

Abstract
Glycogen synthase (GS) and glycogen phosphorylase (GP) are the key enzymes that control, respectively, the synthesis and degradation of glycogen, a multi-branched glucose polymer that serves as a form of energy storage in bacteria, fungi and animals. An abnormal glycogen metabolism is associated with several human diseases. Thus, GS and GP constitute adequate pharmacological targets to modulate cellular glycogen levels by means of their selective inhibition. The compound 1,4-dideoxy-1,4-imino-D-arabi-nitol (DAB) is a known potent inhibitor of GP. We studied the inhibitory effect of DAB, its enantiomer LAB, and 29 DAB derivatives on the activity of rat muscle glycogen phosphorylase (RMGP) and E. coli glycogen synthase (EcGS). The isoform 4 of sucrose synthase (SuSy4) from Solanum tuberosum L. was also included in the study for comparative purposes. Although these three enzymes possess highly conserved catalytic site architectures, the DAB derivatives analysed showed extremely diverse inhibitory potential. Subtle changes in the positions of crucial residues in their active sites are sufficient to discriminate among the structural differences of the tested inhibitors. For the two Leloir-type enzymes, EcGS and SuSy4, which use sugar nucleotides as donors, the inhibitory potency of the compounds analysed was synergistically enhanced by more than three orders of magnitude in the presence of ADP and UDP, respectively. Our results are consistent with a model in which these compounds bind to the subsite in the active centre of the enzymes that is normally occupied by the glucosyl residue which is transferred between donor and acceptor substrates. The ability to selectively inhibit the catalytic activity of the key enzymes of the glycogen metabolism may represent a new approach for the treatment of disorders of the glycogen metabolism.
Introduction
Glycosyltransferases (GTs) are a class of enzymes (EC 2.4) involved in the biosynthesis of oligo- and polysaccharides and glycoconjugates, which are vital for all living systems. GTs catalyse the transfer of saccharide moieties from a glycosyl donor to an acceptor molecule forming glycosidic linkages, both regio- and stereospecifically. GTs have been classified in three different ways according to: (i) the type of donor substrate, (ii) the relative anomeric configuration of the donor substrate and product, and (iii) the protein three dimensional fold. Thus, in terms of the glycosyl donor, GTs are divided in three classes:1 (a) Leloir-type GTs, which use sugar nucleotides as activated monosaccharide donors; (b) non-Leloir-type GTs, which employ sugar phosphates, pyrophosphates or polyprenol phosphates as donors; and (c) transglycosidases, which require non-activated sugars, like lactose and starch.
During the formation of a new glycosidic bond, the anomeric configuration of the transferred sugar in the product can be either retained or inverted with respect to the donor substrate. Thereby, enzymes catalysing glycosyl group transfer are classified as retaining or inverting according to the relative stereochemistry of substrates and reaction products. Reported crystallographic structures of both retaining and inverting GTs have revealed that independent of the stereochemical course of the reaction, all GTs can adopt only one of the two general folds, designed as GT-A and GT-B. The GT-A fold consists of an α/β/α sandwich that resembles the Rossmann fold, while the architecture of the GT-B enzymes consists of two separate Rossmann domains with a connecting linker region and the catalytic centre located between the domains.2,3
We are interested in the development of GT inhibitors and, more precisely, in molecules capable of regulating the activity of the two GTs involved in the glycogen metabolism: glycogen phosphorylase (GP) and glycogen synthase (GS). Glycogen, a multi-branched polymer of α-1,4 and α-1,6-linked glucose units, constitutes the principal storage form of glucose in the animal kingdom and serves as a buffer for glucose needs. GP catalyses the phosphorolytic cleavage of α-1,4 glycosidic bonds, using inorganic phosphate as a co-substrate, to release glucose-1-phosphate (Glc-1P) as the reaction product. Although the biological action of GP is degradation of glycogen, it is classified as a non-Leloir GT (family GT35, according to the Cazy classification based on structural similarities3), since in vitro this enzyme can also catalyse the reverse reaction: the addition of glucose units to glycogen using Glc-1-P as a donor. Interest in the inhibition of GP has considerably increased with the rise in obesity and associated diseases such as type II diabetes. A drug able to specifically reduce blood hyperglycaemia arising in part from excessive glycogen degradation, which is a characteristic of diabetic patients, would constitute a strong therapeutic tool.
GS is a Leloir-type GT that catalyses the successive addition of α-1,4-linked glucose moieties to the non-reducing end of glycogen, using adenosine 5′-diphosphoglucose (ADPG) from plant and bacterial enzymes (GT5 family), or uridine 5′-diphosphoglucose (UDPG) from animal and fungal enzymes as donors. In mammals, glycogen accumulates mainly in the liver and muscle, but it is also produced in the brain, although its levels are low compared to the other two tissues. In the brain, glycogen is normally stored in astrocytes and most neurons do not accumulate this polysaccharide under normal conditions4,5 although they have an active glycogen metabolism.6 Aberrant glycogen accumulation in neurons is observed in neurodegenerative diseases such as the Lafora disease.7 The presence of abnormal glycogen deposits in neurons of mouse and Drosophila models of the Lafora disease (LD) leads to several neuronal losses, locomotion defects and a considerably reduced life span.8 Thus, GS inhibitors may represent adequate pharmacological tools for the treatment of LD. In summary, GP and GS are potential targets for the treatment of diseases related to disorders of the glucose and glycogen metabolism.
