

Abstract
Background
A reliable blood-based assay is required to properly diagnose and monitor Alzheimer’s disease (AD). Many attempts have been made to develop such a diagnostic tool by measuring amyloid-β oligomers (AβOs) in the blood, but none have been successful in terms of method reliability. We present a multimer detection system (MDS), initially developed for the detection of prion oligomers in the blood, to detect AβOs.
Methods
To characterize Aβ in the blood, plasma was spiked with synthetic amyloid-β (Aβ) and incubated over time. Then, the MDS was used to monitor the dynamic changes of AβO levels in the plasma.
Results
Increasing concentrations of AβOs were observed in the plasma of patients with AD but not in the plasma of normal control subjects. The plasma from patients with AD (n = 27) was differentiated from that of the age-matched normal control subjects (n = 144) with a sensitivity of 83.3% and a specificity of 90.0%.
Conclusions
Synthetic Aβ spiked into the blood plasma of patients with AD, but that of not elderly normal control subjects, induced dynamic changes in the formation of AβOs over time. AβOs were detected by the MDS, which is a useful blood-based assay with high sensitivity and specificity for AD diagnosis.
Keywords: Multimer detection system, Alzheimer’s disease, Amyloid-β, Oligomers, Blood biomarker, Synthetic amyloid-β, ELISA, Plasma test
Background
Amyloid-β (Aβ) is a major factor in the pathogenesis of Alzheimer’s disease (AD) [1–3]. Aβ may be involved in the cognitive impairment of memory that leads to AD, particularly in the form of aggregated 42-amino acid isoform of the Aβ peptide (Aβ42), which is a major neurotoxic species among Aβ isomers, including Aβ40, Aβ42, and other truncated forms of Aβ [4–10]. Since the initial identification of these Aβ isomers in bodily fluids, the measurement of Aβ levels in the blood and cerebrospinal fluid (CSF) has been a research priority [11, 12]. Three biomarkers in CSF, specifically Aβ42, total tau, and hyperphosphorylated tau, are widely accepted as AD determinants on the basis of their close correlations with AD pathology [13, 14]. Diagnostic imaging using positron emission tomography (PET) is the preferred method of investigating pathological and functional changes in the brain of patient with AD. 11C- or 18F-labeled Pittsburgh compound B-positron emission tomography (PiB-PET), which involves binding to amyloid depositions, is particularly beneficial for understanding the underlying processes of AD. Therefore, this type of imaging is used both in research and in the clinic to differentiate patients with AD from control subjects and individuals with other types of dementia [15–17].
Several noninvasive diagnostics for AD, based on diverse biomarkers in the saliva, urine, and blood, have been reported and are still in the research phase of development [18–21]. Mapstone et al. [22] identified a panel of ten plasma phospholipids as potential diagnostic biomarkers of AD, which included lysophosphatidylcholine, phosphatidylcholine metabolites, and acylcarnitine metabolites. This plasma lipid panel predicted AD conversion, suggesting 90% sensitivity and 85% specificity in differentiating an at-risk group from the cognitively intact group. Despite encouraging results, low positive predictive values limited the clinical usefulness of this panel as a screening tool in subjects aged 70–80 years or younger. In another study, significant differences in soluble CD40 (sCD40) and sCD40 ligand (sCD40L) levels in plasma were observed between AD cases and control subjects. sCD40 was approximately three times higher in patients with AD than in control subjects with sensitivity and specificity of 68% and 84%, respectively. Similarly, concentrations of sCD40L were 2.27 times higher in AD cases than in control subjects with sensitivity and specificity of 51% and 76%, respectively [23]. A biomarker panel of cortisol, pancreatic polypeptide, insulin-like growth factor binding protein 2, β2-microglobulin, vascular cell adhesion molecule 1, carcinoembryonic antigen, matrix metalloproteinase 2, CD40, macrophage inflammatory protein 1α, superoxide dismutase, and homocysteine was shown to significantly increase in plasma from patients with AD. In addition, apolipoprotein E (ApoE), epidermal growth factor receptor, hemoglobin, calcium, zinc, interleukin (IL)-17, and albumin were revealed to be decreased in patients with AD. Cross-validated accuracy measures from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL) cohort reached a mean (SD) of 85% (3.0%) for sensitivity and specificity and 93% (3.0) for the AUROC. A second validation using the Alzheimer’s Disease Neuroimaging Initiative cohort showed accuracy measures of 80% (3.0%) for sensitivity and specificity and 85% (3.0) for the AUROC [24]. Eighteen signaling blood proteins in hematopoiesis, immune responses, apoptosis, and neuronal supports were suggested to differentiate patients with AD from control subjects with close to 90% accuracy and also to identify patients who would convert from mild cognitive impairment to AD 2–6 years later [25].
Among these, methods employing blood-based biomarkers have been focused on the detection of amyloid-β oligomers (AβOs) and other surrogate biomarkers of AD [26–31]. Experimental cross-sectional analyses undertaken to detect AβOs in the plasma have demonstrated limited and inconclusive results [26–28]. Other blood-based surrogate biomarkers, including ApoE, inflammatory markers (IL-8, IL-1a), Aβ autoantibodies, total serum cholesterol, and microRNAs (specifically miR-9, miR-29a, miR-29b, miR101, miR-125b, miR-132, miR-134, and miR-181c), have also demonstrated variability as disease correlates [29–31]. Although there have been difficulties in developing methods for AD diagnosis using blood-based biomarkers, a reliable and reproducible blood-based assay is still needed for clinical use [32].
A multimer detection system (MDS) was originally developed to detect prion oligomers in the blood of scrapie-infected animals. MDS is a sandwich enzyme-linked immunosorbent assay (ELISA) that preferentially detects oligomers over monomers by creating steric hindrance between capturing and detection antibodies that are specific to a unique/overlapping epitope [33].
In the present study, MDS for AD was developed to detect AβOs using two different antibodies against the N-terminus of Aβ. Initially, MDS was unable to differentiate AβOs in the blood of patients with AD from those of normal control subjects. Synthetic Aβ was then spiked into the plasma of patients with AD and control subjects. Using MDS, the dynamic changes of AβO formation were detected in the spiked plasma of patients with AD but not in the spiked plasma of control subjects. Therefore, in this study, we evaluated the dynamic changes of AβO levels in the plasma of patients with AD compared with those of normal age-matched control subjects.
