606. doi: 10.1039/C1SC00235J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pham M. V., Ye B., Cramer N. Angew. Chem., Int. Ed. 2012;51:10610. doi: 10.1002/anie.201206191. [DOI] [PubMed] [Google Scholar]; (f) Xu X., Liu Y., Park C. M. Angew. Chem., Int. Ed. 2012;51:9372. doi: 10.1002/anie.201204970. [DOI] [PubMed] [Google Scholar]; (g) Hyster T. K., Knörr L., Ward T. R., Rovis T. Science. 2012;338:500. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Ye B., Cramer N. Science. 2012;338:504. doi: 10.1126/science.1226938. [DOI] [PubMed] [Google Scholar]; (i) Quiñones N., Seoane A., García-Fandiño R., Mascareñas J. L., Gulías M. Chem. Sci. 2013;4:2874. [Google Scholar]; (j) Zhao D., Nimphius C., Lindale M., Glorius F. Org. Lett. 2013;15:4504. doi: 10.1021/ol402053n. [DOI] [PubMed] [Google Scholar]; (k) Yu D., de Azambuja F., Gensch T., Daniliuc C. G., Glorius F. Angew. Chem., Int. Ed. 2014;53:9650. doi: 10.1002/anie.201403782. [DOI] [PubMed] [Google Scholar]; (l) Hyster T. K., Dalton D. M., Rovis T. Chem. Sci. 2015;6:254. doi: 10.1039/c4sc02590c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Rakshit S., Grohmann C., Besset T., Glorius F. J. Am. Chem. Soc. 2011;133:2350. doi: 10.1021/ja109676d. [DOI] [PubMed] [Google Scholar]; (n) Shi Z., Boultadakis-Arapinis M., Koester D. C., Glorius F. Chem. Commun. 2014;50:2650. doi: 10.1039/c4cc00029c. [DOI] [PubMed] [Google Scholar]; (o) Wang H., Glorius F. Angew. Chem., Int. Ed. 2012;51:7318. doi: 10.1002/anie.201201273. [DOI] [PubMed] [Google Scholar]
- For Rh(iii)-catalyzed formal [4 + 2]-cyclization of imines and their derivates with alkynes, see: ; (a) Guimond N., Fagnou K. J. Am. Chem. Soc. 2009;131:12050. doi: 10.1021/ja904380q. [DOI] [PubMed] [Google Scholar]; (b) Too P. C., Wang Y. F., Chiba S. Org. Lett. 2010;12:5688. doi: 10.1021/ol102504b. [DOI] [PubMed] [Google Scholar]; (c) Wang Y., Toh K. K., Lee J., Chiba S. Angew. Chem., Int. Ed. 2011;50:5927. doi: 10.1002/anie.201101009. [DOI] [PubMed] [Google Scholar]; (d) Too P. C., Chua S. H., Wong S. H., Chiba S. J. Org. Chem. 2011;76:6159. doi: 10.1021/jo200897q. [DOI] [PubMed] [Google Scholar]; (e) Hyster T. K., Rovis T. Chem. Commun. 2011;47:11846. doi: 10.1039/c1cc15248c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhang X., Chen D., Zhao M., Zhao J., Jia A., Li X. Adv. Synth. Catal. 2011;353:719. [Google Scholar]; (g) Jayakumar J., Parthasarathy K., Cheng C. H. Angew. Chem., Int. Ed. 2012;51:197. doi: 10.1002/anie.201105755. [DOI] [PubMed] [Google Scholar]; (h) Kim D. S., Park J. W., Jun C. H. Chem. Commun. 2012;48:11334. doi: 10.1039/c2cc36699a. [DOI] [PubMed] [Google Scholar]; (i) Sim Y. K., Lee H., Park J. W., Kim D. S., Jun C. H. Chem. Commun. 2012;48:11787. doi: 10.1039/c2cc36956g. [DOI] [PubMed] [Google Scholar]; (j) Dong W., Wang L., Parthasarathy K., Pan F., Bolm C. Angew. Chem., Int. Ed. 2013;52:11573. doi: 10.1002/anie.201304456. [DOI] [PubMed] [Google Scholar]; (k) Chuang S., Gandeepan P., Cheng C. H. Org. Lett. 2013;15:5750. doi: 10.1021/ol402796m. [DOI] [PubMed] [Google Scholar]; (l) Muralirajan K., Cheng C. H. Chem.–Eur. J. 2013;19:6198. doi: 10.1002/chem.201300922. [DOI] [PubMed] [Google Scholar]; (m) Zhao D., Wu Q., Huang X., Song F., Lv T., You J. Chem.