The effects of 1,4-dideoxy-1,4-imino-D-arabinitol (DAB) on GP have been investigated in vitro, in primary cultured rat hepatocytes,9 and in vivo, using ob/ob mice.10,11 DAB was described as a potent GP inhibitor, but its mechanism of action is controversial. Latsis and co-workers9 concluded that DAB is an allosteric inhibitor of GP, while Fosgerau et al.10 suggested that DAB acts as an uncompetitive or a non-competitive inhibitor with respect to glycogen and phosphate, respectively.
Chemical modification of the hydroxymethyl group of DAB and its enantiomer LAB has led to novel amino acid, amino alcohol, and aromatic and piperazin-2-one derivatives with interesting properties as glycosidase inhibitors and with other unprecedented biological activities.12 The synthesis of molecules with new inhibitory profiles in terms of potency and/or selectivity is of special interest in the case of GTs, since there are few known inhibitors.13–16
We have studied the potency, the selectivity and the mechanism of action of DAB, LAB and 29 DAB derivatives (Fig. 1) as inhibitors of the enzymatic activity of three GTs. Rabbit muscle GPa (RMGPa) and Escherichia coli GS (EcGS) were studied as representatives of the enzymes responsible for the degradation and synthesis of glycogen, respectively. Sucrose synthase 4 (SuSy4) from Solanum tuberosum L. is a Leloir GT (GT4 family) that catalyses the transfer of a glucose unit from UDPG to fructose to yield sucrose and UDP. This enzyme, which as RMGPa and EcGS operates through a retaining transfer mechanism, was included in the study as a related member of the large GT superfamily, but which does not act on glycogen.
Fig. 1.

Structures of DAB, LAB and DAB derivatives analysed in this study.
Results and discussion
Inhibitory properties of LAB, DAB and DAB derivatives
The inhibitory potency of DAB, LAB and the 29 DAB derivatives shown in Fig. 1, was tested against purified preparations of commercially available RMGPa and recombinantly produced EcGS and SuSy4.16 First, to establish the optimal conditions of the assay, the kinetic parameters Km and Vmax of the three enzymes for their respective substrates were determined: RMGPa activity was measured in the direction of glycogen phosphorolysis and EcGS and SuSy4 in the direction of synthesis of glycogen and sucrose, respectively (Fig. S-1 and Table S-2, ESI†). The inhibition studies were performed using a saturating concentration of the respective substrates (10 mM Pi and 4 mg mL−1 glycogen for RMGPa; 2 mM ADPG and 4 mg mL−1 glycogen for EcGS; 5 mM UDPG and 30 mM fructose for SuSy4) and 0.5 mM of each of the potential inhibitors.
An initial evaluation revealed that none of the compounds analysed exhibited better inhibitory potential on RMGPa than DAB (Table 1), which showed an IC50 value of 0.93 ± 0.01 μM (Table 2). However, 15, 26 and 28 considerably inhibited EcGS (Table 1), with IC50 values approximately 5-fold lower than that obtained for DAB (Table 2). Finally, 2, 20 and 29 presented remarkable inhibition of SuSy4 (Table 1), with IC50 values approximately 2-fold lower than that of DAB (Table 2). It should be pointed out that GT inhibitors are rare and even rarer are those which do not harness portions of the nucleotide donor.13–16 The glycosidic compounds tested here showed to be very effective inhibitors and exhibited a high degree of selectivity. For instance, under the conditions of the assay, 5 inhibited RGMPa by 98% while left EcGS and SuSy4 activities essentially unchanged. In contrast, 26 inhibited EcGS by more than 70%, while only decreased SuSy4 activity by less than 50% and did not affect RGMPa catalytic activity. DAB, 26 and 2 were selected to pursue further inhibition studies, since they presented the highest inhibitory potency on RGMPa, EcGS and SuSy4, respectively (Table 1).
Table 1.
Inhibition of RMGPa, EcGS and SuSy4 by LAB, DAB and DAB derivatives
| Type of derivative | Inhibitor | Activity (%) | ||
|---|---|---|---|---|
|
| ||||
| RMGPa | EcGS | SuSy4 | ||
| Amino alcohol DAB derivatives | 0 | 100 | 100 | 100 |
| 1 | 56 | 98 | 91 | |
| 2 | 76 | 54 | 9 | |
| 3 | 44 | 98 | 98 | |
| 4 | 70 | 98 | 98 | |
| 5 | 2 | 98 | 98 | |
| 6 | 80 | 96 | 99 | |
| 7 | 32 | 85 | 98 | |
| 8 | 99 | 50 | 47 | |
| Amino acid DAB derivatives | 9 | 98 | 65 | 85 |
| 10 | 75 | 96 | 95 | |
| 11 | 28 | 79 | 84 | |
| 12 | 99 | 93 | 88 | |
| 13 | 22 | 91 | 91 | |
| 14 | 5 | 96 | 92 | |
| 15 | 57 | 31 | 71 | |
| 16 | 99 | 80 | 88 | |
| 17 | 73 | 49 | 61 | |
| 18 | 94 | 89 | 86 | |
| Aromatic DAB derivatives | 19 | 99 | 74 | 75 |
| 20 | 98 | 83 | 16 | |
| 21 | 56 | 75 | 51 | |
| 22 | 98 | 65 | 77 | |
| Piperazin-2-one DAB derivatives | 23 | 93 | 72 | 78 |
| 24 | 98 | 46 | 51 | |
| 25 | 23 | 65 | 72 | |
| 26 | 99 | 28 | 54 | |
| 27 | 99 | 64 | 58 | |
| 28 | 99 | 38 | 72 | |
| 29 | 93 | 64 | 33 | |
| DAB | 0.013 | 72 | 59 | |
| LAB | 10 | 89 | 89 | |
Table 2.