Methods
Clinical data
This study was approved by the institutional review board of Seoul National University Bundang and Chung-Ang University Hospital [B-0905-075-003, B-1202-145-003, C2012048(743), C2013142(1102)]. Pooled plasma samples were collected from 11 patients with AD and 9 elderly normal control subjects, and individual plasma samples of 24 patients with AD and 29 healthy elderly normal control subjects were collected from either Seoul National University Bundang Hospital or Chung-Ang University Hospital (Table 1). Written informed consent was obtained from all patients who participated in this study or from their caregivers. AD cases were each diagnosed with a probable AD amnestic type on the basis of clinical criteria of the National Institute on Aging-Alzheimer’s Association workgroups within a clinical setting with clinical data and follow-up longer than 6 month before inclusion into PiB-PET or CSF studies. Hence, the recruited patients were clinically well-characterized patients with AD, and only they were included in the study. They were diagnosed with AD after initial workup and had not shown any possibility of other neurodegenerative disorders except AD or secondary dementia disorders on the basis of more than 6 months of follow-up. The Mini Mental State Examination, identification of the ApoE phenotype, PET imaging with PiB and 18F-fluorodeoxyglucose, and CSF analysis were performed. The characteristics of all participants are described in Table 1. Fifty-one additional plasma samples from elderly normal control subjects were included to avoid false positivity (Table 2).
Table 1.
Characterization of patients with Alzheimer’s disease and healthy normal control subjects
| AD | Healthy normal control subjects | |
|---|---|---|
| Total sample number | 24 | 29 |
| Sex | ||
| Female | 13 (54.2%) | 16 (55.2%) |
| Male | 11 (45.8%) | 13 (44.8%) |
| Age, years (SD) | 67.6 (±7.4) | 62.4 (±5.7) |
| Education, years (SD) | 13.1 (±3.9) | 13.2 (±3.5) |
| CDR-SOB, mean | 6.35 | 0.03 |
| MMSE score, mean | 17.7 | 29.03 |
| ApoE ε4, % | 47.8 | 21.7 |
| Note test | 1 | 0 |
| Number of plasma samples | 24 | 29 |
| CSF markers | 23 | 28 |
| Aβ42, pg/ml, mean (SD) | 258.6 (±70.8) | 464.8 (±114.4) |
| p-Tau, pg/ml, mean (SD) | 58.6 (±18.6) | 28.0 (±14.3) |
| t-Tau, pg/ml, mean (SD) | 132.1 (±61.8) | 62.1 (±20.3) |
| PiB-PET number | 23 | 28 |
| Mean SUVR | 1.57 | 1.14 |
| FDG-PET number | 18 | 28 |
| Mean SUVR | 0.9 | 1.06 |
Abbreviations: Aβ 42 Amyloid-β 1–42 peptide, AD Alzheimer’s disease, ApoE Apolipoprotein E, CDR-SOB Clinical Dementia Rating Sum of Boxes, CSF Cerebrospinal fluid, FDG 18F-fluorodeoxyglucose, MMSE Mini Mental State Examination, PET Positron emission tomography, PiB 11C-Pittsburgh compound B, p-Tau Phosphorylated tau protein, SUVR Standardized uptake value ratio, t-Tau Total tau protein
Table 2.
Supplementary information on healthy normal control subjects
| Healthy normal control subjects | |
|---|---|
| Total sample number | 51 |
| Sex | |
| Female | 21 (43.1%) |
| Male | 29 (56.9%) |
| Age, years (SD) | 62.25 (±7.89) |
| Education, years (SD) | 10.35 (±3.29) |
| CDR-SOB | 0.03 |
| MMSE score | 28.27 |
CDR-SOB Clinical Dementia Rating Sum of Boxes, MMSE Mini Mental State Examination
Sample preparation
Blood samples were collected in heparin-containing tubes and centrifuged at 850 × g for 30 minutes. The plasma (supernatant) was divided into aliquots and stored at −80 °C until analysis. Plasma samples of patients with AD (n = 11) and elderly normal control subjects (n = 9) were separately pooled for initial method optimization, whereas the remaining samples were assessed individually.
Preparation of synthetic Aβ42
Lyophilized AggreSure β-Amyloid (1–42) peptide (AnaSpec, Fremont, CA, USA) and double-mutant F19S/L34P Aβ42 (mutAβ42; AnyGen Co., Ltd., Gwangju, South Korea) were each dissolved in 50 mM Tris/150 mM NaCl (pH 7.2) at a concentration of 1 mg/ml and then sonicated for 5 minutes to obtain a homogeneous solution. The peptide solution was further diluted with phosphate-buffered saline containing Tween 20 (PBST; Sigma-Aldrich, St. Louis, MO, USA) to a desired concentration of 10 μg/ml. Solutions of diluted peptides were divided into aliquots and kept at −80 °C until further use.
Thioflavin T assay
Aβ aggregation was monitored using a thioflavin T (ThT) assay kit following the suggested protocol of the manufacturer (AnaSpec). Ninety microliters of test sample and 10 μl of 2 mM ThT solution were added to each well of a 96-well plate (Thermo Fisher Scientific, Waltham, MA, USA), and the plates were incubated for different lengths of time. Then, changes in ThT fluorescence intensity were detected by measuring excitation and emission wavelengths of 440 nm and 484 nm, respectively, using a multispectrophotometer (Victor 3TM; PerkinElmer, Waltham, MA, USA) with 15 seconds of shaking before reading and analysis.
TEM
AβO, protofibrils, and fibrils were characterized by TEM at various incubation times (0, 1, 3, 6, 24, and 48 h). Five microliters of each sample was applied to carbon-coated TEM grids that had previously been glow-discharged for 3 minutes in the air and immediately negatively stained (~5 seconds) with 2% uranyl acetate. Excess solution was removed with blotting paper. Image acquisition was carried out using a Philips CM10 transmission electron microscope (Philips Research, Eindhoven, The Netherlands) with an accelerating voltage of 80 kV.