–Eur. J. 2013;19:6239. doi: 10.1002/chem.201300155. [DOI] [PubMed] [Google Scholar]; (n) Lee H., Sim Y. K., Park J. W., Jun C. H. Chem.–Eur. J. 2014;20:323. doi: 10.1002/chem.201302699. [DOI] [PubMed] [Google Scholar]; (o) Zhao D., Lied F., Glorius F. Chem. Sci. 2014;5:2869. [Google Scholar]; (p) Neely J. M., Rovis T. J. Am. Chem. Soc. 2013;135:66. doi: 10.1021/ja3104389. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Shi Z., Koester D. C., Boultadakis-Arapinis M., Glorius F. J. Am. Chem. Soc. 2013;135:12204. doi: 10.1021/ja406338r. [DOI] [PubMed] [Google Scholar]
- For Rh(iii)-catalyzed formal [4 + 2]-cyclization of compounds with the OH group with alkynes, see: ; (a) Shimizu M., Hirano K., Satoh T., Miura M. J. Org. Chem. 2009;74:3478. doi: 10.1021/jo900396z. [DOI] [PubMed] [Google Scholar]; (b) Mochida S., Shimizu M., Hirano K., Satoh T., Miura M. Chem.–Asian J. 2010;5:847. doi: 10.1002/asia.200900639. [DOI] [PubMed] [Google Scholar]; (c) Morimoto K., Hirano K., Satoh T., Miura M. J. Org. Chem. 2011;76:9548. doi: 10.1021/jo201923d. [DOI] [PubMed] [Google Scholar]; (d) Park Y., Seo J., Park S., Yoo E. J., Lee P. H. Chem.–Eur. J. 2013;19:16461. doi: 10.1002/chem.201302652. [DOI] [PubMed] [Google Scholar]; (e) Li Q., Yan Y., Wang X., Gong B., Tang X., Shi J., Xu H. E., Yi W. RSC Adv. 2013;3:23402. [Google Scholar]; (f) Seo J., Park Y., Jeon I., Ryu T., Park S., Lee P. H. Org. Lett. 2013;15:3358. doi: 10.1021/ol401407v. [DOI] [PubMed] [Google Scholar]; (g) Unoh Y., Hashimoto Y., Takeda D., Hirano K., Satoh T., Miura M. Org. Lett. 2013;15:3258. doi: 10.1021/ol4012794. [DOI] [PubMed] [Google Scholar]; (h) Qi Z., Wang M., Li X. Chem. Commun. 2014;50:9776. doi: 10.1039/c4cc03627a. [DOI] [PubMed] [Google Scholar]
- For Rh(iii)-catalyzed formal [4 + 2]-cyclization of compounds with a directing heterocyclic ring with alkynes, see: ; (a) Morimoto K., Hirano K., Satoh T., Miura M. Org. Lett. 2010;12:2068. doi: 10.1021/ol100560k. [DOI] [PubMed] [Google Scholar]; (b) Huang J., Dong L., Han B., Peng C., Chen Y. Chem.–Eur. J. 2012;18:8896. doi: 10.1002/chem.201201207. [DOI] [PubMed] [Google Scholar]; (c) Zhang G., Yang L., Wang Y., Xie Y., Huang H. J. Am. Chem. Soc. 2013;135:8850. doi: 10.1021/ja404414q. [DOI] [PubMed] [Google Scholar]; (d) Wang N., Li B., Song H., Xu S., Wang B. Chem.–Eur. J. 2013;19:358. doi: 10.1002/chem.201203374. [DOI] [PubMed] [Google Scholar]; (e) Luo C., Gandeepan P., Cheng C. Chem. Commun. 2013;49:8528. doi: 10.1039/c3cc45004j. [DOI] [PubMed] [Google Scholar]; (f) Huang J., Zhang Q., Qu C., Sun X., Dong L., Chen Y. Org. Lett. 2013;15:1878. doi: 10.1021/ol400537b. [DOI] [PubMed] [Google Scholar]; (g) Dong L., Huang J., Qu C., Zhang Q., Zhang W., Han B., Peng C. Org. Biomol. Chem. 2013;11:6142. doi: 10.1039/c3ob41177j. [DOI] [PubMed] [Google Scholar]; (h) Iitsuka T., Hirano K., Satoh T., Miura M. Chem.–Eur. J. 2014;20:385. doi: 10.1002/chem.201303847. [DOI] [PubMed] [Google Scholar]