IC50 values of DAB and selected derivatives as inhibitors of RMGPa, EcGS and SuSy4
| Enzyme | Inhibitor | IC50 (μM) |
|---|---|---|
| RMGPa | DAB | 0.93 ± 0.1 |
| EcGS | 15 | 346.1 |
| 26 | 249.0 ± 0.1 | |
| 28 | 298.2 ± 0.1 | |
| DAB | 1320 ± 10 | |
| SuSy4 | 2 | 131.3 ± 0.1 |
| 20 | 96.8 ± 0.1 | |
| 29 | 172.5 ± 0.1 | |
| DAB | 246.5 ± 0.1 |
The double reciprocal plot of RMGPa activity, varying the concentrations of both substrates converged to a negative value (Fig. 2A), which indicates a sequential binding mechanism of the two substrates, and excludes the ping-pong mechanism.
Fig. 2.

Double reciprocal plot of RMGPa activity: In the absence of inhibitor and varying concentrations of both substrates (A). Under pseudo-single substrate conditions and in the presence of varying concentrations of DAB (B and C). Values are the mean of three independent experiments.
Double reciprocal plots under pseudo-single substrate conditions and in the presence of varying concentrations of DAB (Fig. 2B and C) showed that this molecule acts as a competitive inhibitor with respect to both Pi and glycogen. Consistent with earlier studies,17 these results indicate that RMGPa, as a typical non-Leloir GT, follows a rapid equilibrium random bi–bi mechanism, in which the substrates bind independently from one another.18 Our results can therefore be interpreted assuming that DAB, as a glucomimetic compound, can combine with the free enzyme or with the RMGPa–Pi complex, and that its binding to the active site precludes binding of glycogen.
Similarly, the double reciprocal plots of the EcGS (Fig. 3A) and SuSy4 (Fig. 3E) activities at varying concentrations of both substrates converged to negative values, indicating that these enzymes also follow a sequential non ping-pong mechanism. Pseudo-single substrate kinetics in the presence of varying concentrations of 26 for EcGS (Fig. 3B) and 2 for SuSy4 (Fig. 3F) showed that these molecules were competitive inhibitors of the respective glucose-diphosphonucleotide donors, ADPG and UDPG, similar to the behaviour exhibited by ADP and UDP, respectively (Fig. 3D and H). In contrast, 26 and 2 acted as non-competitive inhibitors of the EcGS and SuSy4 acceptors, glycogen and fructose, respectively (Fig. 3C and G). Taken together these results dictate that both Leloir GTs follow an ordered bi–bi kinetic mechanism, where the sugar donor (ADPG or UDPG) binds first to the enzyme, followed by the sugar acceptor (glycogen or fructose), which can only bind to the NDPG–enzyme complex.18–20 The inhibition of both DAB derivatives is brought about by binding to the respective free enzymes, presumably mimicking the glucose moiety of the donor substrates, and thus precluding NDPG binding to the active site. Since the inhibitors compete with NDPG for the free enzyme, but cannot displace the respective acceptors, although their binding prevents the turnover reaction, 26 and 2 show competitive inhibition with respect to NDPG and non-competitive inhibition with respect to the sugar acceptors.18 The inhibition constants determined in these experiments are shown in Table 3.
Fig. 3.

Double reciprocal plots of EcGS (A, B, C, D) and SuSy4 (E, F, G, H) activities: in the absence of inhibitors and at varying concentrations of both substrates (A and E), under pseudo-single substrate conditions and in the presence of varying concentrations of 26 (B and C), 2 (F and G), ADP (D) or UDP (H). Values are the mean of three independent experiments.
Table 3.