Sodium dodecyl sulfate-PAGE and immunoblotting
The aggregation state of Aβ was also analyzed by sodium dodecyl sulfate-PAGE followed by Western blotting. Synthetic peptide samples were electrophoresed on a 10–20% Tris-Tricine precast gel (Bio-Rad Laboratories, Hercules, CA, USA) and visualized by Coomassie blue staining (Bio-Rad Laboratories). After electrophoresis, the proteins were transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories), which was blocked with 2% Block Ace (Bio-Rad Laboratories) in Tris-buffered saline containing Tween 20 (TBST; Sigma-Aldrich) for 1 h at room temperature (RT) under conditions to reduce nonspecific binding. The membrane was incubated for 1 h at RT with a horseradish peroxidase (HRP)-conjugated FF51 antibody (FF51-HRP antibody; PeopleBio Inc., Seoul, South Korea) diluted in 0.4% Block Ace in TBST. Proteins bound to the antibody were visualized with 3,3′,5,5′-tetramethylbenzadine reagent (Sigma-Aldrich).
MDS for Alzheimer’s disease
A modified MDS was used to measure AβOs. With this method, epitope-overlapping antibodies specific for the N-terminus of Aβ were used to capture and detect the Aβ antigen in its multimeric or oligomeric form. Because MDS was initially developed to detect prion oligomers using prion antibodies, over 100 sets of antibodies against Aβ were screened (data not shown). In addition, in-house Aβ antibodies were developed. The mouse monoclonal antibody 6E10 (BioLegend, San Diego, CA, USA) and an in-house FF51-HRP antibody were chosen to detect AβOs in our modified MDS, owing to their sensitivity and specificity. The epitopes for these antibodies overlap at the N-terminus of Aβ. The FF51 antibody specifically recognizes amino acid residues 1–4 of Aβ.
To use MDS, the 6E10 antibody was coated overnight at 4 °C in the wells of a 96-well black plate (Thermo Fisher Scientific) at a dilution of 3 μg/ml in carbonate-bicarbonate buffer (Sigma-Aldrich). The plates were blocked for 2 h with 0.4% Block Ace (100 μl) at RT. After washing three times with PBS (Sigma-Aldrich), the plate was stored at 4 °C until use. Prior to the assay, aliquots of plasma samples were thawed at 37 °C for 15 minutes. Ten microliters of plasma, 4.04 μl of HBR-1, a HAMA blocker (Scantibodies Laboratory, Santee, CA, USA), and PBST were mixed. We spiked the synthetic Aβ42 into plasma mixture and incubated it at 37 °C for the indicated durations.
The plasma sample mixture and serially diluted standards were added to each well of the plate in a total volume of 100 μl. The plates were incubated at RT for 1 h. After washing three times with TBST, the FF51-HRP antibody in TBST containing 0.4% Block Ace was added to the wells, and the plate was incubated for 1 h at RT. To increase the sensitivity of detection, 100 μl/well of enhanced chemiluminescence substrate solution (Rockland Immunochemicals Inc., Limerick, PA, USA) was used, and the luminescent signal was detected and quantified using a Victor 3TM multispectrophotometer.
Measurement of Aβ monomers (Aβ40 and Aβ42)
Sandwich ELISAs were performed to measure Aβ40 and Aβ42 monomer levels. Aβ40 monomers were captured with the 11A50 antibody (specific for the C-terminus of Aβ40) and detected with the 1E11 antibody conjugated to biotin. Aβ42 monomers were captured with the 12F4 antibody (specific for the C-terminus of Aβ42) and detected with the 1E11 antibody conjugated to biotin.
Statistics
Statistical evaluations were performed using the Mann-Whitney U test followed by the calculation of two-tailed p values to determine the significance between groups.
Results
Measuring dynamic changes of AβO levels with MDS
Aβ42 was characterized by gel electrophoresis, Western blotting, and TEM before it was spiked into plasma samples (Fig. 1). On the basis of Coomassie blue staining of dissolved Aβ42, a smear band containing monomers and low-molecular-weight oligomers ranging between 4 and 18 kDa in size was detected, as shown in Fig. 1a. A double-mutant, Aβ42 (F19S/L34P; mutAβ42), was used as a monomeric Aβ control because this mutant has significantly reduced aggregation potential as shown by Western blotting, which yielded a specific band with an approximate molecular weight of 4–5 kDa. The specificities of the wild-type and mutant monomer bands were verified by MDS, as shown in Fig. 1b. MDS was capable of detecting AβOs composed of Aβ42 in a concentration-dependent manner employing half serial dilutions from 100 ng to 3.13 ng, whereas no signal was detected when using mutAβ42. Thus, MDS specifically recognizes AβOs but not Aβ monomers.
Fig. 1.