- For other types of Rh(iii)-catalyzed formal [4 + 2]-cyclization reactions, see: ; (a) Karthikeyan J., Haridharan R., Cheng C. H. Angew. Chem., Int. Ed. 2012;51:12343. doi: 10.1002/anie.201206890. [DOI] [PubMed] [Google Scholar]; (b) Senthilkumar N., Parthasarathy K., Gandeepan P., Cheng C. H. Chem.–Asian J. 2013;8:2175. doi: 10.1002/asia.201300356. [DOI] [PubMed] [Google Scholar]; (c) Kim D. S., Park J. W., Jun C. H. Adv. Synth. Catal. 2013;355:2667. [Google Scholar]; (d) Liu X., Li G., Song F., You J. Nat. Commun. 2014;5:5030. doi: 10.1038/ncomms6030. [DOI] [PubMed] [Google Scholar]
- Selected reports of Rh(iii)-catalyzed [4 + 3] cycloadditions based on C–H activation, see: ; (a) Shi Z., Grohmann C., Glorius F. Angew. Chem., Int. Ed. 2013;52:5393. doi: 10.1002/anie.201301426. [DOI] [PubMed] [Google Scholar]; (b) Cui S., Zhang Y., Wu Q. Chem. Sci. 2013;4:3421. [Google Scholar]; (c) Cui S., Zhang Y., Wang D., Wu Q. Chem. Sci. 2013;4:3912. [Google Scholar]
- After isolation, these two isomers may be separated. However, we were not able to determine the ratio by analyzing the 1H NMR spectra of the crude products (see the ESI file)
- Crystal data: 3aa·H2O: C17H23NOSi·H2O, M W = 303.47, monoclinic, space group P21/n, final R indices [I > 2σ(I)], R 1 = 0.1006, wR 2 = 0.2419; R indices (all data), R 1 = 0.1392, wR 2 = 0.2672; a = 7.8451(6) Å, b = 10.8055(10) Å, c = 41.681(3) Å, β = 92.188(5)°, V = 3530.7(5) Å3, T = 293(2) K, Z = 8, reflections collected/unique 15620/6440 (R int = 0.0483), number of observations [>2σ(I)]: 4214, parameters: 401. ESI.
- For details, see the ESI file
- Simmons E. M., Hartwig J. F. Angew. Chem., Int. Ed. 2012;51:3066. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- ; for a review on the regioselectivity of allene insertion, see: ; (a) Zeng R., Fu C., Ma S. J. Am. Chem. Soc. 2012;134:9597. doi: 10.1021/ja303790s. [DOI] [PubMed] [Google Scholar]; (b) Ye B., Cramer N. J. Am. Chem. Soc. 2013;135:636. doi: 10.1021/ja311956k. [DOI] [PubMed] [Google Scholar]; (c) Zeng R., Wu S., Fu C., Ma S. J. Am. Chem. Soc. 2013;135:18284. doi: 10.1021/ja409861s. [DOI] [PubMed] [Google Scholar]; (d) Bai T., Ma S., Jia G. Coord. Chem. Rev. 2009;253:423. [Google Scholar]; (e) Zeng R., Ye J., Fu C., Ma S. Adv. Synth. Catal. 2013;355:1963. [Google Scholar]; (f) Zhang Y., Skucas E., Krische M. J. Org. Lett. 2009;11:4248. doi: 10.1021/ol901759t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Crabtree R. H., The Organometallic Chemistry of The Transition Metals, Wiley-VCH, Weinheim, 5th edn, 2009, pp. 192–194. [Google Scholar]; (b) Spessard G. O. and Miessler G. L., Organometallic Chemistry, Oxford University Press, 2nd edn, 2009, pp. 186–191. [Google Scholar]
- (a) Fleming I. Org. React. 1989;35:501. [Google Scholar]; (b) Chai G., Zeng R., Fu C., Ma S. Eur. J. Org. Chem. 2013:148. [Google Scholar]
- (a) Machacek M. R., Trost B. M. Angew. Chem., Int. Ed. 2002;41:4693. doi: 10.1002/anie.200290019. [DOI] [PubMed] [Google Scholar]; (b) Faulk B. D., Machacek M. R., Trost B. M. J. Am. Chem. Soc. 2006;128:6745. doi: 10.1021/ja060812g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the recent theoretical studies concerning CMD processes, see: ; (a) Davies D. L., Donald S. M. A., Macgregor S. A. J. Am. Chem. Soc. 2005;127:13754. doi: 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]; (b) Lafrance M., Rowley C. N., Woo T. K., Fagnou K. J. Am. Chem. Soc. 2006;128:8754. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]; (c) Lafrance M., Fagnou K. J. Am. Chem. Soc. 2006;128:16496. doi: 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]; (d) García-Cuadrado D., Braga A. A. C., Maseras F., Echavarren A. M. J. Am. Chem. Soc. 2006;128:1066. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]; (e) Garcia-Cuadrado D., de Mendoza P., Braga A. A. C., Maseras F., Echavarren A. M. J. Am. Chem. Soc. 2007;129:6880. doi: 10.1021/ja071034a. [DOI] [PubMed] [Google Scholar]; (f) Gorelsky S. I., Lapointe D., Fagnou K. J. Am. Chem. Soc. 2008;130:10848. doi: 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]; (g) Sun H.-Y., Gorelsky S. I., Stuart D. R., Campeau L.-C., Fagnou K. J. Org. Chem. 2010;75:8180. doi: 10.1021/jo101821r. [DOI] [PubMed] [Google Scholar]; (h) Chen B., Hou X.-L., Li Y.-X., Wu Y.-D. J. Am. Chem. Soc. 2011;133:7668. doi: 10.1021/ja201425e. [DOI] [PubMed] [Google Scholar]; (i) Zhang S., Shi L., Ding Y. J. Am. Chem. Soc. 2011;133:20218. doi: 10.1021/ja205294y. [DOI] [PubMed] [Google Scholar]; (j) Tang S.-Y., Guo Q.-X., Fu Y. Chem.–Eur. J. 2011;17:13866. doi: 10.1002/chem.201101587. [DOI] [PubMed] [Google Scholar]; (k) Santoro S., Liao R.-Z., Himo F. J. Org. Chem. 2011;76:9246. doi: 10.1021/jo201447e. [DOI] [PubMed] [Google Scholar]; (l) Boutadla Y., Davies D. L., Macgregor S. A., Poblador-Bahamonde A. I. Dalton Trans. 2009:5887. doi: 10.1039/b905469c. [DOI] [PubMed] [Google Scholar]
- Xu L., Zhu Q., Huang G., Cheng B., Xia Y. J. Org. Chem. 2012;77:3017. doi: 10.1021/jo202431q. [DOI] [PubMed] [Google Scholar]
- (a) Brayshaw S. K., Sceats E. L., Green J. C., Weller A. S. Proc. Natl. Acad. Sci. U. S. A. 2007;104:6921. doi: 10.1073/pnas.0609824104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McBee J. L., Escalada J., Tilley T. D. J. Am. Chem. Soc. 2009;131:12703. doi: 10.1021/ja9035169. [DOI] [PubMed] [Google Scholar]; (c) Vyboishchikov S. F., Nikonov G. I. Organometallics. 2007;26:4160. [Google Scholar]
- Frisch M. J., et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009.
- (a) Zhao Y., Truhlar D. G. Theor. Chem. Acc. 2008;120:215. [Google Scholar]; (b) Truhlar D. G. J. Am. Chem. Soc. 2008;130:16824. doi: 10.1021/ja808927h. [DOI] [PubMed] [Google Scholar]; (c) Zhao Y., Truhlar D. G. Acc. Chem. Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- For reviews of density-functional methods, see: ; (a) Parr R. G. and Yang W., Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989. [Google Scholar]; (b) Ziegler T. Chem. Rev. 1991;91:651. [Google Scholar]; (c) Density Functional Methods in Chemistry, ed. J. Labanowski and J. Andzelm, Springer, Berlin, 1991. [Google Scholar]
- Hehre W. J., Ditchfield R., Pople J. A. J. Chem. Phys. 1972;56:2257. [Google Scholar]
- (a) Hay P. J., Wadt W. R. J. Chem. Phys. 1985;82:270. [Google Scholar]; (b) Wadt W. R., Hay P. J. J. Chem. Phys. 1985;82:284. [Google Scholar]
- (a) Barone V., Cossi M. J. Phys. Chem. A. 1998;102:1995. [Google Scholar]; (b) Cossi M., Rega N., Scalmani G., Barone V. J. Comput. Chem. 2003;24:669. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]; (c) Takano Y., Houk K. N. J. Chem. Theory Comput. 2005;1:70. doi: 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.