Observed Ki values of DAB, 26 and 2 and type of inhibition with respect to the different substrates in the reactions catalysed by RMGPa, EcGS and SuSy4, respectively. Values are the mean ± s.e. of three independent experiments
| Target enzyme | Inhibitor | Substrate | Type of Inhibition | Ki |
|---|---|---|---|---|
| RMGPa | DAB | Pi | Competitive | 76 ± 6 nM |
| Glycogen | Competitive | 40 ± 5 nM | ||
| EcGS | 26 ADP | ADPG | Competitive | 346 ± 28 μM |
| Glycogen | Non-competitive | 739 ± 10 μM | ||
| ADPG | Competitive | 87 ± 6 μM | ||
| SuSy4 | 2 UDP | ADPG | Competitive | 353 ± 55 μM |
| Glycogen | Non-competitive | 399 ± 20 μM | ||
| UDPG | Competitive | 145 ± 48 μM |
The IC50 (Table 2) and the Ki values (Table 3) of DAB, 26 and 2 in the inhibition of RGMPa, EcGS and SuSy4, respectively, confirmed the potency of these inhibitors. The IC50 of DAB (0.93 μM) is three orders of magnitude smaller than the Km values of RMGPa for its natural substrates Pi (Km = 1.41 mM) in the phosphorolysis direction or Glc-1-P (Km = 0.92 mM) in the glycogen synthesis direction (Table S-2, ESI†). For the two Leloir-type enzymes these differences are not so large, but the IC50 values of 26 (249 μM) and 2 (131 μM) are still comparable to the Km values of the substrates of the respective enzymes, EcGS (Km for ADPG = 420 μM) and SuSy4 (Km for UDPG = 69 μM) (Table S-2, ESI†).
Next, synergy of DAB derivatives in combination with the non-sugar portion of the corresponding donor substrates (Pi for RGMPa, ADP for EcGS and UDP for SuSy4) was evaluated for the inhibition of the three enzymes. Synergy is defined as an interaction between two inhibitors of an enzyme such that the presence of one inhibitor decreases the dissociation constant of the other (and vice versa). Several studies described such type of synergistic inhibition in GTs. For instance, Errey and co-workers15 reported the inhibition exerted by bisubstrate-like molecules that mimic the natural disaccharide product on the E. coli trehalose-6-phosphate synthase, a retaining Leloir GT that catalyses the transfer of a glucose moiety from UDPG to glucose-6-phosphate to yield tre-halose-6-phosphate. The best inhibitor, validoxylamine-6′-phosphate, showed an IC50 of 5.3 mM, but addition of 0.15 mM UDP (a concentration around UDP’s own Ki) decreased this value to 41 μM (ca. 100-fold improvement). Other examples are the synergistic inhibition of the inverting GT α-1,3-fucosyltransferase, also by a bisubstrate analogue, which required the presence of GDP,21 or the increased affinity of RMGP for nojirimycin tetrazole22 or 5-gluconolactone23 in the presence of inorganic phosphate. In all these cases the authors concluded that the ternary complexes (enzyme-inhibitor-NDP or enzyme-inhibitor-Pi) were transition state analogues of the reactions catalysed by the respective enzymes.
To study the possible synergistic effect of Pi on the inhibition of DAB on RGMPa, we had to determine the enzyme activity in the glycogen synthesis direction (Fig. S-1 and Table S-2, ESI†), since inorganic phosphate is a substrate in the glycogenolytic direction. When we measured the IC50 values of DAB as an inhibitor of the glycogen phosphorolytic activity of RGMPa in the presence of increasing concentrations of Pi (Fig. 4A and B), we observed a modest 7-fold enhancement at 0.3 mM Pi. However, with the two Leloir-type GTs, the addition of ADP or UDP, respectively, led to a remarkable synergistic enhancement of the inhibition exerted by 26 on EcGS and 2 on SuSy4 activities. In the case of EcGS, the inhibition of 26 improved approximately 1000-fold, yielding an IC50 of 0.22 μM at 0.15 mM ADP, which is around ADP’s own Ki (Fig. 4C and D). For SuSy4, the IC50 value of 2 decreased 8800-fold, at 0.15 mM UDP, again a concentration around UDP’s own Ki (Fig. 4E and F).
Fig. 4.

Sigmoidal plot of the percentage of RMGPa (A), EcGS (C) and SuSy4 (E) activity at varying concentrations of DAB (A), 26 (C) or 2 (E). RGMPa and EcGS activities were measured in the direction of glycogen synthesis, and SuSy4 in the direction of fructose synthesis, in the presence of several concentrations of the respective non-sugar component of the donor substrates: Pi for RGMPa (A), ADP for EcGS (C) and UDP for SuSy4 (E). IC50 values calculated from these plots are shown in panels B (DAB, RGMPa), D (26, EcGS) and F (2, SuSy4). Values of tables are mean ± s.e. of three independent samples.
Docking of 26 into the donor site of E. coli glycogen synthase
The inhibition behaviour of 26 on EcGS activity and the observed synergistic effect of ADP strongly suggested that 26 acts as a glucose mimic and binds to the glucosyl subsite of the donor substrate in the active centre of the enzyme. To visualise complexation of 26 within the active site of EcGS, computer-aided docking of this molecule to the available X-ray structure of EcGS was performed. The crystal structure of the EcGS–ADP–glucose ternary complex (PDB 3GUH)24 was used for docking 26 to the site occupied by the glucose moiety of the sugar nucleotide donor.