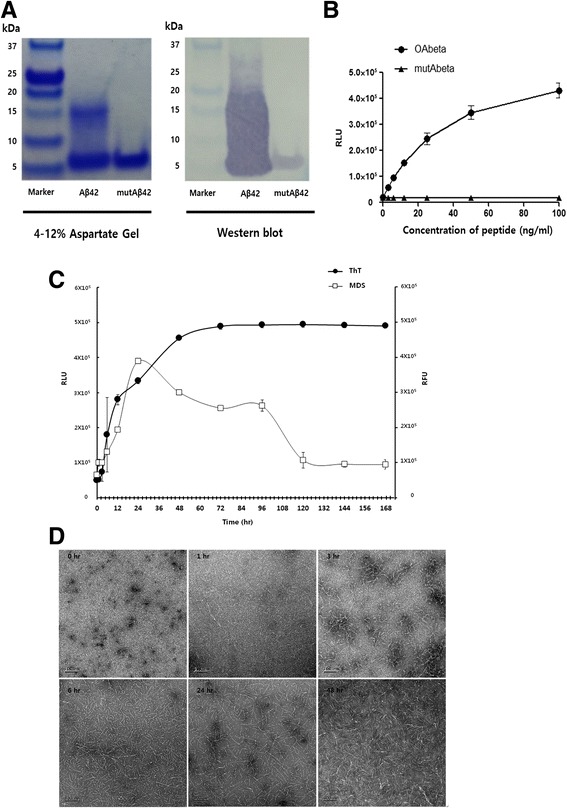
Detection of synthetic amyloid-β 1–42 peptide (Aβ42). a Freshly dissolved synthetic Aβ42 and double-mutant Aβ42 (mutAβ42) were visualized on the 4–12% aspartate PAGE gel by Coomassie blue staining and on the Western blot by FF51-horseradish peroxidase (HRP) antibody. b Standard curve for multimer detection system (MDS). Synthetic peptides were serially diluted and measured by 6E10/FF51-HRP MDS assay. Change of Aβ42 over different times (c and d). Aβ42 was incubated at 37 °C at the indicated time points. To measure amyloid-β oligomer (AβO), an MDS assay was used (c), and fibrillization of Aβ42 over time was measured by thioflavin T (ThT) binding assay (d). TEM images over time by Aβ42 in buffer vehicle are shown. Data are mean ± SD. RLU Relative luminescence units, RFU Relative fluorescence units
As shown in Fig. 1c, changes in Aβ42 oligomer levels over an incubation period of 144 h were monitored using an MDS and the ThT assay. The MDS detected a continual increase in Aβ42 oligomer levels from 0 to 24 h after the start of incubation, followed by a decline until 120 h, at which point the levels remained relatively stable. Conversely, the ThT assay showed an increase in Aβ42 oligomer levels from 0 to 48 h, at which point changes in β-sheet formation were observed. TEM (Fig. 1d) revealed a wide range (1–5 nm) of AβO diameters, with few protofibrils observed. These findings support the MDS results obtained at the start of incubation (0 h) in terms of the formation of AβOs. Within 1 h after the start of incubation, the Aβ42 monomers readily formed large, spherical AβOs ranging from 10 to 15 nm in size, and numerous protofibrils were observed (lengths of 50–80 nm). A TEM image at 3 h revealed the elongation of AβOs to form protofibrils, and significant amounts of large AβOs and protofibrils were observed at 6 h by TEM. A substantial decline in the MDS signal was observed during the time interval of 24–48 h after the start of incubation. TEM revealed the predominance of protofibrils and fibrils (over 120 nm in length) at 24 h, whereas AβOs were rarely observed, and the continuous maturation of protofibrils resulted in an increase in Aβ42 fibrils at 48 h. On the basis of these findings, the MDS sensitively and specifically detects oligomeric and protofibril forms of Aβ, permitting their quantification, whereas the ThT assay is not sensitive and was incapable of detecting increases in Aβ fibril levels, including diverse types of amyloid fibrils [34].
Plasma is cleared of Aβ through several intricate mechanisms of aggregation or sequestration [35–38]. Therefore, pooled samples of plasma from patients with AD or from normal control subjects were spiked with different concentrations of Aβ42 to compare differences in Aβ42 recovery (Fig. 2a and b). AβO levels were reduced in accordance with the Aβ42 concentrations used for spiking when compared with AβO levels in a solution spiked with buffer. Lower concentrations of spiked Aβ42 yielded smaller differences in the formation of AβOs in the pooled plasma of patients with AD compared with that of control subjects. For subsequent experiments, 10 ng/ml Aβ42 was chosen for spiking into plasma because this concentration yielded the smallest measurable difference in the recovery rate of Aβ when comparing plasma from patients with AD with that of normal control subjects. Eleven plasma samples from patients with AD and nine from elderly normal control subjects were separately pooled for each group. Pooled samples were incubated at 37 °C after spiking with Aβ42 (10 ng/ml), and the presence of oligomers was measured using MDS at various time points after the start of incubation. As shown in Fig. 2b, both groups exhibited a gradual decline in oligomer levels over 48 h of incubation; the two groups demonstrated similar levels over this time period. After 48 h of incubation, distinct dynamic changes were observed between the plasma from patients with AD and that of elderly normal control subjects. Larger increases in AβO levels were observed in the plasma from patients with AD after 48 h of incubation, and levels continually increased throughout the rest of the incubation period. In contrast, AβO levels in the plasma from elderly normal control subjects gradually decreased until 72 h after the start of incubation, then rebounded with a considerable increase until 144 h. The largest differences in AβO levels between plasma from patients with AD and plasma from elderly normal control subjects were observed after 144 h of incubation following spiking with Aβ. Changes in Aβ40 and Aβ42 levels after spiking with Aβ42 were also measured by performing 11A50/1E11-biotin ELISA and 12F4/1E11-biotin ELISA. Aβ40 and Aβ42 levels remained relatively unchanged over the incubation period (Fig. 2c and d), whereas significant increases in the oligomer forms, as measured by MDS, were observed in the plasma from patients with AD after 48 h of incubation but not in plasma from elderly normal control subjects. Additional experiments were then performed to confirm whether the differential changes in the Aβ forms were discernible in individual plasma samples.
Fig. 2.
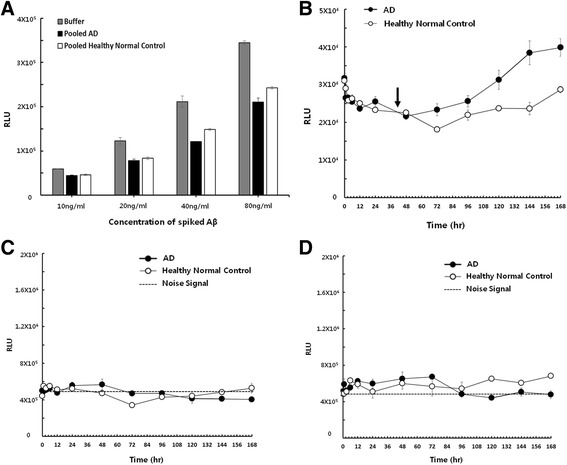
Changes of amyloid-β oligomers (AβOs) and amyloid-β (Aβ) monomers in pooled plasma of patients with Alzheimer’s disease (AD) and healthy normal control subjects. a After being spiked with different concentrations of Aβ42 in buffer vehicle, pooled AD plasma, and pooled healthy normal control plasma, oligomers were measured using a multimer detection system (MDS) at 0 h. b Pooled plasma samples were incubated at 37 °C after being spiked with 10 ng/ml Aβ42. Changes of Aβ42 levels at various time points were measured by MDS. Levels of Aβ40 (c) and Aβ42 (d) after being spiked with 10 ng/ml Aβ42 in pooled human plasma. Pooled plasma samples were incubated at 37 °C after 10 ng/ml Aβ42 were spiked. Data are mean ± SD. RLU Relative luminescence units. Arrow indicated no change in oligomer levels after an incubation period of 24 h
Plasma samples corresponding to individuals included in the pooled groups (patients with AD and elderly normal control subjects) were spiked with Aβ42. Then, MDS was used to monitor changes in AβO levels during incubation from 0 to 144 h (Fig. 3a and b). Each sample was also tested in the absence of synthetic Aβ for comparison. As shown in Fig. 3b1, AβO levels in plasma samples from patients with AD and elderly normal control subjects overlapped without a significant difference (p = 0.6761) at 0 h, regardless of spiking. In contrast, after 144 h of incubation, distinct oligomer levels were detected in Aβ42-spiked samples from the AD and control groups (p < 0.01) on the basis of MDS measurements (Fig. 3b2). Plasma samples from the AD group also demonstrated higher AβO levels than normal control plasma, although this difference was significant but the p value was < 0.05 (Fig. 3a2). Similar to the results of the pooled plasma experiment, Aβ dynamics were evident from 48 to 144 h in individual samples from both groups. As shown in Fig. 3c, substantial increases in oligomer levels were confirmed in the majority of plasma samples from patients with AD, whereas samples from normal control subjects demonstrated no significant increase. No increases in oligomer levels were observed with synthetic Aβ42 in buffer solution after an incubation time of 24 h.
Fig. 3.
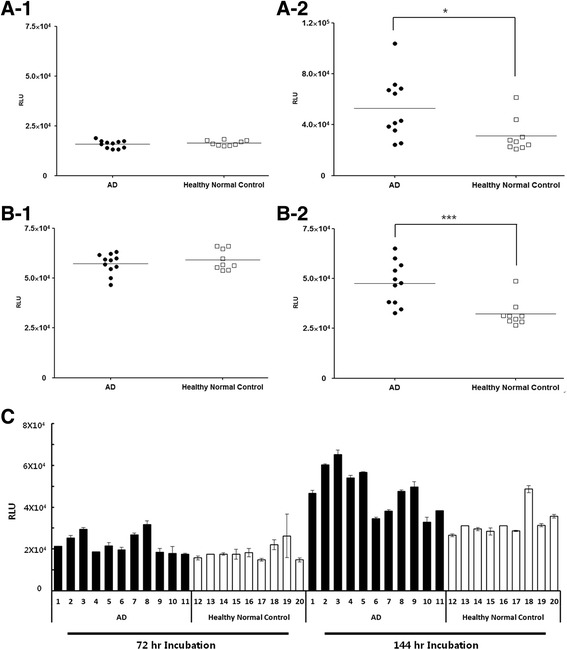
Effect of amyloid-β 1–42 peptide (Aβ42) spiking and incubation in individual subjects (Alzheimer’s disease [AD] and healthy normal control subjects). Each sample was incubated under the indicated conditions. To measure amyloid-β oligomers, a multimer detection system (MDS) assay was used. (a1) 0 ng/ml Aβ42 at 0 h, (a2) 0 ng/ml Aβ42 at 144 h, (b1) 10 ng/ml Aβ42 at 0 h, and (b2) 10 ng/ml Aβ42 at 144 h. (c) Dynamic changes of Aβ42 incubation in individual subjects at 72 h and 144 h. Each sample was incubated for 72 h and 144 h after being spiked with 10 ng/ml Aβ42 and measured by MDS. *p < 0.05; ** p < 0.01. RLU Relative luminescence units
Differential dynamic changes in Aβ levels in plasma of patients with AD versus that of elderly normal control subjects
To evaluate and verify our findings on a larger scale, 24 plasma samples from clinically well-characterized cases of AD and 80 from elderly normal control subjects were examined after spiking with synthetic Aβ (10 ng/ml) and incubating for 144 h. The dynamic changes of oligomer formation were measured using MDS, and oligomer levels were different between the AD and control groups with a sensitivity of 83.33%, a specificity of 90.00%, an AUC of 0.8969, and a p value < 0.0001 (Fig. 4).
Fig. 4.
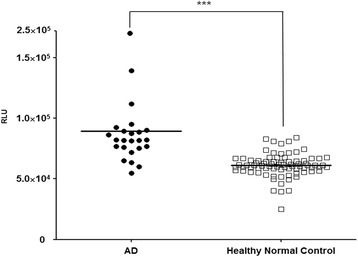
Effect of amyloid-β 1–42 peptide (Aβ42) spiking and incubation in large scale (Alzheimer’s disease [AD] and healthy normal control subjects). Each sample was incubated for 144 h after being spiked with 10 ng/ml Aβ42. To measure amyloid-β oligomers, a multimer detection system assay was used. ***p < 0.001 versus healthy normal control values. RLU Relative luminescence units
Discussion
MDS was initially developed to detect prion disease. This method consists of a sandwich ELISA that exclusively detects oligomeric forms of antigens and relies on two different epitope-overlapping antibodies to capture and detect antigens by creating steric hindrance over a specific epitope [33, 39]. MDS was modified to detect AβOs for AD diagnosis using two epitope-overlapping Aβ antibodies specific for the N-terminus amino acids 1–18 of Aβ42.
We monitored the formation of AβO, protofibrils, and fibrils from synthetic Aβ42 in PBST buffer employing two methods: a ThT assay and MDS (Fig. 1). Aβ oligomerization was closely correlated between these two assays. When the time-course changes of synthetic Aβ in PBST buffer were monitored by MDS, ThT assay, and TEM, the MDS detected a continuous increase of Aβ42 oligomer levels from 0 to 24 h from the start of incubation, followed by a decline until 120 h. Conversely, an increase of fluorescent signal in the ThT assay was correlated with an increase of Aβ42 fibril formation, which was confirmed by TEM imaging (Fig. 1c and d). The MDS signal decreased at that time of fibrillization; thus, it would be reasonable to assume that MDS detected specifically oligomeric and protofibril forms of Aβ. During the early phase of incubation, MDS revealed decreasing AβO levels accompanied by the formation of protofibrils and fibrils, but the levels detected by the ThT assay were stable. Thus, MDS is able to detect AβOs during the early stage of Aβ oligomerization. Decreasing AβO levels, as measured by MDS, in conjunction with the formation of protofibrils and fibrils is potentially explained by two hypotheses. First, MDS has a higher sensitivity for oligomers than for protofibrils or fibrils. Second, the total number of oligomers decreases over time. After full oligomerization, the spiked Aβ converts to protofibrils, resulting in decreased MDS signals.