The best docked conformation of 26 showed no steric clashes and several favourable interactions with active site residues of EcGS (Fig. 5), which could explain its large inhibitory potential. The C2-hydroxyl group of the five-membered ring of 26 makes a hydrogen bond with the backbone amide of Cys379 (3.0 Å), while the C3-hydroxyl group is within the hydrogen bonding distance of the Gly380 backbone amide (2.8 Å) and one oxygen atom of the proximal phosphate of ADP (2.1 Å). The backbone carbonyl group of His161 is at 2.7 Å of the amine group of the six-membered ring of 26. The carbonyl group of that ring can form a hydrogen bond with the Leu19 backbone amide group (2.4 Å) and one imidazole nitrogen of the His161 side chain (2.8 Å). Finally, the guanidinium group of 26 is at a hydrogen bonding distance of the hydroxyl group of the Thr16 side chain (2.6 Å).
Fig. 5.
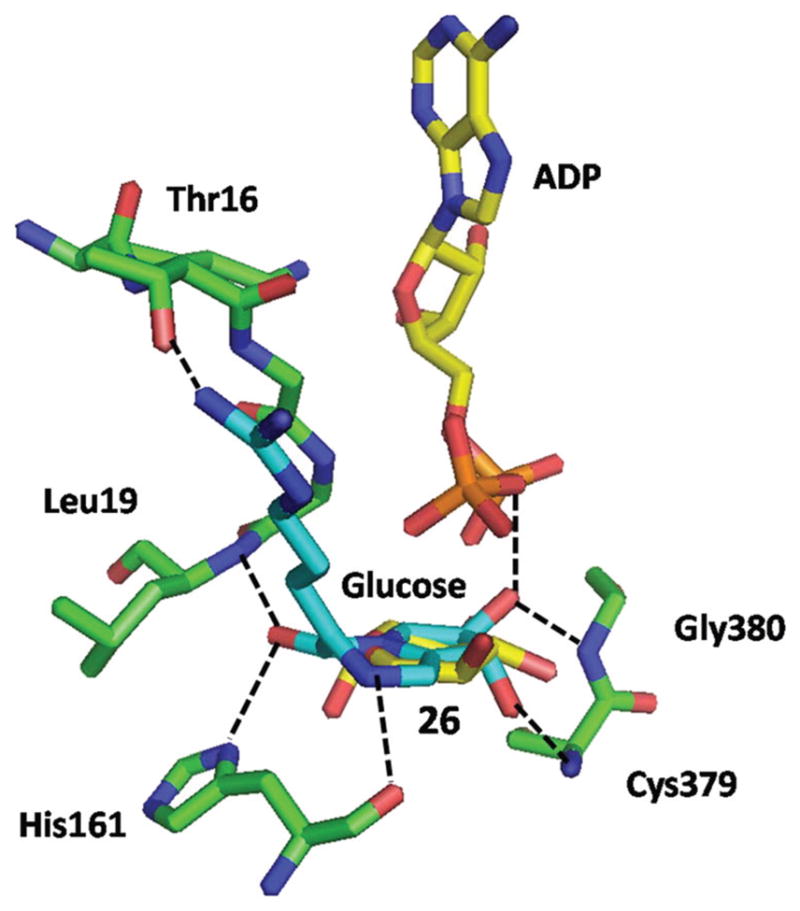
Docking of 26 into the active site of EcGS. Compound 26 is shown in light blue, ADP and glucose are shown in yellow and amino acid residues in green. Nitrogen atoms are coloured dark blue, oxygen atoms are shown in red and phosphorous atoms in orange. Hydrogen bonds are shown as broken black lines.
Mechanistic insights into retaining GTs
Glycogen synthase and glycogen phosphorylase, as all retaining GT-B enzymes, share a common catalytic mechanism.25 These enzymes possess a similar overall fold and critical residues and their interactions with the substrates in the reaction centre are preserved. However, they differ in the kinetic mechanism: EcGS, as SuSy4, two Leloir-type enzymes, follow an ordered bi–bi mechanism, while the non-Leloir-GT RMGPa follows a rapid equilibrium random bi–bi mechanism. This can be explained by the closure movement of the two domains that GT-B Leloir transferases experience upon binding the sugar nucleotide donor, to acquire the active conformation,24–27 which only then can bind the sugar acceptor. GP does not experience such a rearrangement and binds both substrates in a random order. The donor substrate-triggered closure of the catalytic crevice could be the reason for the very large synergistic enhancement of the inhibitory potency of 26 on EcGS activity caused by the presence of ADP (Fig. 4C and D). Inhibitor 26 can bind to the free enzyme, but its affinity for EcGS is largely increased by ADP, since the simultaneous presence of the two compounds could drive the “open to closed” conformational change leading to the establishment of numerous favourable interactions between the ligands and the enzyme, as shown in the docking experiment (Fig. 5). On the other hand, it has been suggested for other GTs, that the synergistic enhancement of the inhibition caused by the presence of the diphosphonucleotide moiety of the glycosil donor is due to the fact that the ternary complexes (enzyme-inhibitor-NDP) are transition state analogues of the reactions catalysed by these enzymes.15,21 In these cases, however, the inhibitors used were bisubstrate-like molecules, harnessing covalently linked portions of both substrates. Since 26 does not possess these structural characteristics, the very large synergistic effect on EcGS inhibition observed upon addition of ADP could alternatively be due to the formation of the quaternary complex with the acceptor substrate (EcGS-26-ADP-glycogen), which could act as a transition state analogue. Identical arguments can be used for the synergistic increase of the inhibitory effect of 2 on SuSy4, caused by the presence of UDP (Fig. 4E and F). The observation that the inhibition of RMGPa by DAB is only modestly improved by the presence of Pi (Fig. 4A and B) does not help to resolve this question. As stated before, GP does not experience the donor substrate-induced closure of the catalytic crevice observed in the Leloir-type GTs, so the simultaneous presence of DAB and inorganic phosphate should not enhance the interactions between the substrates and the enzyme. On the other hand, it has been reported that only inhibitors with an sp2 hybridised anomeric carbon atom, such as nojirimycin tetrazole22 or 5-glucono-lactone,23 are effective transition state analogues of the GP catalysed reaction in the presence of Pi.