When synthetic Aβ42 is spiked into plasma, the plasma composition may dictate and interfere with Aβ oligomerization. Difficulties in detecting spiked synthetic Aβ42 via routine ELISA may be attributable to the presence of many different interfering factors in the plasma, which bind to the spiked synthetic Aβ42 and thus reduce its detection. These factors include naturally occurring Aβ autoantibodies, albumin, fibrinogen, immunoglobulin, ApoJ, ApoE, transthyretin, α2-macroglobulin, serum amyloid P component, plasminogen, and amylin [35–38, 40]. In addition, these molecules in bodily fluids could also inhibit Aβ fibrillization. Our hypothesis was that the composition of those components in blood from patients with AD would be different from that in healthy control subjects. If the same amount of Aβ were spiked into AD and control plasma samples, a different phenomenon would be observed between the two groups.
In this study, when equal amounts of synthetic Aβ42 were spiked into plasma, MDS signals for AβOs declined from 0 to 48 h in both groups (Fig. 2b), potentially due to AβO binding to interfering factors. Although binding affinities and Aβ epitopes likely vary among different binding factors, high concentrations in the plasma would likely result in the scavenging of spiked synthetic Aβ42. Binding of these factors to Aβ may naturally influence the normal functions and sequestration of Aβ, leading to clearance and reduced oligomerization potential in elderly normal persons [35, 37].
Forty-eight hours after spiking with Aβ42, AβO levels measured by MDS began to increase in the plasma from patients with AD but not in that of normal control subjects (Fig. 2b). Patients with AD may exhibit different binding profiles based on their plasma composition in the context of spiked synthetic Aβ42, which in turn may increase oligomerization potential and decrease sequestration capacity. Alternatively, the characteristics of endogenous plasma Aβ may differ between the two groups, permitting the dynamic changes of Aβ oligomerization to be detected.
It is challenging work to detect crude AβOs in plasma because the concentrations of Aβ in blood are very low. Furthermore, the concentrations of AβOs would be a subset of total Aβ in blood. The size of 4.5 kDa could be another reason why the MDS failed to measure the endogenous plasma Aβ from patients with AD. As shown in Fig. 3, the MDS did not discriminate between patients with AD and healthy normal control subjects without incubation conditions. Even though incubation of plasma samples without spiking external Aβ made slight differences between AD and normal control samples, the difference was not significant.
However, even at ultralow concentrations, the formation of AβO in the blood of patients with AD may be initiated via incubation with spiked synthetic Aβ42. The first 48 h of incubation represent a slow nucleation-dependent oligomerization phase during which ultralow concentrations of AβO nuclei are required to bind to spiked synthetic Aβ42. The period after 48 h and up to 144 h represents a rapid-growth phase for the formation of oligomers, protofibrils and fibrils, surpassing the critical detection limit [34, 41–43]. As previously mentioned, the interfering factors may be saturated with spiked Aβ; hence, they may not affect to MDS signal even after 48 h of incubation. In addition, the binding interactions between Aβ and another Aβ may be stronger than other interfering factors for oligomerization. In previous studies, by using protein misfolding cyclic amplification technology, researchers were able to differentiate AβO levels by catalyzing the misfolding and amplification of Aβ aggregates by spiking Aβ42 into the CSF of patients with AD and control individuals [44]. However, spiking Aβ into the plasma to differentiate AβO has not previously been published.
We observed the phenomenon that MDS signals of control subjects were still stable while we spiked the same large amount of synthetic peptide into both AD and normal plasma samples, even after the identical incubation step. Currently, we do not know the exact cause of the phenomenon. Oligomerization of Aβ could be influenced by potential factors in plasma of patients with AD but not plasma samples from normal control. The concentrations of these potential factors could be different in the disease state, but they may not be present in the normal state.
We detected differential AβO dynamic changes in the blood of patients with AD and normal control subjects, but a direct correlation between blood and brain pathology remains uncharacterized. The properties of Aβ plaques in the brain may differ from those in the blood because Aβ in the blood also originates from amyloid precursor protein metabolism in skeletal muscle, organs, skin, and peripheral cells [45, 46]. However, on the basis of previous reports, Aβ peptides cross the blood-brain barrier, resulting in elevated Aβ levels in the CSF and plasma during intracerebroventricular injection of synthetic Aβ42 monomers into normal imprinting control region mice [47, 48]. It will be interesting to identify the correlation between AβO concentrations in the plasma and amyloid plaque deposition in the brains of patients with AD.
Conclusions
Spiked synthetic Aβ42 induced differential dynamic changes in AβO levels in the plasma of patients with AD compared with that of normal control subjects, as detected by MDS. These observations appear to support our hypothesis that the plasma composition and/or characteristics of endogenous Aβ in patients with AD versus normal healthy persons are different. To our knowledge, there have been no published reports involving the spiking of Aβ into plasma. The characterization of differential Aβ oligomerization dynamic changes may contribute to the development of blood-based biomarkers for AD. However, further studies are required to elucidate the mechanisms underlying the formation of AβOs. Longitudinal studies undertaken during the predementia stage of AD should also be carried to assess clinical applications for the early detection and monitoring of this disease.