Conclusions
DAB and its derivatives presented here structurally resemble the glucose moiety of the sugar nucleotide donor of the GTs studied. These compounds, which in all cases were found to be competitive inhibitors with respect to the donor substrate, showed vastly different inhibitory potencies. This observation indicates that subtle differences in the arrangement of crucial residues in the active sites among these enzymes are sufficient to discriminate among structural variations of the inhibitors. The high potency and the selectivity of the DAB derivatives make them promising GT inhibitors and open up the possibility of further development of drugs that could specifically act on glycogen metabolising enzymes, or other GTs, and whose biological effects could be modulated through variations of the chemical modification of the hydroxymethyl group of DAB.
Experimental
Synthesis of DAB derivatives
DAB, LAB and the DAB derivatives (1–29) were synthesised as described previously.12
Expression and purification of recombinant enzymes
Recombinant EcGS and SuSy4 were overexpressed in E. coli and purified as described previously.16
Kinetic analysis
The kinetic data were plotted as initial velocity (μM s−1) versus substrate concentration. The kinetic constants were acquired by fitting the data to the Michaelis–Menten equation by a nonlinear least square regression using the program OriginPro 8.0. Alternatively, data were linearized by representing the inverses of initial rates versus inverses of substrate concentrations and fitted to a straight line.
Assay of rabbit muscle glycogen phosphorylase activity
Glycogen phosphorylase activity was determined in the direction of oligosaccharide synthesis (assay A) or in the degradation (phosphorolysis) direction (assays B).
Assay A
The production of inorganic phosphate from soluble glycogen and glucose-1-phosphate was measured using the method described by Saheki et al.28 with slight modifications. A reaction mixture (50 μL) containing 200 mM HEPES buffer (pH 7.0), 4 mg mL−1 glycogen, 10 mM glucose-1-phosphate and an appropriate amount of enzyme was incubated at 30 °C for 4 min. The reaction was stopped by boiling for 1 min. After tempering the sample at room temperature, 50 μL of 2% ammonium molybdate in 50 mM H2SO4 and 50 μL of 2% ascorbic acid in 0.10% KHSO4 were added to the mixture. The resulting mixture was incubated at 37 °C for 30 min and the absorbance was measured at 700 nm using a Bio-Rad Benchmark Plus Microplate Spectrophotometer. One unit of enzyme activity is defined as the amount of the enzyme producing 1 μmol of phosphate in 1 min at 30 °C.
Assay B
Glycogen phosphorylase activity was measured in the direction of glycogenolysis by determination of NADPH in an assay coupled to phosphoglucomutase and glucose-6-phosphate dehydrogenase as described by Schinzel and Palm29 with minor modifications. The enzyme activity was assayed at pH 7.0 and 30 °C in a 200 mM HEPES buffer containing NADP+ (2.2 mM), MgCl2 (1 mM), glucose 1,6-bisphosphate (5 mM), glucose-6-phosphate dehydrogenase (1 unit), phos phoglucomutase (1 unit), 10 mM NaH2PO4, 4 mg mL−1 glycogen and an appropriate amount of enzyme. The reaction was started by the addition of the enzyme and the increase in absorbance at a wavelength of 340 nm, due to the formation of NADPH, was followed continuously using a Bio-Rad Benchmark Plus Microplate Spectrophotometer for 60 min at 30 °C. One unit of enzyme activity is defined as the amount of enzyme catalysing the formation of 1 μmol of glucose-1-phosphate in 1 min at 30 °C.
Assay of E. coli glycogen synthase activity
Glycogen synthase activity was measured in the glycogen synthesis direction.
Rate measurement in the absence of ADP
The production of ADP was determined according to the enzyme-coupled method reported by Morell and Copeland.30 The reaction mixture (100 μL) was composed of 200 mM HEPES buffer (pH 7.0), 4 mg mL−1 glycogen, 2 mM ADP-glucose, 0.7 mM phosphoenolpyruvic acid, 0.6 mM NADH, 50 mM KCl, 13 mM MgCl2, pyruvate kinase (7.5 U), lactate dehydrogenase (15 U) and the enzyme. The decrease in absorbance of NADH at 340 nm was measured at 30 °C using a Bio-Rad Benchmark Plus Microplate Spectrophotometer. EcGS activity is proportional to the rate of NADH oxidation where one molecule of NADH is oxidized for each molecule of ADP formed. One unit of enzyme activity is defined as the amount of the enzyme producing 1 μmol of ADP in 1 min at 30 °C.
Rate measurement in the presence of ADP
EcGS activity was assayed spectrophotometrically by coupling the formation of ADP to the reactions of pyruvate kinase and lactate dehydrogenase in a stopped assay format using the method described by Errey et al. with few modifications.15 The decrease in absorbance of NADH at 340 nm was measured using a spectrophotometer and compared to control samples that contained all reaction components except EcGS. The amount of ADP formed was identical to that of NADH oxidized to NAD+. The reaction mixtures (50 μL) containing 200 mM HEPES buffer (pH 7.0), 4 mg mL−1 glycogen, 2 mM ADP-glucose, inhibitor, ADP and the enzyme were incubated at 30 °C for 15 minutes and the reactions were stopped by heating the samples in boiling water for 1 minute. The control samples, which included the same components except EcGS, were also prepared and treated in the same way. After stopping the reaction, 50 μL of the assay solution containing phosphoenolpyruvic acid, NADH, pyruvate kinase and lactate dehydrogenase was added to each reaction sample and also the control sample. The resulting mixtures were incubated at 37 °C for 30 min, and the absorbance at 340 nm of wavelength was measured using a Bio-Rad Benchmark Plus Microplate Spectrophotometer. The amount of ADP formed in the reaction was obtained by subtracting absorbance of the reaction samples from that of the corresponding control samples. When ADP was used as an inhibitor, absorbance corresponding to a fixed amount of the inhibitor ADP could be subtracted out and newly-formed ADP could be determined. One unit of enzyme activity is defined as the amount of enzyme catalysing the formation of 1 μmol of ADP in 1 min at 30 °C.
Assay of sucrose synthase activity
Sucrose synthase activity was measured at 30 °C in the sucrose synthesis direction.
Rate measurement in the absence of UDP
The method described above was used, in which the UDP produced by the enzyme is coupled to NADH oxidation via pyruvate kinase and lactate dehydrogenase. The reaction mixture (100 μL) contained 200 mM HEPES buffer (pH 7.0), 5 mM UDP-glucose, 30 mM fructose 0.7 mM phosphoenolpyruvic acid, 0.6 mM NADH, 50 mM KCl, 13 mM MgCl2, pyruvate kinase (7.5 U), lactate dehydrogenase (15 U) and the enzyme. One unit of enzyme activity is defined as the amount of the enzyme producing 1 μmol of UDP in 1 min at 30 °C.
Rate measurement in the presence of UDP
Rate measurement of sucrose synthase activity in the presence of UDP was determined by coupling the formation of UDP to the reactions of pyruvate kinase and lactate dehydrogenase in a stopped assay format as described above. The enzyme activity was assayed at pH 7.0 and 30 °C in a 200 mM HEPES buffer containing 5 mM UDP-glucose, 30 mM fructose, inhibitor, UDP and the enzyme. One unit of enzyme activity is defined as the amount of enzyme catalysing the formation of 1 μmol of UDP in 1 min at 30 °C.
Docking analysis
The structure of 26 was energy minimized by the Hartree–Fock ground state method with the 3-21G basis set using the program Gaussian 03 W version 6.0. The coordinates and refinement restraint files for 26 were prepared using PRODRG of the CCP4 program suite 6.3.0. Computer-aided docking studies were performed using Coot 0.7.0. Docking studies were based on the published X-ray structure of EcGS (PDB 3GUH).24
Supplementary Material
Acknowledgments
This work was supported by the Dirección General de Investigación, Ministerio de Ciencia y Tecnología, Spain, grants CTQ2009-07359, (to P. C.), CTQ2006-00743 (to J. C. F.) and BFU2011-30554 (to J. J. G.), by AGAUR, the Generalitat de Catalunya, Spain, grant 2009SGR-1176 (to J. J. G.) and by the CIBER de Diabetes y Enfermedades Metabólicas Asociadas. M. Díaz-Lobo is grateful for a doctoral grant (BES-2009-029354) from the Ministerio de Ciencia e Investigación.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob01543c
Notes and references
- 1.Leloir LF. Science. 1971;172:1299–1303. doi: 10.1126/science.172.3990.1299. [DOI] [PubMed] [Google Scholar]
- 2.Ünligil UM, Rini JM. Curr Opin Struct Biol. 2000;10:510–517. doi: 10.1016/s0959-440x(00)00124-x. [DOI] [PubMed] [Google Scholar]