Funding
This work was supported by grants from the Korean Health Technology R&D Project (HI14C3331) through the Korean Health Industry Development Institute (KHIDI), the Korean Ministry of Health & Welfare, and the National Research Foundation of Korea (NRF-2017R1A2B4012636).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Abbreviations
- Aβ
Amyloid-β
- Aβ42
Amyloid-β 1–42 peptide
- AβO
Amyloid-β oligomer
- AD
Alzheimer’s disease
- Apo
Apolipoprotein
- CDR-SOB
Clinical Dementia Rating Sum of Boxes
- CSF
Cerebrospinal fluid
- ELISA
Enzyme-linked immunosorbent assay
- FDG
18F-fluorodeoxyglucose
- HRP
Horseradish peroxidase
- IL
Interleukin
- MDS
Multimer detection system
- miR
MicroRNA
- MMSE
Mini Mental State Examination
- PBST
Phosphate-buffered saline containing Tween 20
- PET
Positron emission tomography
- PiB
11C-Pittsburgh compound B
- p-Tau
Phosphorylated tau protein
- RFU
Relative fluorescence unit
- RLU
Relative luminescence unit
- RT
Room temperature
- sCD40
Soluble CD40
- sCD40L
Soluble CD40 ligand
- SUVR
Standardized uptake value ratio
- TBST
Tris-buffered saline containing Tween 20
- TEM
Transmission electron microscopy
- ThT
Thioflavin T
- t-Tau
Total tau protein
Authors’ contributions
SSAA planned, organized and designed all experiments and results, including the writing of the manuscript. BL planned, organized and designed all experiments and results, including the writing of the manuscript. JSY planned and performed all experiments and results, including the writing of the manuscript. KL planned and performed all experiments and results. GJK planned and performed all experiments and results. RL planned and organized all experiments. SWK planned and organized all experiments. SK planned and organized all experiments. YHP planned and collected clinical samples. MJW planned and collected clinical samples. YSY planned and collected clinical samples. YCY planned and collected clinical samples. SYK planned and organized all experiments and results, collected clinical samples, and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by institutional review board of Seoul National University Bundang Hospital and Chung-Ang University Hospital [B-0905-075-003, B-1202-145-003, C2012048(743), C2013142(1102)]. Written informed consent was obtained from all patients who participated in this study or from their caregivers.
Consent for publication
All authors consented to publication of this paper.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Seong Soo A. An, Email: seongaan@gachon.ac.kr
Byoung-sub Lee, Email: bumb777@gmail.com.
Ji Sun Yu, Email: babyface0216@gmail.com.
Kuntaek Lim, Email: kuntaek@gmail.com.
Gwang Je Kim, Email: gwang.je@gmail.com.
Ryan Lee, Email: ryanlee0612@gmail.com.
Shinwon Kim, Email: swayers@gmail.com.
Sungmin Kang, Email: smkang924@gmail.com.
Young Ho Park, Email: kumimesy@gmail.com.
Min Jeong Wang, Email: wangmj98@gmail.com.
Young Soon Yang, Email: astro76@naver.com.
Young Chul Youn, Email: neudoc@gmail.com.
SangYun Kim, Phone: 82-31-787-7462, Email: neuroksy@snu.ac.kr.
References
- 1.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Jan A, Adolfsson O, Allaman I, Buccarello AL, Magistretti PJ, Pfeifer A, et al. Aβ42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Aβ42 species. J Biol Chem. 2011;286(10):8585–96. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 6.Kittelberger KA, Piazza F, Tesco G, Reijmers LG. Natural amyloid-β oligomers acutely impair the formation of a contextual fear memory in mice. PLoS One. 2012;7(1):e29940. doi: 10.1371/journal.pone.0029940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poling A, Morgan-Paisley K, Panos JJ, Kim EM, O’Hare E, Cleary JP, et al. Oligomers of the amyloid-β protein disrupt working memory: confirmation with two behavioral procedures. Behav Brain Res. 2008;193(2):230–4. doi: 10.1016/j.bbr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci. 2005;8(1):79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira ST, Vieira MN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life. 2007;59(4-5):332–45. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- 10.Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–57. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 11.Kummer MP, Heneka MT. Truncated and modified amyloid-β species. Alzheimers Res Ther. 2014;6(3):28. doi: 10.1186/alzrt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi YJ, Chae S, Kim JH, Barald KF, Park JY, Lee SH. Neurotoxic amyloid β oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow. Sci Rep. 2013;3:1921. doi: 10.1038/srep01921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cedazo-Minguez A, Winblad B. Biomarkers for Alzheimer’s disease and other forms of dementia: clinical needs, limitations and future aspects. Exp Gerontol. 2010;45(1):5–14. doi: 10.1016/j.exger.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–44. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 15.Yang L, Rieves D, Ganley C. Brain amyloid imaging—FDA approval of florbetapir F18 injection. N Engl J Med. 2012;367(10):885–7. doi: 10.1056/NEJMp1208061. [DOI] [PubMed] [Google Scholar]
- 16.Ewers M, Sperling RA, Klunk WE, Weiner MW, Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 2011;34(8):430–42. doi: 10.1016/j.tins.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nordberg A, Rinne JO, Kadir A, Langstrom B. The use of PET in Alzheimer disease. Nat Rev Neurol. 2010;6(2):78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Cui Y, Yang J, Zhang J, Yuan D, Wei Y, et al. Combining serum and urine biomarkers in the early diagnosis of mild cognitive impairment that evolves into Alzheimer’s disease in patients with the apolipoprotein E4 genotype. Biomarkers. 2015;20(1):84–8. doi: 10.3109/1354750X.2014.994036. [DOI] [PubMed] [Google Scholar]
- 19.Ma L, Chen J, Wang R, Han Y, Zhang J, Dong W, et al. The level of Alzheimer-associated neuronal thread protein in urine may be an important biomarker of mild cognitive impairment. J Clin Neurosci. 2015;22(4):649–52. doi: 10.1016/j.jocn.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Ma L, Wang R, Han Y, Sheng S, Zhu J, Ji Z, et al. Development of a novel urine Alzheimer-associated neuronal thread protein ELISA kit and its potential use in the diagnosis of Alzheimer’s disease. J Clin Lab Anal. 2015;30(4):308–14. doi: 10.1002/jcla.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bermejo-Pareja F, Antequera D, Vargas T, Molina JA, Carro E. Saliva levels of Abeta1-42 as potential biomarker of Alzheimer’s disease: a pilot study. BMC Neurol. 2010;10:108. doi: 10.1186/1471-2377-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mapstone M, Cheema AK, Fiandaca MS, Zhong X, Mhyre TR, MacArthur LH, et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med. 2014;20(4):415–8. doi: 10.1038/nm.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ait-ghezala G, Abdullah L, Volmar CH, Paris D, Luis CA, Quadros A, et al. Diagnostic utility of APOE, soluble CD40, CD40L, and Aβ1–40 levels in plasma in Alzheimer’s disease. Cytokine. 2008;44(2):283–7. doi: 10.1016/j.cyto.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC, Lam CP, et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol. 2012;69(10):1318–25. doi: 10.1001/archneurol.2012.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 26.Xia W, Yang T, Shankar G, Smith IM, Shen Y, Walsh DM, Selkoe DJ. A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol. 2009;66(2):190–9. doi: 10.1001/archneurol.2008.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjorkqvist M, Ohlsson M, Minthon L, Hansson O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS One. 2012;7(1):e29868. doi: 10.1371/journal.pone.0029868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang T, Hong S, O’Malley T, Sperling RA, Walsh DM, Selkoe DJ. New ELISAs with high specificity for soluble oligomers of amyloid β-protein detect natural Aβ oligomers in human brain but not CSF. Alzheimers Dement. 2013;9(2):99–112. doi: 10.1016/j.jalz.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyman BT, Smith C, Buldyrev I, Whelan C, Brown H, Tang MX, Mayeux R. Autoantibodies to amyloid-β and Alzheimer’s disease. Ann Neurol. 2001;49(6):808–10. doi: 10.1002/ana.1061. [DOI] [PubMed] [Google Scholar]
- 30.Irizarry MC. Biomarkers of Alzheimer disease in plasma. NeuroRx. 2004;1(2):226–34. doi: 10.1602/neurorx.1.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thambisetty M, Lovestone S. Blood-based biomarkers of Alzheimer’s disease: challenging but feasible. Biomark Med. 2010;4(1):65–79. doi: 10.2217/bmm.09.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hampel H, Shen Y, Walsh DM, Aisen P, Shaw LM, Zetterberg H, et al. Biological markers of amyloid β-related mechanisms in Alzheimer’s disease. Exp Neurol. 2010;223(2):334–46. doi: 10.1016/j.expneurol.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An SSA, Lim KT, Oh HJ, Lee BS, Zukic E, Ju YR, et al. Differentiating blood samples from scrapie infected and non-infected hamsters by detecting disease-associated prion proteins using multimer detection system. Biochem Biophys Res Commun. 2010;392(4):505–9. doi: 10.1016/j.bbrc.2010.01.053. [DOI] [PubMed] [Google Scholar]
- 34.Biancalana M, Koide S. Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta. 2010;1804(7):1405–12. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang YJ, Zhou HD, Zhou XF. Clearance of amyloid-β in Alzheimer’s disease: progress, problems and perspectives. Drug Discov Today. 2006;11(19-20):931–8. doi: 10.1016/j.drudis.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Sehlin D, Söllvander S, Paulie S, Brundin R, Ingelsson M, Lannfelt L, et al. Interference from heterophilic antibodies in amyloid-β oligomer ELISAs. J Alzheimers Dis. 2010;21(4):1295–301. doi: 10.3233/JAD-2010-100609. [DOI] [PubMed] [Google Scholar]
- 37.Kuo YM, Emmerling MR, Lampert HC, Hempelman SR, Kokjohn TA, Woods AS, et al. High levels of circulating Aβ42 are sequestered by plasma proteins in Alzheimer’s disease. Biochem Biophys Res Commun. 1999;257(3):787–91. doi: 10.1006/bbrc.1999.0552. [DOI] [PubMed] [Google Scholar]
- 38.Kuo YM, Kokjohn TA, Kalback W, Luehrs D, Galasko DR, Chevallier N, et al. Amyloid-β peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem Biophys Res Commun. 2000;268(3):750–6. doi: 10.1006/bbrc.2000.2222. [DOI] [PubMed] [Google Scholar]
- 39.Lim K, Kim SY, Lee B, Segarra C, Kang S, Ju Y, et al. Magnetic microparticle-based multimer detection system for the detection of prion oligomers in sheep. Int J Nanomed. 2015;10(Spec Iss):241–50. doi: 10.2147/IJN.S88377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ono K, Noguchi-Shinohara M, Samuraki M, Matsumoto Y, Yanase D, et al. Blood-borne factors inhibit Alzheimer’s β-amyloid fibril formation in vitro. Exp Neurol. 2006;202(1):125–32. doi: 10.1016/j.expneurol.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 41.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73(6):1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 42.Ghosh P, Kumar A, Datta B, Rangachari V. Dynamics of protofibril elongation and association involved in Aβ42 peptide aggregation in Alzheimer’s disease. BMC Bioinform. 2010;11(Suppl 6):S24. doi: 10.1186/1471-2105-11-S6-S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeong JS, Ansaloni A, Mezzenga R, Lashuel HA, Dietler G. Novel mechanistic insight into the molecular basis of amyloid polymorphism and secondary nucleation during amyloid formation. J Mol Biol. 2013;425(10):1765–81. doi: 10.1016/j.jmb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C. Detection of misfolded Aβ oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell Rep. 2014;7(1):261–8. doi: 10.1016/j.celrep.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 45.Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, et al. Amyloid β peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 2009;5(1):18–29. doi: 10.1016/j.jalz.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arai H, Lee VM, Messinger ML, Greenberg BD, Lowery DE, Trojanowski JQ. Expression patterns of β-amyloid precursor protein (β-APP) in neural and nonneural human tissues from Alzheimer’s disease and control subjects. Ann Neurol. 1991;30(5):686–93. doi: 10.1002/ana.410300509. [DOI] [PubMed] [Google Scholar]
- 47.Clifford PM, Zarrabi S, Siu G, Kinsler KJ, Kosciuk MC, Venkataraman V, et al. Aβ peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain Res. 2007;1142:223–36. doi: 10.1016/j.brainres.2007.01.070. [DOI] [PubMed] [Google Scholar]
- 48.Cho SM, Kim HV, Lee S, Kim HY, Kim W, Kim TS, et al. Correlations of amyloid-β concentrations between CSF and plasma in acute Alzheimer mouse model. Sci Rep. 2014;4:6777. doi: 10.1038/srep06777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.