- 3.Coutinho PM, Deleury E, Davies GJ, Henrissat B. J Mol Biol. 2003;328:307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- 4.Cataldo AM, Broadwell RD. J Neurocytol. 1986;15:511–524. doi: 10.1007/BF01611733. [DOI] [PubMed] [Google Scholar]
- 5.Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. J Neurosci. 2000;20:6804–6810. doi: 10.1523/JNEUROSCI.20-18-06804.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, Martínez-Pons C, Milán M, Guinovart JJ. J Cereb Blood Flow Metab. 2014;34:945–955. doi: 10.1038/jcbfm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallès-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, Serafin A, Cañas X, Soriano E, Delgado-García JM, Gruart A, Guinovart JJ. EMBO Mol Med. 2011;3:667–681. doi: 10.1002/emmm.201100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duran J, Tevy MF, Garcia-Rocha M, Calbó J, Milán M, Guinovart JJ. EMBO Mol Med. 2012;4:719–729. doi: 10.1002/emmm.201200241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latsis T, Andersen B, Agius L. Biochem J. 2002;368:309–316. doi: 10.1042/BJ20021070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fosgerau K, Westergaard N, Quistorff B, Grunnet N, Kristiansen M, Lundgren K. Arch Biochem Biophys. 2000;380:274–284. doi: 10.1006/abbi.2000.1930. [DOI] [PubMed] [Google Scholar]
- 11.Walls AB, Sickmann HM, Brown A, Bouman SD, Ransom B, Schousboe A, Waagepetersen HS. J Neurochem. 2008;105:1462–1470. doi: 10.1111/j.1471-4159.2008.05250.x. [DOI] [PubMed] [Google Scholar]
- 12.Concia AL, Gómez L, Bujons J, Parella T, Vilaplana C, Cardona PJ, Joglar J, Clapés P. Org Biomol Chem. 2013;11:2005–2021. doi: 10.1039/c3ob27343a. [DOI] [PubMed] [Google Scholar]
- 13.Takayama S, Chung SJ, Igarashi Y, Ichikawa Y, Sepp A, Lechler RI, Wu J, Hayashi T, Siuzdak G, Wong CH. Bioorg Med Chem. 1999;7:401–409. doi: 10.1016/s0968-0896(98)00249-1. [DOI] [PubMed] [Google Scholar]
- 14.Compain P, Martin OR. Bioorg Med Chem. 2001;9:3077–3092. doi: 10.1016/s0968-0896(01)00176-6. [DOI] [PubMed] [Google Scholar]
- 15.Errey JC, Lee SS, Gibson RP, Martinez Fleites C, Barry CS, Jung PMJ, O’Sullivan AC, Davis BG, Davies GJ. Angew Chem, Int Ed. 2010;49:1234–1237. doi: 10.1002/anie.200905096. [DOI] [PubMed] [Google Scholar]
- 16.Díaz-Lobo M, Garcia-Amorós J, Fita I, Velasco D, Guinovart JJ, Ferrer JC. Org Biomol Chem. 2015;13:7282–7288. doi: 10.1039/c5ob00796h. [DOI] [PubMed] [Google Scholar]
- 17.Gold AM, Johson RM, Tseng JK. J Biol Chem. 1970;10:2564–2572. [PubMed] [Google Scholar]
- 18.Bisswanger H. Enzyme Kinetics. Principles and Methods. 2. WILEY-VCH Verlag GmbH & Co; KGaA, Weinhem: 2008. p. 301. [Google Scholar]
- 19.Yu M, Magalhaes MLB, Cook PF, Blanchard JS. Biochemistry. 2006;45:14788–14794. doi: 10.1021/bi061621t. [DOI] [PubMed] [Google Scholar]
- 20.Schäfer WE, Rohwer JM, Botha FC. Eur J Biochem. 2004;271:3971–3977. doi: 10.1111/j.1432-1033.2004.04288.x. [DOI] [PubMed] [Google Scholar]
- 21.Qiao L, Murray BW, Shimazaki M, Schultz J, Wong CH. J Am Chem Soc. 1996;118:7653–7662. [Google Scholar]
- 22.Mitchell EP, Withers SG, Ermert P, Vasella AT, Garman EF, Oikonomakos NG, Johnson LN. Biochem J. 1996;35:7341–7355. doi: 10.1021/bi960072w. [DOI] [PubMed] [Google Scholar]
- 23.Gold AM, Legrad E, Sanchez G. J Biol Chem. 1971;246:5700–5706. [PubMed] [Google Scholar]
- 24.Sheng F, Jia X, Yep A, Preiss J, Geiger JH. J Biol Chem. 2009;284:17796–17807. doi: 10.1074/jbc.M809804200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buschiazzo A, Ugalde JE, Guerin ME, Shepard W, Ugalde RA, Alzari PM. EMBO J. 2004;23:3196–3205. doi: 10.1038/sj.emboj.7600324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson RP, Turkenburg JP, Charnock SJ, Lloyd R, Davies GJ. Chem Biol. 2002;9:1337–1346. doi: 10.1016/s1074-5521(02)00292-2. [DOI] [PubMed] [Google Scholar]
- 27.Zheng Y, Anderson S, Zhang Y, Garavito RM. J Biol Chem. 2011;286:36108–36118. doi: 10.1074/jbc.M111.275974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saheki S, Takeda A, Shimazu T. Anal Biochem. 1985;148:277–281. doi: 10.1016/0003-2697(85)90229-5. [DOI] [PubMed] [Google Scholar]
- 29.Schinzel R, Palm D. Biochemistry. 1990;29:9956–9962. doi: 10.1021/bi00494a028. [DOI] [PubMed] [Google Scholar]
- 30.Morell M, Copeland L. Plant Physiol. 1985;78:149–154. doi: 10.1104/pp.78.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.